

Abstract
Alzheimer’s disease (AD) is a protein misfolding disease. Early hypothesis of AD pathology posits that 39–43 AA long misfolded amyloid beta (Aβ) peptide form a fibrillar structure and induce pathophysiological response by destabilizing cellular ionic homeostasis. Loss of cell ionic homeostasis is believed to be either indirectly due to amyloid beta-induced oxidative stress or directly by its interaction with the cell membrane and/or activating pathways for ion exchange. Significantly though, no Aβ specific cell membrane receptors are known and oxidative stress mediated pathology is only partial and indirect. Most importantly, recent studies strongly indicate that amyloid fibrils may not by themselves cause AD pathology. Subsequently, a competing hypothesis has been proposed wherein amyloid derived diffusible ligands (ADDLs) that are large Aβ oligomers (~>60kD), mediate AD pathology. No structural details, however, of these large globular units exist nor is there any known suitable mechanism by which they would induce AD pathology. Experimental data indicate that they alter cell viability by non-specifically changing the plasma membrane stability and increasing the overall ionic leakiness. The relevance of this non-specific mechanism for AD-specific pathology seems limited. Here, we provide a viable new paradigm: AD pathology mediated by amyloid ion channels made of small Aβ oligomers (trimers to octamers). This review is focused to 3D structural analysis of the Aβ channel. The presence of amyloid channels is consistent with electrophysiological and cell biology studies summarized in companion reviews in this special issue. They show ion channel-like activity and channel-mediated cell toxicity. Amyloid ion channels with defined gating and pharmacological agents would provide a tangible target for designing therapeutics for AD pathology.
Keywords: Amyloid beta, Ion Channel, 3D structure, membrane associated conformations, channel mediated cell degeneration
1. Ion Channel hypothesis
Alzheimer’s disease (AD) belongs to a class of degenerative diseases commonly referred to as the protein misfolding disease that includes many other neurodegenerative diseases (such as Huntington’s, Parkinson’s, familial British and Danish Dementia’s) as well as certain systemic diseases (such as type II Diabetes and amyloidosis). A common feature of these protein misfolding diseases is that an abnormal folding of peptides favors conversion of these proteins from a native, often soluble form, to a nonnative, often insoluble structure [1–4]. Consistent with this scenario, the main pathological hallmark in AD brain is the progressive deposition of amyloid plaques: the bundles of amorphous Aβ fibers. One of the original dogmas regarding the protein misfolding (or conformational) diseases is that these fibrillar Aβ (that make the amyloid plaques) induce pathophysiological response due to activation of a signaling pathway that subsequently destabilize cellular ionic homeostasis. On the other hand, recent studies lead us to believe that these amyloid plaques might not be the cause of the disease [5–13] - amyloid toxicity could be reproduced without the presence of any amyloid fibers but by only small or large oligomeric amyloids. Also, in amyloid animal models, there are AD-like symptoms without any significant level of amyloid plaques.
Large oligomeric amyloids have attracted considerable attention recently and a new dogma has been proposed wherein large amyloid oligomers that are primarily the Amyloid derived diffusible ligands (ADDLs) are responsible for amyloid-induced toxicity [9]. These large amyloid oligomers have been isolated using antibodies made against ADDLs; and these antibodies by themselves have been effective in prevention of amyloid cell toxicity. There are certain caveats with this line of work. First of all, as their name implies, they are the diffusible “ligands” and yet there is no specific “receptor”, let alone the cell membrane receptors. For these ADDLs no specific cell receptors have been identified. Second, it is unclear, how these large, most likely unstructured, globs of amyloid would selectively alter cell membrane permeability. Recent biophysical studies on these ADDLs indicate that they allow non-specific movement of ions across lipid bilayers [14] by thinning the membrane and altering the dielectric properties. This appears to be something like an amyloid hammer thinning the plasma membrane by its shear weight when it interacts with the membrane. Third, it will be extremely difficult to rule out the presence of small oligomers in the ADDL preparations, in spite of their purification using anti-ADDL antibody and filtration of larger oligomers and fibers. ADDLs are formed from a prolonged incubation of small oligomeric peptides.
Small oligomeric amyloids (e.g., monomeric to octameric amyloid beta complex) have also been shown to reproduce amyloid toxicity that were previously thought to be due to amyloid fibers and plaques [3, 6, 8, 11–13]. Our study as well as studies by many other investigators indicate that Aβ undergo conformational changes during its interaction with cell membrane such that they assume specific functional properties consistent with an ion channel and form specific 3D channel-like structures supporting the channel activity. These ion channels which normally favor cationic transport would destabilize the cell ionic homeostasis. This loss of ionic homeostasis predominantly enhances cellular calcium concentration that in turn would induce cell toxicity. Significantly, it is possible that large oligomers (ADDLs) are made of small oligomers (trimers, tetramers, hexamers, and like) and these small oligomers have channel-like activity and structural feature, thus reconciling the two dogmas.
Based on results from abovementioned recent studies and the discussions to follow in this article, on the possibility that small amyloid oligomers form ion channels, we present an overall amyloid channel hypothesis. Figure 1 summarizes the key feature of this hypothesis. Briefly, Aβ, a 39–43 amino acid long peptide, when released in the medium after its cleavage from the single transmembrane spanning precursor called amyloid precursor protein (APP), undergo multi-step oligomerization, ranging from the formation of small oligomers to large oligomers (commonly referred to as the ADDLs) to protofibrils to fibrils and plaques. All amyloid forms have been reported to alter synaptic information processing and induce neurotoxicity. A common denominator for amyloid beta AD toxicity is the gain of intracellular calcium, the severity of toxicity depends upon the level of calcium loading as well as the cell’s defense capability. How these various amyloid forms induce pathophysiological responses is the matter of intense research. Several mechanisms for Aβ neurotoxic activity have been proposed, such as the activation of a signaling pathway by extracellular aggregates (fibrils, plagues), induction of oxidative stress due to protein aggregates, or recruitment of factors by intracellular aggregates. In some instances, these amyloids, especially large amyloid complexes and amyloid fibers, might not interact with cell membrane to affect their toxic effects. But in most instances, they interact with the cell membrane. Large oligomers (or the ADDLs) are toxic by non-selectively disrupting cell membrane viability, perhaps like a hammer hitting a leaf. Small oligomers (mono- and dimers), on the other hand, allow ion permeability through newly formed ion channels. The following description aims to provide key evidence for this amyloid ion channel paradigm.
Figure 1.

Ion channel hypothesis for AD pathology. A. Aβ, a 39–43 AA long peptide is cleaved from the single transmembrane spanning precursor, called amyloid precursor protein (APP). B. Aβ undergoes multi-step oligomerization, ranging from the formation of small oligomers to large oligomers (commonly referred to as the ADDLs) to protofibrils to fibrils and plaques. In normal physiological conditions, most of the secreted amyloids remain monomers or small oligomers. C. Several mechanisms for neurotoxic activity of Aβ have been proposed, such as activation of a signaling pathway by extracellular aggregates (fibrils, plagues), induction of oxidative stress due to protein aggregates, or recruitment of factors by intracellular aggregates. Large oligomers (or the ADDLs) may be toxic by non-selectively disrupting cell membrane viability, like a hammer hitting a leaf). Small oligomers (mono- and dimers) fold into the membrane forming ion channels. D. A common denominator for AD toxicity is mainly the gain of intracellular calcium, the severity of toxicity depends upon the level of calcium loading as well as the cell’s defense capability. See [4, 14, 26, 47, 48, 50].
2. Globular Amyloids induce cellular degeneration in live cells
In normal cellular metabolism, Aβ is continuously released in pico- to- nanomolar concentrations and are present in both the extracellular and cytoplasmic regions. Globular amyloid beta is reported to induce known AD pathophysiological features, namely, a loss of synaptic degeneration and eventual cell death. Small oligomeric and non-fibrillar (globular) Aβ is reported to cause profound cytoskeletal damage and cell death [6, 11, 13]. In these studies, after online addition of very low concentration of freshly prepared amyloid beta, cell degeneration began within 15–20 minutes, the time too short to form any fibrillar amyloids. Moreover, Aβ-induced cytoskeletal degeneration and cell death was dose-dependent (Figure 2) [6, 11, 13] and Aβ–induced cell degeneration and eventual death can be prevented by anti-Aβ antibody [6, 11, 13] as well as by many physiological and pharmacological agents. Prevention of cellular degeneration due to the presence of antibodies against the amyloids shows that the toxicity is amyloid specific. Figure 3 summarizes results from a study by Lin et al (2001) supporting the channel-mediated toxicity [11]. Significantly, in the presence of physalaemin, a tachykinin, Aβ(1–42) still induced significant cell degeneration suggesting that Aβ(1–42) induced pathophysiological response is not mediated via a previously proposed tachykinin neuropeptide pathway, a potential amyloid beta receptor [15]. Also, MK-801, an NMDA receptor antagonist did not prevent Aβ(1–42)-induced toxicity, On the other hand, in absence of Ca2+ in the medium, Aβ(1–42) did not induce cellular degeneration [6, 13], and Zinc provided the cells with a strong protection against the amyloid toxicity. The final set of controls support the notion that amyloid-induced pathophysiological response requires extracellular calcium and Zinc is a specific antagonist of amyloid-induced cell toxicity. It is commonly believed that the extracellular calcium does not directly modulate amyloid toxicity rather after its cellular uptake.
Figure 2.
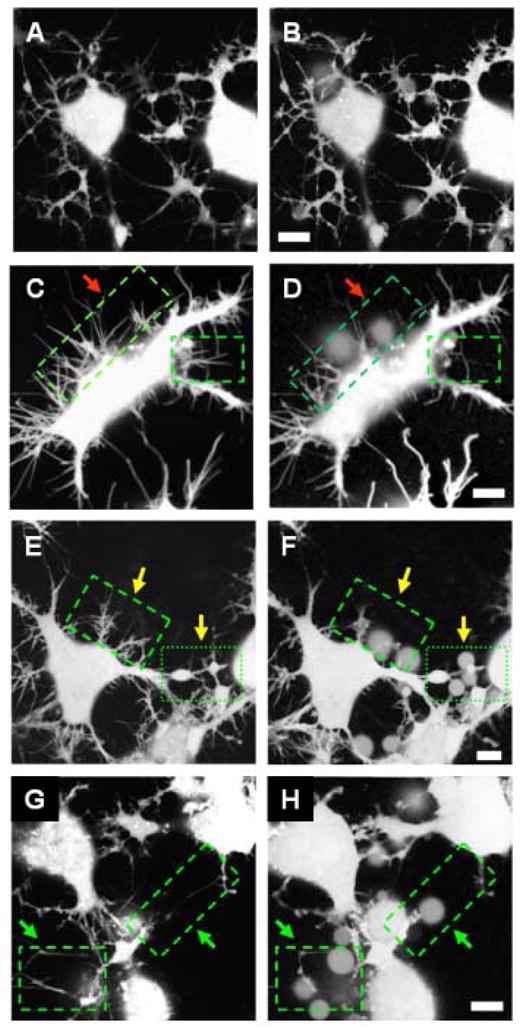
Morphological changes induced in neuronal cells by Aβ(1–42). Cells are loaded with calcein AM. Images A, C, E and G are taken as control before online addition of Aβ(1–42). Images B, D, F and H are taken 45 minutes after addition of Aβ(1–42). No morphological degeneration is observed for cells not treated with Aβ(1–42) (panels A and B). Significant degeneration is observed for cells treated with 50 nM Aβ(1–42) (panel D), indicated by arrows. Loss of cell-cell contact and neurite beading is observed after treatment with 500 nM Aβ(1–42) (panel F), and 5 μM Aβ(1–42) leads to loss of neuronal processes and fragmented neurites [11]. Scale bars 10 μm.
Figure 3.
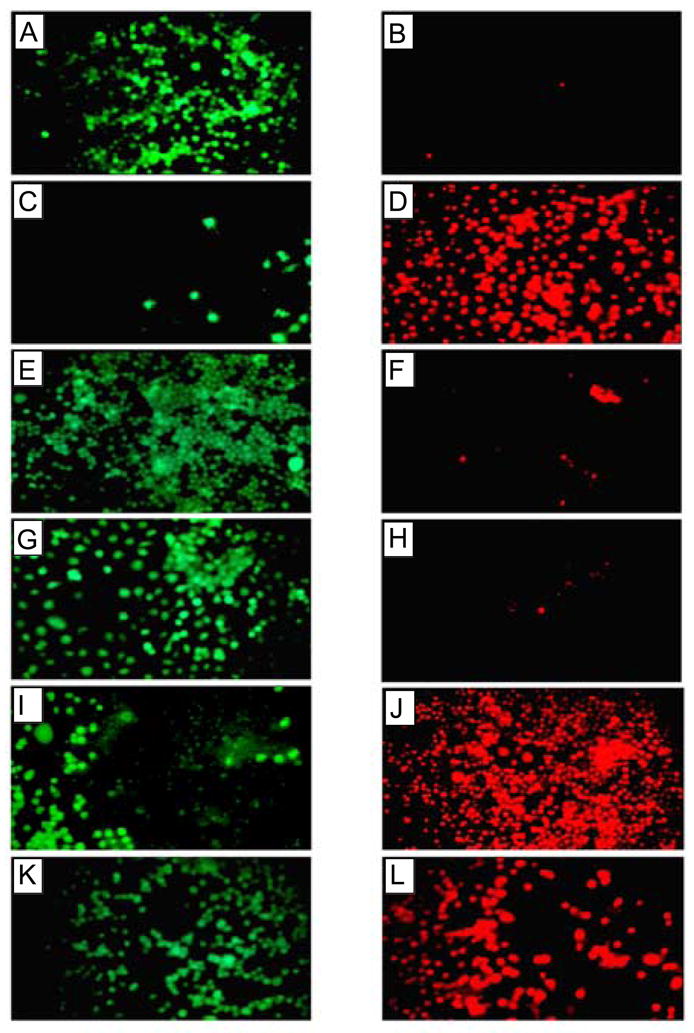
Cell viability assay of neuronal cells treated with Aβ(1–42). Cellular toxicity induced by Aβ(1–42) was blocked by Zn2+ and by removal of extracellular calcium, but not by tachykinin or NMDA antagonist. Fluorescence images of cells treated with calcein (live cells, left panels) and ethidium homodimer 1 (dead cells, right panels) are shown after Aβ(1–42) treatment. Panels A and B show control cells not treated with Aβ(1–42), panels C and D show cells treated with 10 μM Aβ(1–42), panels E and F show cells after Aβ(1–42) treatment in the absence of extracellular calcium, panels G and H show cells after Aβ(1–42) treatment in the presence of 50 μM ZnCl2, panels I and J show cells after Aβ(1–42) treatment in the presence of 20 μM tachykinin (physalaemin), and K and L show cells after Aβ(1–42) treatment in the presence of 20 μM MK-801, an NMDA receptor antagonist [11].
3. Aβ-mediated calcium uptake
Consistent with the abovementioned reasoning, Bhatia et al (2000) and Lin et al (2001) have shown amyloid beta channel-mediated calcium uptake [6, 11]. In these studies, only freshly prepared amyloid beta (without any large oligomers or fibers) were used. Figure 4 shows summary results from one such studies using Calcium Green, a calcium sensitive dye, and laser confocal microscopy. A transient and often repetitive wave of Ca2+ was observed in cells shortly after their treatment with Aβ(1–42) but only when the extracellular medium contained Ca2+.
Figure 4.

Confocal Calcium Green fluorescence images of endothelial cells before (A), and after addition of 0.22 μM (B), and 2.2 μM (C) of Aβ(1–42). Observed Calcium waves are consistent with extracellular calcium uptake and amyloid [6].
These calcium waves were observed for concentrations as low as 0.22 μM Aβ(1–42) and increased with increasing concentration. Such calcium waves require extracellular calcium and the Ca2+ wave pattern is consistent with the possibility that Aβ forms Ca2+-permeable channels in the plasma membrane. The intracellular calcium imaging also showed that Aβ(1–42) causes rapid cellular damage unlikely to be caused by an oxidative mechanism but indeed initiated by calcium uptake via Aβ. Such an increased cellular calcium level can change the local micromechanical properties of cells [16] and induce cytoskeletal reorganization, one of the earliest detectable changes observed in neurodegenerative disorders such as AD [17].
The possibility that the Aβ-mediated cellular Ca2+ uptake is due to amyloid beta forming Ca2+-sensitive ion channels was reported earlier by Kawahara and his colleagues [18, 19]. They had recorded Zinc-sensitive Ca2+ currents from newly created Aβ channels in neuronal membranes. Their results were consistent with the possibility that the Ca2+ current were observed after the interaction of Abeta with the neuronal membrane and forming Abeta ion channels.
4. Membrane associated conformational changes
AFM imaging provides the direct evidence that Aβ at a low (physiological) concentration and in physiologically relevant buffers remain globular and do not form fibrils for hours to several days (Figure 5)[20]. Aggregation properties of monomeric amyloids depend on the preparation and environmental conditions. Zinc accelerates oligomerization to larger sized amyloid aggregates, a process which is reversible by online addition of the Zinc chelator EDTA. Subsequent studies from various laboratories indicate that the aggregation behavior also depends on the pH of the solution as well as the nature of the cations present in the solution [21–24].
Figure 5.

Amyloid beta(1–42) deposited on mica after incubation in PBS for 3 hours (left) and 48 hours (right). Even after long incubation time the majority of peptide is still in globular form, although some fibrils are observed [11].
Amyloids undergo conformational changes when they associate with lipidic membrane and membrane associated conformational changes are different than the one expected of their non-membranous associations. Biochemical and biophysical studies strongly support the notion that a range of misfolded proteins when they interact with lipid membrane make oligomeric complexes [11, 25–27]. In an elegant recent study, Quist et al (2005) reported that globular monomers (and a small fraction of dimers) of soluble amyloid peptides when inserted in lipidic bilayer membrane undergo specific oligomerization regime, i.e., while in solution mostly monomers and sometimes dimers of these amyloids are observed, in the membrane oligomers up to octamers are observed (Figure 6) [26]. The insets in the figure show circular dichroism spectra of the amyloids in solution supporting their varying alpha helical to beta sheet or mixed alpha helical and beta sheet conformations before their insertion into membrane.
Figure 6.

Electrophoresis of British amyloid (ABri), Danish Amyloid (ADan), Alpha-synuclein, Amylin, Serum Amyloid A, and Amyloid Beta (1–40), showing membrane induced oligomerization. In solution monomers, and sometimes dimers (ABri, Amylin, Aβ(1–40)) are observed. In the membrane oligomers up to octamers are observed. The insets show circular dichroism spectra of the amyloids in solution.
In addition to their membrane-induced oligomerization, some preformed oligomeric soluble complexes may also retain their structure after their insertion into a bilayer [11, 28]. In addition, the presence of large oligomeric complexes (such as ADDLs) made up of small oligomers is not rule out. These large oligomers, however alter membrane permeability not through ion channel pathways but by reducing the height of the dielectric barrier provided by the bilayer [14]. Globular amyloids have been shown to induce deleterious effect, by either non specific membrane leakage [29, 30], or by formation of ion channel like structures [11, 25, 26, 31]. As described below, atomic Force Microscopy (AFM) studies have shown the presence of amyloid ion channels in lipid bilayers [11, 25–27]. These and other studies also suggest that amyloid peptide oligomers can form supra molecular structures [19, 25–27, 32–36].
5. Amyloids reconstituted in lipid bilayers exhibit channel like behavior
A series of studies have reported that small oligomeric amyloid have channel-like properties and form channel-like structures [11, 25–27, 31, 33, 37]. Lin et al (1999) examined such possibility by using a permeability assay and compared the results with those obtained by other groups using electrophysiological channel current measurements. Results from 45Ca2+ uptake studies in Lin’s study (figure 7) confirmed such a possibility - the transport of Ca2+ into lipid vesicles reconstituted with Aβ was observed [25].
Figure 7.

Uptake of 45Ca2+ in lipid vesicles reconstituted with Aβ(1–40). Uptake is blocked partially by anti- Aβ(1–40) antibody, and completely blocked by Zinc.
The 45Ca2+ uptake was partially blocked using antibodies raised against the N-terminal region of Aβ(1–40), which shows the Aβ(1–40) specificity of the calcium uptake. Ca2+-uptake was completely blocked by Zinc; Zinc can competitively bind to negatively charged phospholipid head groups. Alternately, Arispe et al (1996) [38] had reported that Zinc blocks Abeta currents by direct interaction with the Abeta molecule itself, most likely by an interaction with the Abeta histidine residues located in the entrance of the amyloid pore [38]. Similar results were obtained in experiments measuring the Ca2+-uptake in vesicles reconstituted with Aβ(1–42) [27]. In these vesicles, 45Ca2+-uptake was strongly reduced using antibodies against the amyloid peptide, and completely prohibited by allowing for Zinc or Tris ions in the medium. However, anti-oxidants did not prevent the calcium uptake, and thus seem to have no significant influence on the Aβ channel mediated ionic exchange [27].
AFM imaging of such lipid vesicles shows that vesicles reconstituted with Aβ have a well-defined vesicular structure and range in size from 200–500 nm along with only a small fraction of larger vesicles. Figure 8 illustrates AFM images of vesicles formed by reconstituted Aβ(1–40). Force dissection by AFM indicates that such vesicles are unilamellar [25]. Similar results were observed for liposomes reconstituted for Aβ(1–42) [27].
Figure 8.

AFM images of liposomes prepared by bath sonication, reconstituted with (left) and without (right) Aβ(1–40) [25].
As discussed previously, a common denominator for amyloid toxicity is an altered cellular calcium level [39] that could be either through modulation of existing calcium channels, or through Aβ-channels. Consistent with the amyloid-channel mediated calcium uptake, when Aβ is reconstituted in artificial lipid bilayers, cation selective currents through the reconstituted bilayers are observed [32, 38]. Similarly, Aβ elicits such currents when incubated with patches excised from hypothalamic GnRH neurons [18].
6. High resolution structure of Amyloid channels
In order to justify the abovementioned reasoning for amyloid channel activity and channel-mediated cell degeneration, it is important to show that small oligomeric amyloids form ion channel structure in purified or reconstituted membrane as well as in whole cell plasma membrane. Examining amyloid channel structure in intact cell membrane is a difficult proposition. For cells with naturally expressing APP, it will be difficult to distinguish amyloid channels made of APP from channels made of amyloid beta or for that matter from any other ion channels present in the cell membrane. Ideally, one would need cells that do not express APP and these cells when incubated with globular amyloids retain their viability and have significant number of amyloid channels. Then, these channels should also be identified unequivocally, and the imaging resolution should be high enough to provide the 3D structural information. Main hurdle for such studies is the lack of high resolution imaging technique for whole cells and the difficulty of proper identification of these channels. Existing high resolution techniques, such as electron microscopy (EM) and X-ray diffraction require a highly crystalline patch of similar structures and extensive sample preparation protocols that are not easily amenable to amyloid studies. AFM integrated with fluorescence microscopy is ideally suited for such studies but needs further advances and capability to provide high resolution information on living cell membrane. On the other hand, AFM has been used to image various ion channels in reconstituted, isolated purified membrane, and in a few occasion in intact cell membrane [11, 26, 40–42].
In a well controlled study of purified amyloids [26], AFM images show that a range of amyloids retained a globular structure when imaged after their adsorption from solution and without any membrane being present. In these studies, significantly, no to very little amyloid fibers were observed (Figure 5). The sizes of the globular features generally ranged from 1–10 nm for different amyloids. The size and volume of these amyloid structures were consistent with their differing molecular weights - small molecular weight amyloids had small diameter and overall volume and large molecular weight amyloids had larger structural features. Moreover, their size distribution was consistent with them being mostly monomer and dimers [6, 11, 13, 22, 25, 28, 43, 44].
The 3D structure of several amyloids after reconstitution in membranes have been imaged using AFM [11, 26, 28, 29, 43, 45]. A variety of amyloids when reconstituted in lipid bilayer membrane has been imaged and AFM images show the donut like features (Figure 9) formed from the globular amyloid structures in solution. Upon closer examination at high resolution, ion channel like structure with a central pore surrounded by a wall made of oligomeric subunits can be observed for all of these amyloids (Figure 10). Subunit arrangements varied from rectangular with four subunits, pentagonal with five subunits, hexagonal with six subunits, to octahedral with eight subunits. Octahedral arrangement was only observed for serum amyloid A and alpha-synuclein, pentamers were mainly observed for amylin, and for Abri resolution was good enough for ultrastructural details only in a few channels [26].
Figure 9.

AFM images of Aβ(1–40) and Aβ(1–42) reconstituted in bilayers. Images are amplitude images acquired in tapping mode. Donut shaped features are observed with sizes ranging from 8–15 nm [11, 26]. The inset shows a larger scale membrane patch.
Figure 10.

High resolution AFM images of different amyloids reconstituted in lipid bilayers. The image sizes are 20 nm. Different conformations and subunit arrangements can be observed, consistent with the results from parallel electrophoresis studies [26].
These results show that soluble amyloids, regardless of their secondary structure (figure 1) can assume supramolecular 3D structures when reconstituted in bilayers. The stoicheometry (number of subunits) of these channels were consistent with the overall molecular weights measured for oligomers in the reconstituted membrane (Figure 6) and electrical recording [26]. On the other hand, it will be difficult to compare the extracellular mass (volume) of each amyloid channels with their molecular weight as the theoretical models of their structures are just beginning to emerge. Moreover, the stability of their channel-like conformations would depend upon several factors, including pH, metal ions, cholesterol, gangliosides [46].
7. Overall Ion Channel Hypothesis and Structural Evidence
Theoretical models for the structure of channels formed by membrane bound Aβ are available (Figure 11, Panel D) [47, 48]. These models suggest that amyloid beta can insert partially at differing angles and can form multimeric channels with a central pore [47]. A recent model for alpha-synuclein–membrane complex also indicates the possibility of fully or partially inserted peptide chains (Figure 11, Panel D)[49]. Such ion channel models were designed as aggregates of identical subunits. For Aβ three types of models were proposed with the pore forming region made up of β-hairpin, the middle helixes, or the hydrophobic C-terminus helixes [48]. The latter two can convert back and forth upon a conformational change, and may explain why different conductance states are observed for single channels [32]. 3D structural features observed in AFM studies are consistent with the theoretical models derived for amyloid beta (Figure 11, Panel D). The theoretical model for alpha-synuclein channel derived from numerical simulation and fitted with EM images [49] also conforms to the 3D channel structure derived from AFM imaging (Figure 11, Panel D). Molecular modeling and molecular dynamics simulations showed that alpha-synuclein homodimers in a head-to-head conformation can incorporate additional alpha-synuclein molecules, leading to the formation of pentameric and hexameric ring like structures (Figure 11, Panel D) [49]. Whether the channel like structure for other amyloids in AFM imaging can conform to membrane-spanning respective channels is yet to be determined and would have to wait till relevant theoretical models are available. Moreover, the structural evidence for these channel structures in neuronal plasma membrane is just beginning to be examined. A recent abstract by S. Inoue supports such a possibility (Inoue, S., unpublished data).
Figure 11.

Structural evidence for amyloid ion channel paradigm and its relevance to AD pathophysiology. An array of studies using combination of AFM, biochemical analysis, fluorescence microscopy and electrical recording supports the ion channel model. A. Small oligomers of Aβ formed by enzymatic cleavage of APP remain globular up to 24 hours of incubation. B. AFM image of globular/monomeric amyloids on mica surface. C. AFM image of an array of ion channels randomly distributed in a lipid bilayer, the inset shows a larger scale bilayer patch. D. Ion channel structure and activity. AFM images of individual ion channels for Aβ (left) and α-Synuclein (right) with a central pore as well as their single channel currents recorded in parallel. Below the electrical recording graphs are shown theoretical models of Aβ and α-Synuclein ion channels as well as their membrane insertion [47–49], with a top view and a cross-sectional view of transmembrane insertion. E. Toxicity by calcium uptake and disruption of ionic homeostasis through Aβ- or pre-existing ion channels ultimately leads to neuronal degeneration.
The 3D structural evidence for amyloid ion channels, compatibility of these structures to known theoretical models and their putative pathway for cell degeneration are summarized in Figure 11. Monomeric amyloid beta derived from its precursor (APP) (Figure 11, Panel A) are oligomeric (but mostly monomers and dimers) in a physiologically relevant concentration and operating environment (pH, temp, ionic balance) when imaged by AFM (Figure 11, Panel B). We provide structural evidence from atomic force microscopy imaging that amyloid beta do not form fibers in physiologically relevant condition at a physiological concentration (Figure 5). Interaction of many amyloids with lipid bilayer alters their conformation to favor formation of small oligomeric complexes (Figure 6) and AFM imaging unequivocally show them forming ion channel-like oligomeric structures with a central pore (Figure 11, Panel C and Panel D top row). In parallel, lipid bilayer ion channel reconstitution electrophysiological studies show a typical channel-like activity including signature single channel conductances (Figure 11, panel D, second row from the top). Theoretical models derived for amyloid channel and alpha-synuclein channels (in the laboratories of Durrell and colleagues and Masliah and colleagues, respectively [47–49] support these ion channel structures. A concern that these small peptides that could insert only partially in the membrane does not seem to undermine the possibility of them making an ion channel - model of amyloid membrane insertions (Figure 11, Panel D bottom row) support the channel model. In its early though convincing phase of channel hypothesis, all amyloids appear to have a common structural feature that is supported by their channel-like electrical activity and permeability of cations.
Cell degeneration studies using non-fibrillar and globular amyloids from various laboratories support the role of these amyloid channels in altering cell behavior. Panel D in figure 11 indicates amyloid channel-induced alteration in cell behavior. Amyloid ion channels would provide a direct pathway between the cell cytoplasm and the extracellular milieu and would mediate specific ion transport [19, 32, 33]. Most significantly, cellular uptake of calcium through these channels would destabilize the cell’s calcium homeostasis. For a small number of amyloid channels as could occur during a transient and/or low concentration of globular amyloids, the cytoplasmic calcium would increase marginally. This will induce localized and subtle effects on synapses and synaptic efficacy. On the other hand, for a sustained and/or high concentration of globular amyloids that would allow formation of a large number of amyloid channels, the cytoplasmic calcium would increase significantly. In this scenario, there would be a widespread neuronal/neuritic dysfunction and eventual degeneration. An amyloid plaque will provide a constant source of local release of globular amyloids resulting in localized and sustained amyloid-induced pathophysiology. The details of this amyloid channel model need to be understood, including the role of various lipids, oxidative molecules, and other membrane associated complements in the channel formation as well as in their stability. Nevertheless, amyloid channels provide a viable structural substrate for testing tangible hypothesis underlying amyloid diseases and designing effective therapeutics.
Acknowledgments
The work summarized in this review is primarily from the laboratory of Ratnesh Lal at the University of California and was supported by grants from NIH, Alzheimer’s Association, and Alzheimer ’s disease program of the Department of Health California.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dobson M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL. Cerebral amyloid angiopathies: A pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003;62:885–898. doi: 10.1093/jnen/62.9.885. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 4.Temussi PA, Masino L, Pastore A. From Alzheimer to Huntington: why is a structural understanding so difficult? Embo J. 2003;22:355–361. doi: 10.1093/emboj/cdg044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pallito MM, Ghanta J, Heinzelman P, Kiessling LL, Murphy RM. Recognition sequence design for peptidyl modulators of beta-amyloid aggregation and toxicity. Biochemistry. 1999;38:3570–3578. doi: 10.1021/bi982119e. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia R, Lin H, Lal R. Fresh and globular amyloid beta protein (1–42) induces rapid cellular degeneration: evidence for A beta P channel-mediated cellular toxicity. Faseb J. 2000;14:1233–1243. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- 7.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo JS, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 8.Gibson G, Gunasekera N, Lee M, Lelyveld V, El-Agnaf OMA, Wright A, Austen B. Oligomerization and neurotoxicity of the amyloid ADan peptide implicated in familial Danish dementia. J Neurochem. 2004;88:281–290. doi: 10.1046/j.1471-4159.2003.02134.x. [DOI] [PubMed] [Google Scholar]
- 9.Gong YS, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, Bures J, Higgins LS. Specific spatial learning deficits become severe with age in beta-amyloid precursor protein transgenic mice that harbor diffuse beta-amyloid deposits but do not form plaques. Proc Natl Acad Sci U S A. 2001;98:14675–14680. doi: 10.1073/pnas.261562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin H, Bhatia R, Lal R. Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. Faseb J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 12.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 13.Zhu YJ, Lin H, Lal R. Fresh and nonfibrillar amyloid beta protein(1–40) induces rapid cellular degeneration in aged human fibroblasts: evidence for A beta P-channel-mediated cellular toxicity. Faseb J. 2000;14:1244–1254. doi: 10.1096/fasebj.14.9.1244. [DOI] [PubMed] [Google Scholar]
- 14.Sokolov Y, Ashot Kozak J, Kayed R, Chanturia A, Glabe CG, Hall JE. Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J Gen Physiol. 2006;128:637–647. doi: 10.1085/jgp.200609533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 16.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. Beta amyloid mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 17.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 18.Kawahara M, Arispe N, Kuroda Y, Rojas E. Alzheimer’s disease amyloid beta-protein forms Zn2+-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys J. 1997;73:67–75. doi: 10.1016/S0006-3495(97)78048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000;275:14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- 20.Parbhu A, Lin H, Thimm J, Lal R. Imaging real-time aggregation of amyloid beta protein (1–42) by atomic force microscopy. Peptides. 2002;23:1265–1270. doi: 10.1016/s0196-9781(02)00061-x. [DOI] [PubMed] [Google Scholar]
- 21.Stine WB, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid beta peptide oligomerization and fibrillogenesis. Journal of Biological Chemistry. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 22.Ding TT, Lee SJ, Rochet JC, Lansbury PT. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 23.Goldsbury C, Frey P, Olivieri V, Aebi U, Muller SA. Multiple assembly pathways underlie amyloid-beta fibril polymorphisms. J Molec Biol. 2005;352:282–298. doi: 10.1016/j.jmb.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM. Dramatic aggregation of Alzheimer Abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 25.Lin H, Zhu YWJ, Lal R. Amyloid beta protein (1–40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry. 1999;38:11189–11196. doi: 10.1021/bi982997c. [DOI] [PubMed] [Google Scholar]
- 26.Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhee SK, Quist AP, Lal R. Amyloid beta protein-(1–42) forms calcium-permeable, Zn2+-sensitive channel. J Biol Chem. 1998;273:13379–13382. doi: 10.1074/jbc.273.22.13379. [DOI] [PubMed] [Google Scholar]
- 28.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT. alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 29.Green JD, Kreplak L, Goldsbury C, Blatter XL, Stolz M, Cooper GS, Seelig A, Kist-Ler J, Aebi U. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. J Mol Biol. 2004;342:877–887. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 30.Kayed R, Sokolov Y, Edmonds B, MacIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein Mis-folding diseases. J Biol Chem. 2004 doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 31.Arispe N. Architecture of the Alzheimer’s A beta P ion channel pore. J Membr Biol. 2004;197:33–48. doi: 10.1007/s00232-003-0638-7. [DOI] [PubMed] [Google Scholar]
- 32.Arispe N, Pollard HB, Rojas E. Giant Multilevel Cation Channels Formed by Alzheimer-Disease Amyloid Beta-Protein a-Beta-P-(1–40) in Bilayer-Membranes. Proc Natl Acad Sci U S A. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirakura Y, Carreras I, Sipe JD, Kagan BL. Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense. Amyloid-J Protein Fold Disord. 2002;9:13–23. doi: 10.3109/13506120209072440. [DOI] [PubMed] [Google Scholar]
- 34.Kourie JI, Henry CL. Ion channel formation and membrane-linked pathologies of misfolded hydrophobic proteins: The role of dangerous unchaperoned molecules. Clin Exp Pharmacol Physiol. 2002;29:741–753. doi: 10.1046/j.1440-1681.2002.03737.x. [DOI] [PubMed] [Google Scholar]
- 35.Kourie JI, Shorthouse AA. Properties of cytotoxic peptide-formed ion channels. Am J Physiol-Cell Physiol. 2000;278:C1063–C1087. doi: 10.1152/ajpcell.2000.278.6.C1063. [DOI] [PubMed] [Google Scholar]
- 36.Pollard HB, Arispe N, Rojas E. Ion channel hypothesis for Alzheimer amyloid peptide neurotoxicity. Cell Mol Neurobiol. 1995;15:513–526. doi: 10.1007/BF02071314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin MC. The channel hypothesis of Alzheimer’s disease: current status. Peptides. 2002;23:1311–1315. doi: 10.1016/s0196-9781(02)00067-0. [DOI] [PubMed] [Google Scholar]
- 38.Arispe N, Pollard HB, Rojas E. Zn2+ interaction with Alzheimer amyloid beta protein calcium channels. Proc Natl Acad Sci U S A. 1996;93:1710–1715. doi: 10.1073/pnas.93.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattson MP, Cheng B, Davis B, Bryant K, Lieberburg I, Rydel RE. Beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci Res. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Philippsen A, Im W, Engel A, Schirmer T, Roux B, Muller DJ. Imaging the electrostatic potential of transmembrane channels: atomic probe microscopy of OmpF porin. Biophys J. 2002;82:1667–1676. doi: 10.1016/S0006-3495(02)75517-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheuring S, Ringler P, Borgnia M, Stahlberg H, Muller DJ, Agre P, Engel A. High resolution AFM topographs of the Escherichia coli water channel aquaporin Z. EMBO J. 1999;18:4981–4987. doi: 10.1093/emboj/18.18.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thimm J, Mechler A, Lin H, Rhee S, Lal R. Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. J Biol Chem. 2005;280:10646–10654. doi: 10.1074/jbc.M412749200. [DOI] [PubMed] [Google Scholar]
- 43.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT. Neurodegenerative disease - Amyloid pores from pathogenic mutations. Nature. 2002;418:291–291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 44.Srinivasan R, Jones EM, Liu K, Ghiso J, Marchant RE, Zagorski MG. pH-dependent amyloid and protofibril formation by the ABri peptide of familial British dementia. J Mol Biol. 2003;333:1003–1023. doi: 10.1016/j.jmb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Lashuel HA, Hartley DM, Petre BM, Wall JS, Simon MN, Walz T, Lansbury PT. Mixtures of wild-type and a pathogenic (E22G) form of A beta 40 in vitro accumulate protofibrils, including amyloid pores. J Mol Biol. 2003;332:795–808. doi: 10.1016/s0022-2836(03)00927-6. [DOI] [PubMed] [Google Scholar]
- 46.Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-beta peptide with membrane lipid. J Biol Chem. 2003;278:2977–2982. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 47.Mobley DL, Cox DL, Singh RRP, Maddox MW, Longo ML. Modeling amyloid beta-peptide insertion into lipid bilayers. Biophys J. 2004;86:3585–3597. doi: 10.1529/biophysj.103.032342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durell SR, Guy HR, Arispe N, Rojas E, Pollard HB. Theoretical-Models of the Ion-Channel Structure of Amyloid Beta-Protein. Biophys J. 1994;67:2137–2145. doi: 10.1016/S0006-3495(94)80717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsigelny IF, Bar-On P, Sharikov Y, Crews L, Hashimoto M, Miller MA, Keller SH, Platoshyn O, Yuan JXJ, Masliah E. Dynamics of alpha synuclein aggregation and inhibition of pore-like oligomer development by beta synuclein. FEBS J. 2007;274:1862–1877. doi: 10.1111/j.1742-4658.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- 50.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nature Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]