

Abstract
There are no specific approved drugs or vaccines for the treatment or prevention of infectious dengue virus and there are very few compounds known that inhibit the replication of this virus. This communication describes the concise synthesis of two uracil-based multifunctional compounds. One of these compounds (1) has strong activity against dengue virus. It also exhibits low activity against a few other RNA viruses, but is highly active against yellow fever virus, a related flavivirus. It is likely that the mechanism of action of the antiviral activity of this compound is through its inhibition of the enzyme, inosine monophosphate dehydrogenase (IMPDH). Molecular modeling studies reveal that the compound can have specific hydrogen bonding interactions with a number of amino acids in the active site of IMPDH, a stacking interaction with the bound natural substrate, IMP, and the ability to interfere with the binding of NAD+ with IMPDH, prior to the hydration step.
The etiological agents of dengue fever (DF) and dengue hemorrhagic fever (DHF) are four closely related dengue viruses, which are antigenically similar but immunologically distinct serotypes of the family called Flavivirus.1–3 The major vector for these viruses is the mosquito, Aedes aegypti. Estimates make the number of cases of dengue fever as high as 100 million annually, which makes this arthropod-borne viral infection a serious global health problem.4 Dengue virus genome is a single, positive-stranded RNA of about 11 kD with a 5′-cap but without a polyadenylated terminus. After fusion and entry, translation of genomic RNA occurs in infected cells.1,3 Processing of the viral polyprotein is apparently catalyzed by both viral and cellular enzymes. NS proteins are involved in the replication cycle. For example, the NS3 viral protease protein is essential for viral replication.5 The NS5 conserved protein has a methyltransferase motif in the N-terminal domain and an RNA-dependent RNA polymerase in the C-terminal domain. The transferase and polymerase are possible viral targets in antiviral strategies.5–7 However, there are no specific approved drugs or vaccines for the treatment or prevention of DF and DHF. This communication reports on the synthesis and antiviral activity of a novel, uracil-based multifunctional compound, 1, which is a potent inhibitor of the replication of dengue virus. In contrast, its deaza analog, 2, was inactive against this flavivirus (Figure 1).
Figure 1.

Structures of target compounds
Synthesis
Synthesis of the target compound was achieved using the Gould-Jacobs reaction8,9 as the key step (Scheme 1, see footnote 11 for details). Thus, condensation of 6-amino-1,3-dibenzyluracil (5), prepared through the coupling of 3 and 4,10 and diethyl ethoxymethylenemalonate under basic conditions, provided the aminomethylenemalonate 6 (42%). Intermediate 6 was heated in Dowtherm A (heat transfer fluid), which resulted in intramolecular cyclization to produce the pyridopyrimidine ester 7 (74%). Hydrolysis of ester 7 under acidic conditions afforded the target compound 1 as a crystalline solid in 67% yield. The structure of 1 was established by UV, 1H and 13C NMR and HRMS data.11 Compound 2 was synthesized by a related methodology.12
Scheme 1.

Synthesis of antiviral compound 1.
Antiviral Activity and Mechanism
Compound 1 exhibited relatively strong activity against dengue virus type 2 (New Guinea C) with an EC50 of 5.7μg/ml [visual inspection of cytopathic effect (CPE) inhibition] and 6.3μg/ml (neutral red uptake assay) and CC50 of >100 μg/ml producing a TI (therapeutic index) of >18 and >16, respectively, in Vero cells.13 Confirmatory virus yield reduction (VYR) assay gave an EC90 value of 3.8 μg/ml and a CC50 of >100 μg/ml, resulting in a TI of >26. The activity appears to be specific for this compound as the related synthesized compound, 2, that is without the endocyclic nitrogen in the second ring, does not exhibit significant activity against this virus family. Compound 1 represents one of the few compounds that has strong antiviral activity against dengue virus. Interestingly, this compound is moderately active by neutral red assay and highly active (TI > 63) in the virus yield reduction (VYR) assay against yellow fever virus (17D strain), another virus within the Flavivirus genus.
The mechanism of the antiviral activity of compound 1 is likely associated with its ability to inhibit the enzyme, inosine monophosphate dehydrogenase (IMPDH; EC 1.1.1.205). IMPDH catalyses the oxidative conversion of inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) with the involvement of the coenzyme, nicotinamide adenine dinucleotide (NAD+).14–16 IMPDH is an important target enzyme in drug discovery. Consistent with this suggestion is the observation that some nucleosides, which are inhibitors of IMPDH as their monophosphates, have been found to have anticancer, antiviral and immunosuppressive activity.17–22 Inhibition of IMPDH results in the reduction of the GTP pool and linear correlations do exist between GTP inhibition and antiviral activity against RNA viruses.23 The mechanism of the antiviral activity of compound 1 may be associated with its inherent ability to inhibit IMPDH through interference of the formation of ternary complex normally formed between the enzyme, the substrate IMP and the cofactor, NAD+.
In order to investigate the binding of inhibitor 1 with IMPDH in the presence of IMP, extensive molecular modeling studies were carried out using SYBYL 7.2 and GOLD 3.2. Figure 2 shows the preferred docking pose of inhibitor 1 in the active site region of IMPDH.
Figure 2.
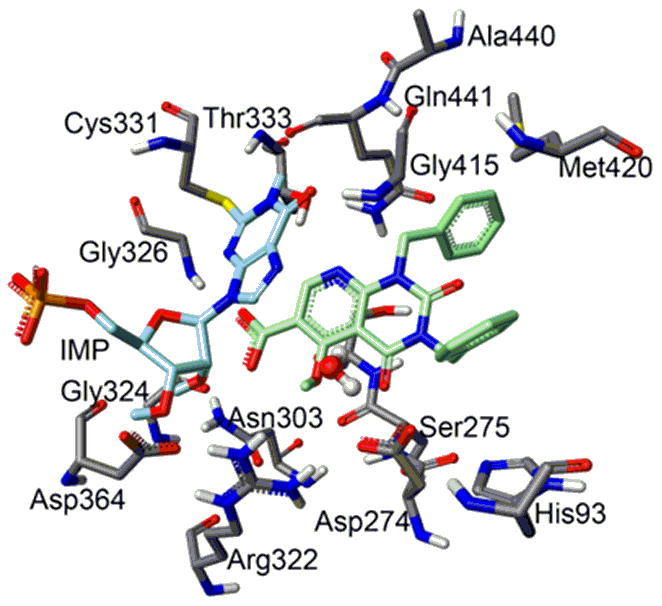
Molecular modeling (SYBYL 7.2, GOLD 3.3) of IMPDH complexed with inhibitor 1 (shown in green) and substrate IMP. The inhibitor interferes with the binding of NAD+ with the enzyme.
The binding allows for favorable stacking interactions between the hypoxanthine base of substrate IMP and the fused heterocyclic ring of the inhibitor. Additionally, the pyridine ring nitrogen of the inhibitor is appropriately situated to form hydrogen bonds with Thr333 and Gln441. This is a key interaction with the enzyme as the absence of this nitrogen (as in compound 2) results in significantly lower RNA antiviral activity. The carboxylate moiety of 1 is positioned to form hydrogen bonds with Asn303 and Gly326. In addition, the “phenolic” OH and the adjacent amide carbonyl may form a hydrogen bonding network with Asp 274 via a water molecule. The two phenyl rings may have stacking or other interactions, one with His 93 and the other with the carbonyl of Gln 441. Our molecular modeling studies confirm the ability of inhibitor 1 to bind in the active site region of the inhibitor. This binding would interfere with the interaction of the cofactor, NAD+, and IMPDH in the formation of the ternary complex with the substrate, which is required for hydration of the hypoxanthine ring.15 Support for our molecular modeling data and interpretation comes from the X-ray crystal structure of the complex of IMPDH, IMP and mycophenolic acid (MPA) together with the mutagenesis and kinetic data for the mechanism of the non-competitive inhibition of IMPDH by MPA.24 It is also of relevance to note that MPA exhibits antiviral activity against dengue,25 yellow fever virus26 and West Nile virus.27 Further validation that the mechanism of the antiviral activity of compound 1 is connected with its ability to inhibit IMPDH came from the observation that addition of guanosine to the cell culture assay medium reversed the antiviral activity, presumably by replenishing the levels of the GTP pool.
In conclusion, we have synthesized a new heterocyclic compound, 1, that shows relatively strong in vitro antiviral activity against dengue virus. This compound is also highly active against another virus of the genus, Flavivirus, the yellow fever virus. In addition, the compound exhibits antiviral activity, albeit low, against other RNA viruses. The mechanism of the antiviral activity is likely associated with inhibition of the enzyme, IMPDH and this is supported by the observation that addition of guanosine to the cell culture medium reverses the antiviral activity. Molecular docking data point to specific hydrogen bonding interactions with a number of amino acids in the active site of IMPDH and to a stacking interaction with the bound natural substrate, IMP. Inhibitor 1 represents one of the few compounds that exhibit significant activity against dengue virus.
Acknowledgments
This research was supported by research awards U19 AI 056540 and NO1 AI30048 from the National Institute of Allergy and Infectious Diseases, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Halstead SB. Science. 1988;239:476. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- 2.Ooi E, Goh K, Gubler DJ. Emerging Infectious Disease. 2006;12:887. doi: 10.3201/10.3201/eid1206.051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas SJ, Strickman D, Vaughn DW. Adv Virus Res. 2003;61:235. doi: 10.1016/s0065-3527(03)61006-7. [DOI] [PubMed] [Google Scholar]
- 4.Clyde K, Kyle JL, Harris E. J Virol. 2006;80:11418. doi: 10.1128/JVI.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melino S, Paci M. FEBS Journal. 2007;274:2986. doi: 10.1111/j.1742-4658.2007.05831.x. [DOI] [PubMed] [Google Scholar]
- 6.Chu JJH, Yang PL. Proc Natl Acad Sci USA. 2007;104:3520. doi: 10.1073/pnas.0611681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rawlinson SM, Pryor MJ, Wright PJ, Jans DA. Current Drug Targets. 2006;7:1623. doi: 10.2174/138945006779025383. [DOI] [PubMed] [Google Scholar]
- 8.Anderson GL. J Heterocyclic Chem. 1985;22:1469–1470. [Google Scholar]
- 9.Heber D, Ravens U, Shulze T. Pharmazie. 1993;48:509–513. [PubMed] [Google Scholar]
- 10.Papesch V, Schroeder EF. J Org Chem. 1951;16:1879–1890. [Google Scholar]
- 11.1,3-Dibenzyl-5-hydroxy-2,4-dioxo-1,2,3,4-tetrahydro-pyrido[2,3-d]pyrimidine-6-carboxylic acid (1): To a solution of 6-amino-1,3-dibenzyluracil (5)10 (615 mg, 2 mmol) and diethyl ethoxymethylenemalonate (541 mg, 2.5 mmol) in 10 mL of anhydrous ethanol was added sodium ethoxide solution [21% in ethanol (0.75 mL, 2 mmol)]. The mixture was heated under reflux for 12 h and the solvent was evaporated. Purification of the resulting residue by flash chromatography (hexanes: ethyl acetate 4:1) gave 400 mg (41.8%) of 6 as a waxy solid. 1HNMR (500 MHz, CDCl3): 11.1 (d, 1H, J=12 Hz), 8.04 (d, 1H, J=12 Hz), 7.28–7.50 (m, 10H), 5.55 (s, 1H), 5.25 (s, 2H), 5.20 (s, 2H), 4.28 (q, 2H, J=7.0 Hz), 4.22 (q, 2H, J=7.0 Hz), 1.33 (t, 3H, J=7.0 Hz), 1.30 (t, 3H, J=7.0 Hz). ESI MS calcd for C26H28N3O6 (M+H)+ 478, found 478. Compound 6 (380 mg) in 3 mL of Dowtherm A was heated under reflux for 1 h. After cooling, the mixture was purified by flash chromatography (hexanes: ethyl acetate 4:1) to afford 255 mg of 7 as a solid (74.2%). 1HNMR (500 MHz, CDCl3): 13.42 (br, s, 1H), 9.02 (s, 1H), 7.47–7.52 (m, 4H), 7.28–7.35 (m, 6H), 5.55 (s, 2H), 5.23 (s, 2H), 4.41 (q, 2H, J=7.0 Hz), 1.41 (t, 3H, J=7.0 Hz). ESI HRMS calcd for C24H22N3O5 (M+H)+ 432.1559, found 432.1555. Compound 7 (86.3 mg, 0.2 mmol) in 6 mL of dioxane and 5 mL of 1 N HCl was heated under reflux for 48 h and then cooled. The solid that crystallized out was collected and washed with a mixture of chloroform and hexanes (1:1, 8 mL) to give 54 mg (66.9%) of the target molecule 1. Mp 248–250°C. 1HNMR (500 MHz, DMSO-d6): 8.84 (s, 1H), 7.21–7.36 (m, 10H), 5.45 (s, 2H), 5.11 (s, 2H). 13CNMR (125 MHz, DMSO-d6): 169.3, 166.5, 164.2, 157.0, 154.2, 150.6, 137.1, 136.7, 128.7, 128.6, 127.9, 127.7, 127.5, 127.4, 110.2, 98.1, 45.9, 44.5. UV (MeOH) 240 nm (ε 40,800), 311 nm (ε 7,600). ESI HRMS calcd for C22H18N3O5 (M+H)+ 404.1246, found 404.1244.
- 12.Data for compound 2: Mp 211–213°C. 1HNMR (CDCl3, 500 MHz): 14.5 (s, 1H), 10.54 (br, s, 1H), 8.27 (d, 1H, J=9.0), 7.55 (d, 2H, J=7.0), 7.31–7.39(m, 6H), 7.22 (d, 2H, J=7.0), 6.75 (d, 1H, J=9.0), 5.38 (s, 2H), 5.33 (s, 2H). 13CNMR (CDCl3, 125 MHz): 166.8, 164.3, 161.4, 150.4, 144.6, 140.7, 135.6, 134.6, 129.4, 129.39, 128.9, 128.5, 128.4, 126.6, 110.7, 106.3, 101.0, 48.4, 45.4. UV (MeOH) 236 nm (ε41,400); 330 nm (ε9,500). ESI HRMS calcd for C23H19N2O5 (M+H)+ 403.1294, found 403.1297.
- 13.Antiviral assays were determined as follows. Inhibitor 1 was prepared as 8 half-log dilutions with a top concentration of 100 μg/ml. DENV-2 (New Guinea C) was prepared at a dose of 10^3 pfu/ml and added to Vero cells shortly after the addition of compound. Visual CPE was recorded, after which cells were stained with neutral red for determination of viability. To verify activity, a virus yield reduction assay was run to determine the concentration of compound necessary to reduce the virus titer by 1 log10 (EC90).
- 14.Sintchak MD, Nimmesgern EI. Immunopharmacology. 2000;47:163. doi: 10.1016/s0162-3109(00)00193-4. [DOI] [PubMed] [Google Scholar]
- 15.Kerr KM, Digits JA, Kuperwasser N, Hedstrom L. Biochemistry. 2000;39:9804. doi: 10.1021/bi0005409. [DOI] [PubMed] [Google Scholar]
- 16.Colby TD, Vanderveen K, Strickler MD, Markham GD, Goldstein BM. Proc Natl Acad Sci (USA) 1999;96:3531. doi: 10.1073/pnas.96.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franchetti P, Grifantini M. Curr Med Chem. 1999;6:599. [PubMed] [Google Scholar]
- 18.Goldstein BM, Colby TD. Curr Med Chem. 1999;6:519. [PubMed] [Google Scholar]
- 19.Shu Q, Nair V. Med Res Rev. 2008;28:219. doi: 10.1002/med.20104. [DOI] [PubMed] [Google Scholar]
- 20.Nair V, Shu Q. Antiviral Chem Chemother. 2007;18:245. doi: 10.1177/095632020701800501. [DOI] [PubMed] [Google Scholar]
- 21.Nair V, Kamboj RC. Bioorg Med Chem Lett. 2003;13:645. doi: 10.1016/s0960-894x(02)01053-3. [DOI] [PubMed] [Google Scholar]
- 22.Nair V, Bonsu E, Gupta M, Story S. Antiviral Res. 2005;65:A67. doi: 10.1081/ncn-200060308. [DOI] [PubMed] [Google Scholar]
- 23.Leyssen P, Balzarini J, DeClercq E, Neyts J. J Virol. 2005;79:1943. doi: 10.1128/JVI.79.3.1943-1947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko MA, Wilson KP. Cell. 1996;85:921. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- 25.Diamond MS, Zachariah M, Harris E. Virology. 2002;304:211. doi: 10.1006/viro.2002.1685. [DOI] [PubMed] [Google Scholar]
- 26.Neyts J, Meerbach A, McKenna P, De Clercq E. Antiviral Res. 1996;30:125. doi: 10.1016/0166-3542(96)89697-5. [DOI] [PubMed] [Google Scholar]
- 27.Morrey JD, Smee DF, Sidwell RW, Tseng C. Antiviral Res. 2002;55:107. doi: 10.1016/s0166-3542(02)00013-x. [DOI] [PubMed] [Google Scholar]