

Abstract
The hepatitis C virus (HCV) NS3 protein is a helicase capable of unwinding duplex RNA or DNA. This study uses a newly developed molecular beacon based helicase assay (MBHA) to investigate how nucleoside triphosphates (NTPs) fuel HCV helicase catalyzed DNA unwinding. The MBHA monitors the irreversible helicase catalyzed displacement of an oligonucleotide-bound molecular beacon so that rates of helicase translocation can be directly measured in real time. The MBHA reveals that HCV helicase unwinds DNA at different rates depending on the nature and concentration of NTPs in solution, such that the fastest reactions are observed in the presence of CTP followed by ATP, UTP, and GTP. 3′-deoxy-NTPs generally support faster DNA unwinding, with dTTP supporting faster rates than any other canonical (d)NTP. The presence of an intact NS3 protease domain makes HCV helicase somewhat less specific than truncated NS3 bearing only its helicase region (NS3h). Various NTPs bind NS3h with similar affinities, but each NTP supports a different unwinding rate and processivity. Studies with NTP analogs reveal that specificity is determined by the nature of the Watson-Crick base-pairing region of the NTP base and the nature of the functional groups attached to the 2′ and 3′ carbons of the NTP sugar. The divalent metal bridging the NTP to NS3h also influences observed unwinding rates, with Mn2+ supporting about 10 times faster unwinding than Mg2+. Unlike Mg2+, Mn2+ does not support HCV helicase catalyzed ATP hydrolysis in the absence of stimulating nucleic acids. Results are discussed in relation to models for how ATP might fuel the unwinding reaction.
Keywords: Viral replication, motor protein, ATPase, RNA, DNA
Hepatitis C virus (HCV) is a single stranded, positive sense RNA virus infecting an estimated 2% of the world’s population.1 The HCV genome is translated into a 3,000 amino acid long polypeptide that is subsequently cleaved into 10 mature structural and non-structural proteins by host and viral proteases. Among the non-structural proteins is NS3, a multifunctional protein with protease, ATPase, and helicase activities. Though the exact role of HCV helicase in the viral lifecycle is unclear, a fully functioning helicase is necessary for HCV RNA replication5 and virulence in vivo, thus making it a possible antiviral drug target.7 The NS3 helicase is an ATP activated motor protein that can disrupt RNA duplexes, DNA duplexes, and even dislodge DNA bound proteins. Recombinant NS3 lacking the protease domain retains ATPase and helicase functionality. This “NS3h” retains most of its ability to separate DNA duplexes, but loses some of its ability to processively unwind RNA because it does not form oligomers like the full-length protein. Numerous studies have helped unveil mechanisms of NS3h and NS3 catalyzed ATP hydrolysis, DNA unwinding, and RNA unwinding, but exactly how ATP hydrolysis enables the protein to move along nucleic acid remains a mystery. Here, we study the HCV helicase as a motor protein using a newly developed high throughput molecular beacon based helicase assay (MBHA)20 to monitor how ATP and other nucleoside triphosphates (NTPs) interact with the protein to fuel its action.
Like other motor proteins,21 HCV helicase presumably functions by switching between different conformational states in an ATP dependent manner. Comparisons of the various HCV helicase atomic structures reveal a clear flexibility in the protein. As first noted by Yao et al., a flexible linker between the two RecA-like motor domains enables the C-terminal RecA-like domain (i.e. “domain 2”) to pivot 3–4° relative to the rest of the protein. Kim et al. have also pointed out that a closure of this inter-domain cleft might occur upon ATP binding. Although this conformational change is used as the molecular basis for most present models that liken the HCV helicase to an inchworm moving along a nucleic acid track, exactly how ATP modulates protein isomerisation is not known, in part, because no structures exist of a nucleotide analog bound to HCV helicase. Comparisons with other helicases that have been crystallized with ATP analogs, like the closely related yellow fever virus NS3 or Dengue virus NS3 indicate that the HCV helicase-bound ATP ribose and base moieties might be exposed to the solvent, whereas the triphosphate moiety might be buried between the motor domains. If this were the case, one would expect that HCV helicase would indiscriminately cleave any NTP, and that all NTPs would fuel unwinding to an equal extent. There is some data, however, that hint that HCV helicase might more intimately contact NTPs.
The first evidence defining a preference of HCV helicase for certain NTPs came from analyses of HCV helicase catalyzed NTP hydrolysis. Suzich et al. noted that HCV helicase cleaves all eight canonical NTPs in the order of preference (d)ATP > (d)CTP > UTP/dTTP > (d)GTP. Others have observed a similar NTPase specificity even with enzymes derived from diverse HCV genotypes. This NTPase specificity is mainly based on a different affinity of the enzyme for each NTP with variations seen mainly in Km, not Vmax. At saturating NTP concentrations, HCV helicase hydrolyzes most NTPs at similar rates.
Unlike the published reports characterizing the specificity of the NS3 NTPase function, there is no clear consensus in the literature regarding the ability of various NTPs to support DNA unwinding. There have only been a few systematic analyses, none of which have even examined a complete panel of the canonical NTPs (ATP, GTP, CTP, UTP, dATP, dGTP, dCTP, dTTP). The existing reports are surprisingly different, with some groups noting a lack of specificity, some noting modest differences and some reporting dramatic differences. For example, when comparing ATP with dATP, Kyono et al. note that both ATP with dATP are used equally well, Wardell et al. report that dATP supports 60% of the unwinding observed with ATP, but Locatelli et al. observe that ATP supports 100 times better unwinding than dATP. Such disagreements in the apparent specificities of the HCV NS3 NTPase and helicase functions has led to debates on whether or not NTP hydrolysis actually fuels the helicase reaction.
This study attempts to clarify how different NTPs fuel HCV helicase catalyzed DNA unwinding and reveals structure activity relationship that could be useful for helicase inhibitor design. The ~100-fold differences between the studies discussed above could be due to the fact that they used different proteins obtained from different HCV strains or genotypes, or because they relied on unwinding assays where excess enzyme was incubated with a small amount of DNA and products were measured at only a single time point. Here we used a diverse collection of recombinant HCV NS3 proteins isolated from a variety of HCV genotypes along with our new MBHA20 to more rigorously analyze the ability of a variety of NTPs to fuel unwinding under both steady state and single turnover conditions. The results show a clear conserved NTP specificity that mimics that seen in certain NTPase assays. Studies with nucleotide analogs reveal that the nature of the NTP base and sugar both influence unwinding rates, and divalent metal ion specificity points out that nucleotides interact with two distinct conformations of the enzyme.
Results
A thorough study of how NTPs fuel the NS3 helicase reaction has been difficult to complete because of the meticulous nature of helicase unwinding assays, most of which require either high protein concentrations or the addition of extra DNA strands to prevent the products from re-annealing after separation. These DNA “capture strands” could influence observed rates by driving the reaction forward or by sequestering active protein. Recently, we developed a new helicase assay that uses molecular beacons to continuously monitor helicase catalyzed unwinding. The new assay (Figure 1(a)) is unique because it is essentially irreversible, allowing data to be collected at limiting protein concentrations and without substrate traps.20 We used this molecular beacon based helicase assay (MBHA) to study how various NTPs activate HCV helicase catalyzed DNA unwinding.
Figure 1.
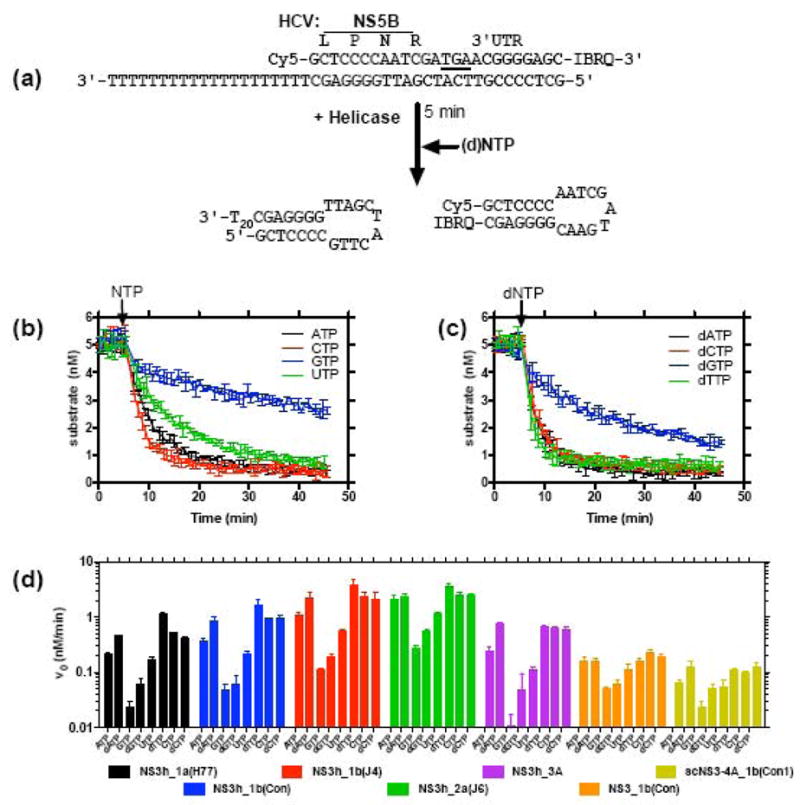
The ability of various NTPs to support HCV helicase catalyzed DNA unwinding. (a) The molecular beacon based helicase assay (MBHA),20 in which HCV helicase separates a molecular beacon from a longer oligonucleotide. Both products form hairpins after separation, making the reaction essentially irreversible. Reactions were performed at 22 °C by first incubating 2 mM MgCl2, 25 nM NS3h and 5 nM HCV-substrate in 25 mM MOPS pH 6.5, and then initiating by adding each NTP to 0.5 mM. (b) Reaction time courses for reactions catalyzed by NS3h encoded by HCV genotype 1b(con1) (NS3h_1b(con1)) in the presence of ATP (black), CTP (red), GTP (blue) and UTP (green). (c) Reaction time courses for dATP (black), dCTP (red), dGTP (blue) and dTTP (green). Arrows in (b) and (c) show when each NTP was added to initiate the reaction. (d) Comparison of initial velocities obtained in reactions fueled by each (d)NTP in the presence of NS3h encoded by HCV genotype 1a(H77), black, 1b(con1), blue, 1b(J4), red, 2a(J6), green, or 3a, purple. Reactions catalyzed by full length NS3 (orange), and a single chain NS3–NS4A construct (yellow, 250 nM enzyme), both from genotype 1b(con1), are shown for comparison. Error bars represent standard deviation of three independent reactions.
The MBHAs used in this study all utilize substrates labeled with the fluorescent dye cyanine 5 (Cy5) and one of two quenching dyes sold by Integrated DNA technology (Corralville, IA): Black Hole Quencher™-2 (BHQ2) or Iowa Black RQ™ (IBRQ). With either combination, very little florescence is observed when the molecular beacon is not annealed to a complementary DNA molecule, and fluorescence increases ~100 fold when the beacon is annealed to complementary DNA. All MBHA substrates were prepared by heating complementary oligonucleotides together, and allowing them to cool slowly to room temperature. Typically, 60–80% of the beacons annealed under these conditions, but nevertheless MBHA substrates were purified to remove free oligonucleotides using polyacrylamide gel electrophoresis. Cy5-based molecular beacons were chosen because Cy5 fluorescence itself does not change upon strand annealing or when protein binds a Cy5-based MBHA substrate. Either event could complicate analyses. Control experiments using similar substrates lacking a quenching dye revealed no change in Cy5 fluorescence upon annealing or dissociation, and no changes in fluorescence were noted when HCV helicase was combined with any of the MBHA substrates used here.20 As has been reported before,20 data from a MBHA can be converted to concentration of substrate remaining simply by calculating the proportion of fluorescence remaining after subtracting background fluorescence (equation (1), Materials & Methods). The resulting substrate concentrations correspond, within error, to concentrations derived by separating reaction products using polyacrylamide gel electrophoresis.
Various NTPs fuel different steady-state rates of HCV helicase catalyzed DNA unwinding
To judge the ability of each (d)NTP to support helicase catalyzed DNA unwinding, MBHAs were performed under conditions where the initial reaction rates were linear with the amount of NS3h present in the reaction mixture. A series of unwinding reactions was performed in the presence of ATP, or with another canonical NTP (Figure 1(b)), or dNTP (Figure 1(c)). Each NTP fueled unwinding, but reaction rates with each NTP differed significantly. Compared with ATP, GTP supported rates that were typically about 5-times slower, while dTTP supported unwinding rates that were about 3-times faster than those seen with ATP. Furthermore, when comparing an NTP to its analogous dNTPs, the dNTP supported 2–3 fold faster unwinding. These results suggest that the structure of not only the NTP base, but also its sugar moiety, influences unwinding rates.
In order to determine if this specificity is conserved among the various HCV strains, the NTP screen was repeated with five different NS3h proteins. To simplify comparisons, initial velocities (vo) were determined from the slope of the initial linear portion of each reaction. We have shown before that HCV helicases derived from different HCV genotypes unwind DNA (or RNA) at different rates and the same differences were again observed using the MBHA (Figure 1(d)). Typically genotype 1b helicases were the most active in unwinding assays while those from genotype 3a were least capable in unwinding assays. Importantly, however, the same specificity was observed in all proteins tested, regardless of their genotypic origin (Fig. 1d).
In a second set of NTP screens, the specificity of NS3h was compared with the full-length NS3 or a single chain protein that mimics the NS3–NS4A complex. Proteins with the protease unwound the substrate more slowly in this assay, but their relative specificity when fueled by the various (d)NTPs was preserved. Even though the relative NTP specificity was similar regardless of whether or not the protease or NS4A were present, both full-length proteins demonstrated somewhat less pronounced differences in unwinding rates when fueled by the various NTPs than NS3h (Figure 1(d)). In other words, the difference in unwinding rates between the best (dTTP) and worst (GTP) activating (d)NTPs was less for protease possessing proteins than for the various NS3h’s lacking a protease domain.
The unique and conserved HCV helicase NTP specificity can be explained by either different affinities for each activator (i.e. a Kd effect) or a different extent of activation (i.e a Vmax effect). To distinguish between these two possibilities, MBHAs were performed at various concentrations of ATP, the best activator (dTTP), and the worst activator (GTP) (Figure 2(a)). Since free metal or free NTP inhibits the HCV helicase, care was taken to perform each assay at the same concentration of free metal. To minimize any possible salt effects, NTP titrations were performed with enough MgCl2 added to each reaction to saturate all NTPs (to form Mg(NTP)2− complexes) and leave only 0.25 mM of free Mg2+. Under these conditions, optimal unwinding was observed at Mg(NTP)2− concentrations between 0.5 and 1 mM for each NTP used. In each case slower unwinding rates were observed as Mg(NTP)2− concentrations were increased above 1 mM. The simplest method to analyze these Mg(NTP)2− titrations would be to simply fit data to the Michaelis-Menten equation while excluding all data obtained at Mg(NTP)2− above 1 mM. Such analyses (dotted lines in Figure 2(a)), suggests that each Mg(NTP)2− supports a very different Vmax for unwinding. Mg(ATP)2− supports a Vmax that is 15 times faster than Mg(GTP)2−, and Mg(dTTP)2- supports a Vmax that is 6.7 times faster than the Vmax supported by Mg(ATP)2−. The apparent Km’s are more similar and vary by less than 7-fold compared to the 106-fold differences seen among the Vmax’s. There are large errors associated with such fits and the curves are poorly restrained, however, because there are few points outside the linear range.
Figure 2.
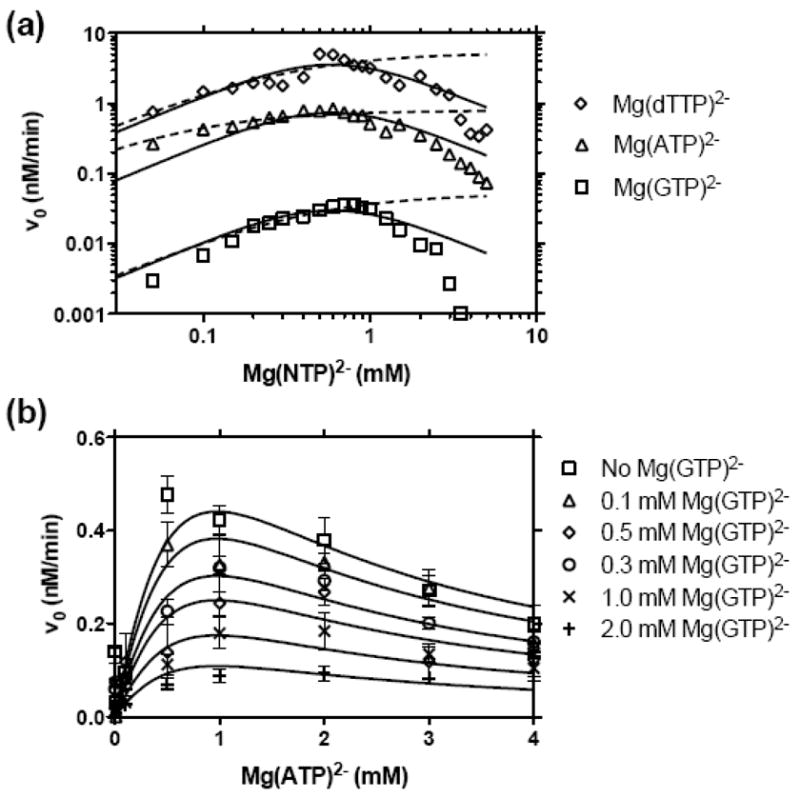
NS3h_1b(con1) catalyzed DNA unwinding at various NTP concentrations. MBHAs were performed as described in Figure 1 except that various concentrations of NTPs were used and MgCl2 was added to each reaction such that only 0.25 mM free Mg2+ was present at the start of each reaction, assuming Mg2+ forms a 1:1 complex with ATP with a high affinity. For example, reactions with 1 mM ATP, contained 1.25 mM total MgCl2, whereas those with 0.2 mM dTTP contained 0.45 mM MgCl2. (a) Comparison of reactions activated by GTP (squares), ATP (triangles), and dTTP (diamonds). The data are globally fit to a model for substrate inhibition (equation (2), solid lines), with a Ka describing NTP activation of 3.8 mM and a Ki describing NTP inhibition of 0.09 mM. In this model each NTP supports a different Vmax: GTP= 0.41 nM/min, ATP = 10 nM/min, and dTTP = 50 nM/min. In an alternate analysis, data for reaction performed at or below 1 mM NTP were fit to the Michaelis-Menten equation (dotted lines) with the following constants: Km (GTP)= 0.41 mM, Vmax (GTP) = 0.05 nM/min, Km (ATP)= 0.07 mM, Vmax (ATP) = 0.79 nM/min, Km (dTTP)= 0.30 mM, Vmax (dTTP) = 5.3 nM/min. (b) Initial rates of NS3h catalyzed DNA unwinding in the presence of various concentrations of ATP and GTP. Data are fit to a model where GTP acts as a non-competitive inhibitor while ATP acts as an activator and substrate inhibitor (equation (3)) with a Ka (ATP) of 1.3 mM, Ki (ATP) of 0.69 mM, Kii (GTP) of 0.65 mM, and a Vmax of 1.6 nM/min.
To attempt to utilize all the data collected (rather than arbitrarily discarding all rates obtained at Mg(NTP)2− concentrations above those needed for peak activity), data were also fit to a model that assumed that NTP could bind both as an activator and as an uncompetitive (i.e. “substrate”) inhibitor (equation (2)). In equation (2), Ka can be thought of as a measure of the affinity for the productive binding event, while Ki can be thought of as the affinity of the (d)NTP for a non-productive binding event. Data fit equation (2) well if all three NTPs were assumed to bind with equal affinities, i.e., Ka’s and Ki’s were shared in a global fit (Figure 2(a), solid lines), but data fit poorly if it were assumed that all three NTPs support the same Vmax (fit not shown). Deviations from this model occur mainly at NTP concentrations above 3 mM, where slower unwinding rates might be attributed to self association of Mg(NTP)2− with itself, making it unavailable for interaction with the enzyme.32 Another explanation for the slower observed unwinding rates at higher NTP concentrations is that they are simply due to higher ionic strengths present in such reactions. NS3h is exquisitely sensitive to salt effects and in MBHAs, NaCl inhibits NS3h catalyzed DNA unwinding with an IC50 of 15 mM (data not shown). Nevertheless, both a global fit to equation (2) and individual Michaelis-Menten plots, support the contention that different NTPs support different unwinding rates, but they bind with similar affinities.
In a second set of reactions, unwinding rates were measured in the presence of Mg(ATP)2− and the poorest activator, Mg(GTP)2−, to determine if Mg(GTP)2− could inhibit Mg(ATP)2− fueled unwinding. The goal was to determine if Mg(GTP)2− and Mg(ATP)2− competed for the same site on the enzyme. Reactions were performed with various amounts of ATP, and with various concentrations of Mg(GTP)2− (Figure 2(b)). The data fit reasonably well to a model where GTP competes for both the activating (Ka) and inhibitory site (Ki) occupied by Mg(ATP)2− (equation (3), Figure 2(b)). In this model, the affinity for Mg(ATP)2− and Mg(GTP)2− were similar. These results support the notion that all NTPs bind to the same site on HCV helicase with similar affinities.
NTP specificity under single-turnover conditions
The above data suggest that the nature of the NTP influences the rate at which HCV helicase can unwind DNA. However, it should be noted that all the above MBHAs were performed under conditions where enzyme molecules were free to re-bind substrates after dissociating. Since others have suggested that the rate limiting step in such “multiple turnover” HCV helicase catalyzed reactions is product release, the above data could simply reflect slower or faster product release in the presence of different NTP fuels, not differences in rates of strand separation. To more directly examine how various NTPs affect unwinding, similar reactions were repeated under single turnover conditions comparing the best (dTTP), and worst (GTP) activators with ATP (Figure 3). Reactions were performed with a stopped-flow device where one syringe contained enzyme and DNA substrate and a second syringe contained NTP and an enzyme trap, the oligonucleotide dT20. If the same amount of dT20 were added with the substrate, no strand separation would occur.20 Thus, any observed separation in this setup would be due only to the action of enzymes bound to the DNA substrate at the beginning of the reaction. The observed rate of the reaction under single turnover conditions reflects several different factors, such astranslocation rate, dissociation rate, and step size. The amplitude of the single-turnover reaction reflects the amount of DNA unwound in a single encounter with the substrate, that isthe helicase “processivity.”34
Figure 3.
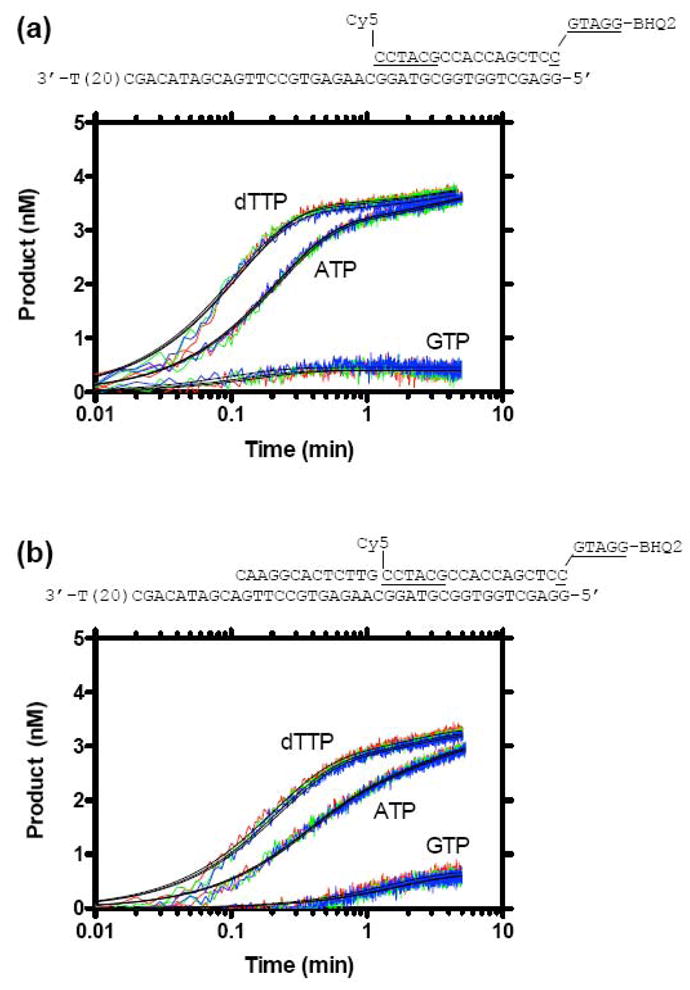
NS3h_1b(con1) catalyzed DNA unwinding in the presence of an enzyme trap (single turnover conditions). Reactions were performed by rapidly mixing 150 μl from syringe A containing 50 mM MOPS pH 6.5, 10 nM DNA-substrate, 50 nM NS3h, 4 mM MgCl2 with 150 μl from syringe B containing 1 mM NTP and 2 μM of the oligocucleotide dT20 (5′-TTTTT TTTTT TTTTT TTTT-3′). Final concentrations were 25 mM MOPS pH 6.5, 5 nM DNA substrate, 25 nM NS3h, 2 mM MgCl2, 0.5 mM NTP and 1 μM dT20. (a) Three independent reactions (red, green, blue lines) in which unwinding of a 17 bp DNA substrate was monitored in the presence of ATP, dTTP, and GTP. (b) Three independent reactions (red, green, blue lines) in which unwinding of a 30 bp DNA substrate was monitored in the presence of ATP, dTTP, and GTP. Data were fit to a two-phase rate equation (equation (4)) to yield the rate constants and amplitudes summarized in Table 1. Curve fits are shown as solid black lines.
When single turnover reactions were performed with a 17 base pair MBHA substrate, clear differences were again apparent among the various NTPs (Figure 3(a)). The data fit best to a two-phase rate equation (equation (4)). All reactions performed with either ATP or dTTP displayed a clear slow (lag) phase. The first order rate constants describing the slow phases (kslow) of both ATP and dTTP fueled reactions were similar, but the amplitudes of the slow phases (Aslow) of ATP fueled reactions were almost twice those for dTTP fueled reactions (Table 1). The fast phases of the ATP and dTTP fueled reactions were similar, both in terms of rate constants (kfast) and reaction amplitudes (Afast). In contrast, when GTP was used to fuel unwinding of a 17 base pair substrate, no slow phase was observed, and while the rate constants describing the fast phases of GTP fueled reactions were similar to those describing ATP and dTTP fueled reactions, the amplitudes of the GTP fueled reactions were 7 times less than those seen in ATP fueled reactions. Thus, differences between the ability of the various NTPs to fuel unwinding appear to be due to different reaction amplitudes.
Table 1.
Rate constants determined from single-turnover MBHAs. Three separate reactions were performed using three different NTP fuels using two different MBHA substrates, as described in Material and Methods and shown in Figure 3. Each parameter is the average of three parameters determined from three fitted reactions. Errors are standard deviations.
| Substrate | Parameter | dTTP | ATP | GTP |
|---|---|---|---|---|
| 17 base pairs | kslow (min−1) | 0.28 ± 0.06 | 0.28 ± 0.05 | ~0a |
| Aslow (nM) | 0.48 ± 0.03 | 0.70 ± 0.04 | ~0a | |
| kfast (min−1) | 9.1 ± 0.4 | 8.4 ± 0.1 | 8.3 ± 1.9 | |
| Afast (nM) | 3.4 ± 0.4 | 3.1 ± 0.02 | 0.42 ± 0.03 | |
| 30 base pairs | kslow (min−1) | 0.50 ± 0.02 | 0.46 ± 0.03 | 0.62 ± 0.08 |
| Aslow (nM) | 0.66 ± 0.02 | 1.2 ± 0.03 | 0.11 ± 0.05 | |
| kfast (min−1) | 4.8 ± 0.3 | 3.1 ± 0.1 | 0.61 ± 0.06 | |
| Afast (nM) | 2.6 ± 0.03 | 1.8 ± 0.03 | 0.58 ± 0.08 |
Reactions with the 17 base pair substrate fueled by GTP fit better to a 1-phase model. In the two-phase model, the slow phase for the 17-base pair GTP fueled reactions was extraordinarily slow and brief (e.g. 1.4 × 10−16 min−1, 1.7 × 10−16 nM).
To examine if single turnover reaction rate constants and amplitudes accurately reflect unwinding, the same reactions were repeated with a longer, 30 base pair DNA substrate, in which a third oligonucleotide was annealed upstream of the molecular beacon (Figure 3(b)). If reaction amplitudes truly reflect processivities, amplitudes should be lower when longer duplexes are used. Indeed, with the 30-bp duplex total reaction amplitudes were lower, but this change was mainly due to lower amplitudes of the fast phase of the reaction. Amplitudes of the slow phase were higher when NS3h unwound the 30-bp substrate. As seen with the shorter substrate, the helicase spent about twice as long in the slow phase when ATP fueled the reaction than when dTTP fueled the reaction. Again the slow phase of the reaction was much shorter when GTP was used as a fuel. Unlike with the shorter substrate, the fast phase of the reaction proceeded with very different rate constants when supported by the three fuels, with GTP supporting the slowest, least processive reactions, and dTTP supporting the fastest most processive reactions. In general, however, the three NTPs influenced both unwinding rates and processivity in a manner proportional to that seen in steady-state MBHAs, demonstrating that the specificity seen in steady state reactions reflects actual differences in how each NTP supports HCV helicase catalyzed DNA unwinding.
The average rate constants for the fast phase of the single turnover MBHA reactions (Table 1) reflect unwinding rates far faster than those observed under multiple turnover conditions, again supporting the notion that product release or another event sets the upper limit for kcat. For example, maximal steady state unwinding rates observed with Mg(ATP)2− were 0.8 nM/min at 25 nM NS3h, and these numbers would reflect a kcat of 0.032 min−1. Similarly, the kcat for the Mg(dTTP)2− fueled reaction were at most 0.2 min−1. These kcat calculations are probably over-estimates since they assume NS3h unwinds as a monomer, which is debatable, but nevertheless these kcat’s are far slower than rate constants describing the fast phase of the unwinding for either the 17-bp or 30-bp substrate (Table 1).
Role of the base and sugar moieties in NTP preference
To examine which portion of the NTP affects HCV helicase catalyzed DNA unwinding, we first examined the ability of a series of purine NTPs to fuel DNA unwinding (Figure 4(a)). All of the NTPs fueled unwinding with rates that were different from those seen with ATP, suggesting that the structure of the base moiety affected the activity of the enzyme. The only modified purine that supported unwinding faster than ATP was an NTP with a methyl group added to the N6 position of ATP, which fueled unwinding at rates 22% faster than those observed with unmodified ATP. All other purine modified NTPs investigated supported slower DNA unwinding than ATP. An NTP with an amino group at the 2 position of ATP supported rates only 78% of those seen with ATP. Notably slower unwinding rates, 47% of those supported by ATP, were seen with an NTP with a methyl group added to the N1 position of ATP. GTP fueled unwinding at rates five times slower than those seen with ATP, and Xanthosine triphosphate (XTP) fueled unwinding at the slowest rates, only 7% of that seen with ATP.
Figure 4.
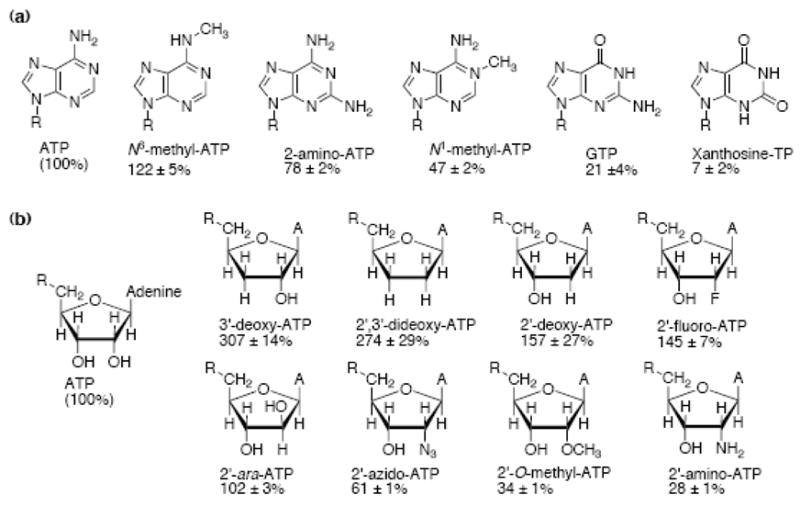
The relative ability of various NTP analogs to support NS3h_1b(con1) catalyzed DNA unwinding. Reactions were performed as described in Figure 1, except that ATP was replaced with various NTP analogs. Initial rates were compared to those obtained under the same conditions with ATP and are reported as a percent relative to ATP, errors are reported as ± one standard deviation. (a) Analogs with modified bases. For N6-Me-ATP, 2-amino-ATP and N1-Me-ATP, n=2, otherwise, n=3. (b) Analogs with modified sugars. For 2′-ara-ATP, 3′-dATP and 2′,3′-ddATP, n=2, otherwise, n=3.
Next, since the sugar moiety also influenced unwinding rates (Figure 1), we analyzed unwinding rates supported by a second set of NTP analogs with sugar modifications (Figure 4(b)). Some compounds in this set are of particular interest because they are under investigation as possible anti-HCV agents. For example, both obligate chain terminators, which lack a 3′-OH, and non-obligate chain terminators where the 2′-C is modified, are potent inhibitors of the HCV NS5B RNA-dependent RNA polymerase.
A comparison of rates supported by NTPs with modified sugars reveals other clear structure activity relationships. NTPs with large functional groups at the 2′ position, such as amino, O-methyl or azido, supported slower unwinding rates: 28, 34 and 61% of the unwinding rate seen with ATP, respectively. In contrast, NTPs with smaller groups such as F or H, in place of the 2′-OH of ATP, supported faster unwinding rates: 145 and 157% of that of ATP, respectively. An NTP where 2′-OH faced the other side of the sugar ring (i.e. an NTP based on arabinose rather than ribose, 2′-ara-ATP) supported practically the same unwinding rates as ATP, indicating that the stereochemistry of the 2′ position has little influence on unwinding rates.
The nature of the functional group present at the ribose 3′ position also affected observed unwinding rates. 3′-dATP fueled unwinding at rates 3-fold faster than ATP. 2′,3′-ddATP supported unwinding rates 2.7 fold faster than ATP- a slight decrease in unwinding rate compared to 3′-dATP, but it should be noted that the rates for 2′-dATP and 2′,3′-ddATP are within error of each other, and may be considered equivalent.
Metal ion specificity suggests that NTPs bind two different enzyme conformations
In addition to screening various NTPs, we used our new MBHA to examine how divalent metal cations influence HCV helicase catalyzed DNA unwinding. Divalent metal cations are absolutely required for HCV helicase catalyzed DNA unwinding. One metal ion bridges the helicase and its NTP substrate and others might modulate interactions with DNA. We found that besides Mg2+, only Mn2+ could fuel unwinding. Other metals, such as Ca2+, Zn2+, and Co2+, did not support HCV helicase catalyzed DNA unwinding (data not shown). Interestingly, when compared with unwinding in the presence of Mg2+, Mn2+ supported 10-times faster rates than Mg2+ regardless of the concentration of metal in solution (Figure 5(a)). A similar preference for Mn2+ has been reported by others but has never been explained on a molecular level. As we have reported before with another unwinding assay, unwinding rates increase while increasing the amount of metal in an ATP fueled reaction mixture, but they subsequently fall at higher concentrations, suggesting that free metal inhibits the reaction (Figure 5(a)).
Figure 5.

Comparison of NS3h_1b(con1) catalyzed DNA unwinding and ATP hydrolysis in the presence of Mg2+ or Mn2+. MBHAs were performed as in Figure 1 except as noted. (a) Reactions containing 0.5 mM ATP were titrated with either MgCl2 (squares) or MnCl2 (triangles). (b) Initial DNA unwinding rates in the presence of MgCl2 or MnCl2 using the 8 canonical NTPs. (c) Reactions were titrated with various amounts of Mn(ATP)2− in a 1:1 ratio, in the presence of 0.25 mM free Mn2+, and compared with the same reactions performed with Mg2+ instead of Mn2+. Data for Mg(ATP)2- are from Figure 2(a). (d) ATP hydrolysis was monitored in MBHA’s containing 25 nM NS3h_1b(con1), 1 mM ATP and either 1.25 mM MgCl2 (squares) ore 1.25 mM MnCl2 (circles). Reactions were performed in triplicate and fit by linear regression to reveal rates of ATP hydrolysis of 629 ± 5.4 nM/min (Mg2+) and 605 ± 4 nM/min (Mn2+).
To examine how Mn2+ might influence NTP specificity, we next repeated the NTP screen with Mn2+ substituting Mg2+. Increased unwinding rates in the presence of Mn2+ were not unique to ATP, and at 0.5 mM all NTPs supported faster unwinding when 2 mM Mn2+ was used instead of 2 mM Mg2+ (Figure 5(b)). The specificity profile remained mostly unchanged in the presence of Mn2+, while the absolute magnitude of the rates increased (Figure 5(b)).
Since a M(ATP)2− complex (rather than ATP alone) activates the unwinding reaction, we next examined the possibility that Mg2+ and Mn2+ formed respective M(ATP)2− complexes that interacted differently with the enzyme. When various concentrations of M(ATP)2− were tested using only 0.25 mM free metal, both Mg(ATP)2− and Mn(ATP)2− activated unwinding in a near-identical fashion with peak rates between 0.5 and 1 mM M(ATP)2− although, again, the rates were proportionately faster in the presence of Mn2+ than Mg2+ at all concentrations of M(ATP)2− (Figure 5(c)).
We next examined if faster unwinding in the presence of Mn2+ might be due to faster ATP hydrolysis in the MBHA. To this end, triplicate MBHAs were performed in the presence of 25 nM NS3h_1b(con1), 1 mM ATP and 1.25 mM MgCl2 or MnCl2. Under such conditions, the MBHA DNA substrate is unwound about 14 times faster in the presence of Mn2+. Surprisingly, however, under the same conditions, ATP was actually hydrolyzed slightly slower in the presence of Mn2+ than it was in the presence of Mg2+, though the rates of ATP hydrolysis were so similar they are within experimental error (Figure 5(d)). Notably, ATP was hydrolyzed far faster than DNA was unwound both in the presence of Mn2+ and Mg2+. In the reactions containing 25 nM NS3h, 1 mM ATP and 1.25 Mg2+, about 1,000 ATP were hydrolyzed for each 25 bp duplex separated. The rates were more comparable with Mn2+, where about 75 ATP were hydrolyzed for each 25 bp duplex separated.
To better understand why Mn2+ supports faster unwinding rates, we next examined how Mn2+ influences rates of ATP hydrolysis under optimal conditions, and we examined the activity of various other similar divalent metal cations in the same assays. ATP hydrolysis was monitored in conditions similar to those used in unwinding assays except that less protein was added, they were performed at 37 °C (rather than 22 °C) and the MBHA substrate was not present. Instead, either no nucleic acid was present (Figure 6(a)) or poly(U) RNA was present (Figure 6(b)). Poly(U) was chosen because its is the best activator of the NS3 ATPase. The two conditions mimic the two states where HCV helicase hydrolyzes ATP the slowest (without nucleic acids) and the fastest (with poly(U)). In the presence of RNA, Mg2+, Mn2+, Co2+, and Zn2+ (but not Ca2+) supported hydrolysis (Figure 6(b)), but in the absence of RNA, only added Mg2+ enhanced hydrolysis whereas all other metals tested (Mn2+, Co2+, and Zn2+, and Ca2+) inhibited the rate of hydrolysis seen in the absence of added metal. In the absence of RNA, increasing Mg2+ increased ATP hydrolysis steadily from a basal rate, but increasing Mn2+ decreased ATPase activity (Figure 6(a)). In other words, Mg2+ stimulated the enzyme’s basal (i.e. activity in the absence of RNA) ATPase activity whereas other metals inhibited basal ATP hydrolysis. As stimulatory metals were added in the presence of RNA, ATP hydrolysis rates increased to maxima, and then fell again at higher metal ion concentrations. As was seen in the presence of the MBHA substrate and unlike in assays monitoring unwinding rates, hydrolysis rates were lower when Mn2+ replaced Mg2+ (Figure 6(b)). We interpret the difference of the ability of Mn2+ and Mg2+ to stimulate ATP hydrolysis as evidence that ATP can interact with two distinct forms of HCV helicase. One form is associated with RNA (or DNA) and hydrolyzes ATP rapidly, while the other enzyme form is not bound to RNA and hydrolyzes ATP slowly.
Figure 6.
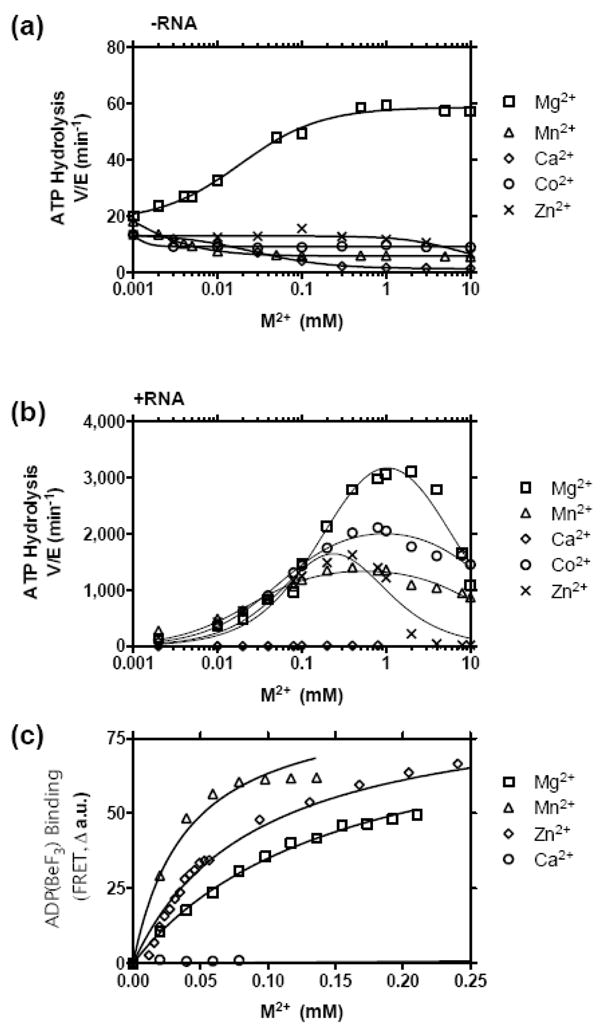
Ability of various metal ions to support NS3h_1b(con1) catalyzed ATP hydrolysis and NTP analog binding. Inorganic phosphate was measured after 15 min at 37 °C in reactions containing 2 mM ATP and either 100 nM (a) or 15 nM (b) NS3h in 25 mM MOPS pH 6.5 with the indicated amounts of metal ions. (a) ATP hydrolysis in the absence of RNA. (b) ATP hydrolysis in the presence of 0.1 mg/ml poly(U) RNA (Sigma, average length 2500 nt). (c) FRET analysis of mantADP(BeF3) binding to HCV helicase. A solution containing 0.5 μM NS3h 1b (con1), 5 mM NaF, 0.5 mM BeF2, 25 mM MOPS pH 6.25, 2 μM mantADP was titrated with various metal ions. Data are fit to a one site binding equation to determine Kd: (FRET=Bmax*[M]/(Kd+[M]) with shared a Bmax of 88 a.u. The following Kd’s were determined: MgCl2 = 153 μM, MnCl2 = 40 μM, and ZnCl2 = 90 μM.
If Mg2+, Mn2+, Co2+, and Zn2+ all bind in the presence of RNA, then it is likely that they also bind in the absence of RNA to form a enzyme-metal-ATP complex. However, from the analysis of ATPase rates in the absence of RNA, it is not clear whether HCV helicase binds Mn2+, Ca2+, Co2+ or Zn2+ coordinated ATP under such conditions. For example, Mn2+ could simply chelate ATP in the absence of RNA, and Mn(ATP)2− might not bind the enzyme. Therefore, to determine whether or not Mg2+, Mn2+, Co2+, and Zn2+ form an enzyme-metal-NTP complex, we examined metal binding using the fluorescent non-hydrolyzable ATP analog mant-ADP(BeF3).40 This assay measures FRET that occurs between the protein and bound mant-ADP. Previously, Lam et al. have shown that FRET in this assay is proportional to the amount of Mg2+ in solution. Substitution of Mg2+ with other divalent cations revealed that both Mn2+ and Zn2+ formed an enzyme-metal-NTP complex, but that Ca2+ did not (Figure 6(c)). Conclusive results could not be obtained with Co2+ because that metal quenched fluorescence at high concentrations. Data fit to a standard one-site binding model to reveal that both Mn2+ and Zn2+ appear to form the complex more tightly than Mg2+ (Figure 6(c)).
Discussion
The NS3 helicase catalyzes a complex reaction requiring energy derived from NTP binding and/or hydrolysis. This study provides a detailed look at how NTPs and divalent metal cations interact with HCV helicase to fuel a DNA unwinding reaction. The NS3 protein displays a clear specificity for (d)NTPs which is conserved among the HCV genotypes. In addition, reactions with a panel of nucleotide analogs have more precisely clarified contacts that are made between the helicase and its NTP fuel, implying intimate and discriminatory contacts between NTP and enzyme. This uniquely conserved fuel specificity could be used as a starting point for the design of nucleotide derivatives or possibly non-nucleoside based inhibitors. The results also allow a further delineation of the mechanism whereby NTPs fuel unwinding.
The simplest scheme for the action of a monomeric motor protein fueled by ATP is one in which the protein switches between two different conformations, which differentially bind the substrate on which the protein moves. Each conformation could theoretically hydrolyze ATP, and at each stage, the protein could theoretically switch between the two conformations. However, to function as a motor, the conformational changes must be coordinated with ATP hydrolysis such that the geometric changes occur only at two stages to provide a “power stroke.” With other motor proteins, it is more clear when the protein changes conformations. For example, kinesin changes conformations after hydrolysis and after phosphate release.42 Although more than one conformation of HCV helicase has been observed in crystal structures, it is not clear if or when any conformational change takes place in the helicase catalyzed unwinding reaction. Below, we interpret our data in light of a simplified model that assumes HCV helicase acts as a monomer and functions as a motor by shifting between only two distinct protein conformations. Although it is recognized that the NS3 helicase can unwind DNA as a monomer, it is also well established that the protein forms dimers and higher order oligomers. In the cell, multiple NS3 subunits exist as part of the viral replicase complex, and the NS3 cellular concentration greatly exceeds the total cellular concentration of HCV RNA. Thus, it needs to be recognized that the model below applies only to a single core functional unit of a complex macromolecular machine, and that additional inter-protein interactions most likely coordinate the basic NS3 motor function with other components of the viral replicase machinery.
The data presented here support a model in which ATP hydrolysis leads to a conformational change, and a second conformational change takes place sometime after product release. We propose a minimum of two HCV helicase conformations. One conformation, which binds DNA/RNA tightly, might resemble the structure of NS3h bound to an oligonucleotide (e.g. PDB 1A1V). The other might resemble a conformation where the protein is free of RNA (e.g. PDB 8OHM), where domain 2 is rotated away from domain 1 to produce a larger cleft between the RecA-like motor domains. In a manner similar to the Brownian motor model proposed by Levin et al., we propose that the “open” form of the enzyme is free to slide along the substrate (DNA or RNA) while the closed form is tightly bound to DNA. A coordinated switch between the conformations would allow the protein to move like a Brownian engine. During unwinding, ATP could interact with either form, but a conformational change must occur during a single binding cycle, or the enzyme would not function as a molecular motor. Hydrolysis by either form without a conformational change would be unproductive. Tight ATP binding to the open form would prevent the enzyme from re-binding to the substrate, while tight binding to the closed form would prevent the protein from translocating.
The main line of evidence supporting ATP binding to two different NS3h conformations comes from metal ion specificity studies. The more efficient NS3h catalyzed DNA unwinding in the presence of Mn(ATP)2− could be explained by the fact that Mn(ATP)2− interacts with the open conformation of the enzyme in a manner different from how Mg(ATP)2− interacts with the open form. ATP appears to bind HCV helicase regardless of whether Mg2+ or Mn2+ is the divalent metal cation in solution. As evidence, both Mg2+ and Mn2+ supported ATP hydrolysis in the presence of RNA (Figure 6(b)), i.e., when the protein is in the closed conformation. While only Mg2+ stimulates hydrolysis when HCV helicase is not bound to RNA, i.e., in the open conformation (Figure 6(a)), both metals support the formation of the ternary enzyme-metal-nucleotide complex (Figure 6(c)). Thus, it appears that both the closed and open forms can bind Mn(ATP)2− or Mg(ATP)2−, but the open form binds Mn(ATP)2− in a manner such that Mn(ATP)2− is hydrolyzed at least 100 times slower than Mg(ATP)2−.
The fact that Mn2+ suppresses the basal rate of HCV helicase catalyzed ATP hydrolysis is also important because it reveals that in the presence of Mn2+, NS3h more closely resembles more efficient helicases and nucleic acid motor proteins. Most helicases hydrolyze ATP very slowly in the absence of stimulating nucleic acids, and it has long been recognized that HCV helicase is unusual because it rapidly hydrolyzes ATP in the absence of nucleic acid. In other words, in the presence of Mg2+, nucleic acid only stimulates HCV helicase 10–100 fold, whereas nucleic acid stimulates ATP hydrolysis catalyzed by similar model helicases by 1,000 fold or more.45 ATP hydrolysis in the absence of nucleic acid is clearly unproductive and might yield to stalling of a motor protein. For example, it is conceivable that ATP binding to the open form of HCV helicase switches the protein to the closed conformation, whereas ATP hydrolysis by the closed form switches the protein to the open conformation. ATP hydrolysis by the open form might bypass a key switch to the closed conformation and lock the enzyme in a state that binds DNA poorly. Such a model would explain how suppression of basal ATP hydrolysis (by Mn2+) would allow the protein to more efficiently use the energy stored in ATP to move along DNA. In support of more efficient ATP utilization in the presence of Mn2+, comparisons of steady state unwinding rates and ATP hydrolysis reveal that NS4h hydrolyzes about 40 ATP in the time it takes to separate a single base pair of duplex DNA when Mg2+ is the only divalent cation present, but NS3h hydrolyzes only 3 ATP in the time needed to separate a base pair in the presence of Mn2+.
Even though our discovery that Mn2+ suppresses HCV catalyzed ATP hydrolysis in the absence of RNA is new and noteworthy, it must be recognized that this clear difference in ATP hydrolysis simply correlates with more efficient unwinding in the presence of Mn2+. No direct link has been demonstrated in this study, and exactly how this differential binding of Mn(ATP)2− leads to faster unwinding relative to Mg(ATP)2− is not clear at this time. Alternate explanations for more efficient HCV helicase catalyzed DNA unwinding in the presence of Mn2+ exist that do not relate to ATP binding or hydrolysis. Mn2+ interacts not only with ATP but also with the DNA substrate and Mn2+ might facilitate faster unwinding rates simply by altering the affinity of the enzyme for the DNA substrate or products. To thoroughly examine these possibilities and determine exactly how Mn2+ assists unwinding one could conceivably investigate binding and unwinding in the presence of Mn2+ using MBHA substrates, but the experiments would require faster stopped-flow instruments than the one used in this study.
Besides providing further evidence for a nucleotide induced conformational change, the above data reveal how different NTPs and NTP analogs interact with the enzyme. The data reveal that the enzyme makes critical contacts with both the base and the sugar while the NTP fuels unwinding. Specifically, the Watson-Crick edge of the purine ring and the 2′ and 3′ groups of the ribose appear to contact the enzyme at some stage of the reaction.
Comparison of NTPs with different purines reveals that the enzyme prefers NTPs where the N1 is not protonated. As evidence, all NTPs lacking hydrogen at position N1 of the purine ring (ATP, N6-methyl-ATP and 2-amino-ATP) supported faster unwinding than NTPs with hydrogen at the purine N1 (N1-methyl-ATP, xanthosine-TP and GTP). The modification to ATP with the single biggest impact was the presence of a methyl group at the N1 position of ATP. N1-methyl ATP supported unwinding rates less than half of those seen with ATP. Further changes to ATP at either the 2 or 5 positions caused a further decrease in the ability to support unwinding. Thus, we conclude that the single greatest factor affecting rates of unwinding fueled by purine NTPs seems to be the hydrogen bond donor or acceptor ability of the nitrogen at position N1 on the purine ring, with other Watson-Crick base-pairing positions having less relative impact on NTP fueled unwinding rates.
We propose that contacts with the base are made primarily when the enzyme is in the open conformation because a similar NTP specificity is observed when the ATPase rate is measured in the absence of RNA and added magnesium. Under such conditions, NTPs bind weakly to the enzyme and subtle differences in specificity can be more easily detected at NTP concentrations needed to detect hydrolysis. In the absence of RNA and metal ions, the protein hydrolyzes ATP fairly rapidly (kcat ~ 0.3 s−1), but it hydrolyses GTP at barely detectable rates. Analysis of NS3h catalyzed hydrolysis of purine NTP derivatives in the absence of RNA and added metals reveals that the presence of hydrogen at the N1 position is again critical for the protein-NTP interaction. Thus, it appears that NTPase activity in the absence of RNA directly correlates with NS3h catalyzed DNA unwinding rates, suggesting that contact with the NTP base takes place when the enzyme switches from the closed to the open conformation during the power stroke.
In studies comparing NTPs with various sugars, the modification to ATP producing the largest impact on unwinding activation was the removal of the 3′-OH group, resulting in a tripling of unwinding rates compared with ATP. The second largest impact on unwinding rates is the size of the group at the 2′ position, with smaller groups (F, H) supporting faster unwinding rates and larger groups (NH2, OH, OCH3, N3) supporting slower unwinding rates than ATP. Overall, it is appears that size of the 2′ group, rather than the specific chemical nature of the group, is most important at the 2′ position of the ribose, while the presence or absence of a 3′-OH has the single biggest impact on the ability to fuel unwinding.
We should refrain from speculating about when the enzyme might contact the sugar of the NTP because unlike NTP purine specificity, HCV helicase specificity with regard to the sugar is not consistent regardless of the conditions used. For example, while all the NS3h proteins tested clearly preferred dATP to ATP by 2:1, this difference was much less pronounced when full-length NS3 was used instead of NS3h (Figure 1(d)), or when Mn2+ replaced Mg2+ (Figure 5(b)). The observed NTP specificity seen here with full length NS3 are similar to the preferences reported by Wardell et al., who also used full-length NS3, but it is still hard to reconcile our data with those of Locatelli et al., who reported a more dramatic difference between unwinding activation by ATP and dATP and poor unwinding with dTTP.
One last note regarding NTP specificity is the interesting observation that the preference of HCV helicase for dTTP is not unique to this class of proteins; the helicase encoded by E. coli bacteriophage T7 has long been known to prefer dTTP as a fuel for its helicase action.
Hepatitis C virus is responsible for profound morbidity and mortality worldwide, and new HCV treatments are desperately needed. Few HCV helicase inhibitors have been reported, but given our observation that NTP specificity profiles are conserved across HCV genotypes, one might want to renew efforts in developing helicase inhibitors as antiviral agents.48 Also relevant to HCV drug development efforts is the observation that HCV helicase can utilize a wide variety of compounds to fuel its action. Just as Heck et al. have previously observed with ribavirin triphosphate, NS3 helicase utilizes the triphosphate metabolites of HCV antivirals that act ultimately as inhibitors of the NS5B RNA-dependent RNA polymerase (Figure 4(b)). Since the HCV helicase has the ability to hydrolyze both obligate and non-obligate chain terminators, the possibility exists that a nucleoside triphosphate analog intended to inhibit the NS5B polymerase might not affect its intended target due to the conversion to its respective nucleoside diphosphate by NS3 helicase. The new MBHA could be used to refine lead compounds intended to target NS5B by finding derivatives that are not hydrolyzed by NS3. Alternatively, new screens might be worthwhile to discover nucleoside derivatives or even non-nucleosides that function as antivirals solely by inhibiting HCV helicase.
Materials and Methods
Proteins
Cloning, expression, and purification of the various recombinant HCV helicase constructs have been previously described: NS3h_1a(H77), NS3h_1b(J4), NS3h_2a(J6), NS3h_3a, NS3h_1b(con1), NS3_1b(con1), and scNS3-4A_1b(con1). Briefly, recombinant protein was expressed in E. coli Rosetta (DE3) cells (Novagen) transformed with an appropriate plasmid. One liter cultures were harvested, lysed by sonication, and purified by sequentially using Ni-NTA column chromatography, ammonium sulfate precipitation, gel filtration column chromatography, and ion exchange column chromatography. Protein concentrations were determined from absorbance at 280 nm using the following extinction coefficients: NS3h_1a(H77) = 46.2 mM−1 cm−1, NS3h_1b(J4)= 52.5 mM−1 cm−1, NS3h_2a(J6)= 52.3 mM−1 cm−1, NS3h_3a= 57.2 mM−1 cm−1, NS3h_1b(con1)= 51.8 mM−1 cm−1, NS3_1b(con1)= 64.8 mM−1 cm−1, scNS3-4A_1b(con1)= 64.8 mM−1 cm−1.
Helicase substrates
Helicase substrates were prepared by combining DNA oligonucleotides (Integrated DNA Technologies, Coralville, IA) at a 1:1 molar ratio to a concentration of 20 μM in 10 mM Tris HCl pH 8.5, placing in 95 °C water, and allowing them to cool to room temperature for 1 hr. The partially-duplex MBHA substrates possessing a 3′ ssDNA tail were then purified of free oligonucleotides (ssDNA) by mixing DNA 6:1 with 6X loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol FF, 40% sucrose) and separating with 20% non-denaturing PAGE at a constant 200 V for 1 hr. The Cy5 labeled duplex DNA and ssDNA bands were clearly visible without need for further staining. The MBHA substrate was excised, crushed, and soaked in 2 volumes of elution buffer (0.5 M ammonium acetate, 1 mM EDTA, pH 8.0) at 37 °C for 3 hours to elute the substrate. The supernatant was removed, the gel fragments rinsed with an additional volume of elution buffer, and this supernatant combined with the previous elution to give a solution of purified partially-duplex DNA. Concentrations of the final purified MBHA substrates were determined by measuring absorbance at 648 nm and using the extinction coefficient for Cy5 (250 mM−1 cm−1). Similar concentrations were confirmed using the approximate extinction coefficient for partially duplex DNA (1 A260 = 33 μg/ml DNA). Purification yielded an approximately 1 μM stock MBHA substrate solution (~50% recovery) that was diluted to 100 nM solutions in 10 mM Tris HCl pH 8.5 and stored at −80 °C until needed.
Molecular beacon based helicase assay (MBHA)
The details of the MBHA and how it mimics the standard helicase assays have been previously described.20 Briefly, unless otherwise noted, each 100 μL unwinding assay contained 25 mM MOPS pH 6.5, 2 mM MgCl2, 25 nM NS3h, 5 nM MBHA substrate, was initiated with 0.5 mM ATP, and was carried out at 22 °C. For experiments comparing various NTP concentrations, MgCl2 was added such that 0.25 mM free Mg2+ was present in solution. Data were collected on a Varian Cary Eclipse fluorescence spectrophotometer (Palo Alto, CA) equipped with a microplate reader. Stopped flow reactions were performed with an Applied Photophysics RX.2000 rapid kinetics accessory (Leatherhead, UK). Syringe A was loaded with 50 mM MOPS pH 6.5, 4 mM MgCl2, 50 nM NS3h, and 10 nM substrate. Syringe B was loaded with 1.0 mM ATP and 2 μM dT20. Reactions were started by rapidly mixing the contents of syringe A and B in a 1:1 ratio such that final concentrations of all reagents were identical to those noted above for the standard helicase assay, with the addition of 1 μM dT20 to serve as an enzyme “trap” and force single-turnover conditions.
ATP hydrolysis assay
A modified “malachite green” assay was used to measure ATP hydrolysis. 50 μL reactions contained 25 mM MOPS pH 6.5, 5 mM MgCl2, 2 mM ATP, 0.1 mg/mL poly(U) and were initiated by adding 5–100 nM NS3h. Reactions were incubated for 15 minutes at 37 °C then stopped by mixing 40 μL or the reaction with 200 μL of malachite green reagent (3 volumes of 0.045% (w/v) malachite green: 1 volume of 4.2% ammonium molybdate in 4N HCl: 0.05 volumes of 0.1% Tween 20). After 10 s, 25 μL of 34% sodium citrate was added to each reaction, color was allowed to develop for 20 minutes. A630 was proportional to the concentration of phosphate produced, and was used to calculate free phosphate using a standard curve.
mantADP(BeF3) binding assay
Formation of a 2′-O-(N-methylanthraniloyl)adenosine 5′-diphosphate (mantADP)-BeF3-helicase complex was monitored using Förster resonance energy transfer (FRET) with the same technique that was previously described. Specifically, a solution containing 0.5 μM NS3h_1b(con1),p 2 μM mantADP, 0.2% Tween-20, 25 mM MOPS pH 6.5, 5 mM NaF, and 0.5 mM BeF2 was titrated with mantADP. FRET was monitored by exciting the sample at 280 nm and monitoring emitted light a 443 nm.
Data Analysis
The concentration of substrate remaining at each time point in any MBHA was calculated using equation (1):
| (1) |
In equation (1), [S]0 is the concentration of MBHA substrate at the beginning of the reaction, BKG is the fluorescence of a blank sample containing all the reagents except for the MBHA substrate, Fo is the fluorescence before ATP addition, and F if the fluorescence at any given time point after ATP addition.
To determine initial velocities in MBHAs, datasets of fluorescence decay after ATP addition were either truncated after 2 min and fit to linear regression or fit to a single-phase exponential decay equation, and the first derivative of the resulting curve taken at time zero. Similar results were obtained with both methods. Initial velocities were fit directly to equations using non-linear regression with Graphpad Prism 5.0 (San Diego, CA).
Activation of unwinding as a function of NTP concentration (Figure 2(a), Figure 5(c)) was fit to an equation describing the NTP as both an activator and as an uncompetitive “substrate” inhibitor.
| (2) |
In equation (2), [A] is activator ((d)NTP) concentration, Ka is a dissociation constant describing binding of NTP to free enzyme, and Ki is a dissociation constant describing the binding of a second (inhibitory) NTP to the enzyme.
Inhibition by GTP (Figure 2(b)) was described by an equation where ATP acts as both an activator and an inhibitory substrate while GTP acts as a non-competitive inhibitor:
| (3) |
In equation (3), [A] is ATP concentration, [G] is GTP concentration, Ka is a dissociation constant describing binding of ATP to free enzyme, and Ki is a dissociation constant describing the binding of a second (inhibitory) ATP to the enzyme, and Kii is a dissociation constant describing enzyme-GTP binding.
Single turnover MBHAs (Figure 3) were analyzed by first calculating the concentration of free molecular beacon (product) by subtracting substrate remaining at each time point (calculated with equation (1)) from total substrate concentration in each reaction (5 nM). Product concentrations were fit to time with a two-phase rate equation:
| (4) |
In equation (4), Aslow is the amplitude of the slow phase of the reaction, kslow is a first order rate constant describing the slow phase, Afast is the amplitude of the fast phase of the reaction, and kfast is a first order rate constant describing the fast phase of the reaction.
Acknowledgments
We would like to thank Dr. Fred Jaffe, Ryan, S. Rypma, Gagandeep Narula, and Sukaliani Banik for valuable technical assistance. The National Institutes of Health grants AI052395 and MH085690 supported this work.
Abbreviations used
- HCV
hepatitis C virus
- MBHA
molecular beacon helicase assay
- NS3
nonstructural protein 3
- NS3h
an NS3 fragment lacking its protease domain
- (d)NTP
(deoxy) nucleoside triphosphate
- a.u
arbitrary fluorescence units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McHutchison JG. Understanding hepatitis C. Am J Manag Care. 2004;10:S21–9. [PubMed] [Google Scholar]
- 2.Hahm B, Han DS, Back SH, Song OK, Cho MJ, Kim CJ, Shimotohno K, Jang SK. NS3–4A of hepatitis C virus is a chymotrypsin-like protease. J Virol. 1995;69:2534–2539. doi: 10.1128/jvi.69.4.2534-2539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzich JA, Tamura JK, Palmer-Hill F, Warrener P, Grakoui A, Rice CM, Feinstone SM, Collett MS. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J Virol. 1993;67:6152–6158. doi: 10.1128/jvi.67.10.6152-6158.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gwack Y, Kim DW, Han JH, Choe J. Characterization of RNA binding activity and RNA helicase activity of the hepatitis C virus NS3 protein. Biochem Biophys Res Commun. 1996;225:654–659. doi: 10.1006/bbrc.1996.1225. [DOI] [PubMed] [Google Scholar]
- 5.Lam AM, Frick DN. Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J Virol. 2006;80:404–411. doi: 10.1128/JVI.80.1.404-411.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J Virol. 2000;74:2046–2051. doi: 10.1128/jvi.74.4.2046-2051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frick DN. The hepatitis C virus NS3 protein: a model RNA helicase and potential drug target. Curr Issues Mol Biol. 2007;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 8.Kim DW, Gwack Y, Han JH, Choe J. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem Biophys Res Commun. 1995;215:160–166. doi: 10.1006/bbrc.1995.2447. [DOI] [PubMed] [Google Scholar]
- 9.Tai CL, Chi WK, Chen DS, Hwang LH. The helicase activity associated with hepatitis C virus nonstructural protein 3 (NS3) J Virol. 1996;70:8477–8484. doi: 10.1128/jvi.70.12.8477-8484.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris PD, Byrd AK, Tackett AJ, Cameron CE, Tanega P, Ott R, Fanning E, Raney KD. Hepatitis C virus NS3 and simian virus 40 T antigen helicases displace streptavidin from 5′-biotinylated oligonucleotides but not from 3′-biotinylated oligonucleotides: evidence for directional bias in translocation on single-stranded DNA. Biochemistry. 2002;41:2372–2378. doi: 10.1021/bi012058b. [DOI] [PubMed] [Google Scholar]
- 11.Frick DN, Rypma RS, Lam AM, Gu B. The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J Biol Chem. 2004;279:1269–1280. doi: 10.1074/jbc.M310630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preugschat F, Averett DR, Clarke BE, Porter DJ. A steady-state and pre-steady-state kinetic analysis of the NTPase activity associated with the hepatitis C virus NS3 helicase domain. J Biol Chem. 1996;271:24449–24457. doi: 10.1074/jbc.271.40.24449. [DOI] [PubMed] [Google Scholar]
- 13.Locatelli GA, Gosselin G, Spadari S, Maga G. Hepatitis C virus NS3 NTPase/helicase: different stereoselectivity in nucleoside triphosphate utilisation suggests that NTPase and helicase activities are coupled by a nucleotide-dependent rate limiting step. J Mol Biol. 2001;313:683–694. doi: 10.1006/jmbi.2001.5088. [DOI] [PubMed] [Google Scholar]
- 14.Frick DN, Banik S, Rypma RS. Role of divalent metal cations in ATP hydrolysis catalyzed by the hepatitis C virus NS3 helicase: magnesium provides a bridge for ATP to fuel unwinding. J Mol Biol. 2007;365:1017–1032. doi: 10.1016/j.jmb.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levin MK, Gurjar M, Patel SS. A Brownian motor mechanism of translocation and strand separation by hepatitis C virus helicase. Nat Struct Mol Biol. 2005;12:429–435. doi: 10.1038/nsmb920. [DOI] [PubMed] [Google Scholar]
- 16.Myong S, Bruno MM, Pyle AM, Ha T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science. 2007;317:513–516. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sikora B, Chen Y, Lichti CF, Harrison MK, Jennings TA, Tang Y, Tackett AJ, Jordan JB, Sakon J, Cameron CE, Raney KD. Hepatitis C virus NS3 helicase forms oligomeric structures that exhibit optimal DNA unwinding activity in vitro. J Biol Chem. 2008;283:11516–11525. doi: 10.1074/jbc.M708125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serebrov V, Pyle AM. Periodic cycles of RNA unwinding and pausing by hepatitis C virus NS3 helicase. Nature. 2004;430:476–480. doi: 10.1038/nature02704. [DOI] [PubMed] [Google Scholar]
- 19.Dumont S, Cheng W, Serebrov V, Beran RK, Tinoco IJ, Pyle AM, Bustamante C. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature. 2006;439:105–108. doi: 10.1038/nature04331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belon CA, Frick DN. Monitoring helicase activity with molecular beacons. BioTechniques. 2008;45:433–40. 442. doi: 10.2144/000112834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vale RD, Milligan RA. The way things move: looking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- 22.Yao N, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 23.Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Bera AK, Kuhn RJ, Smith JL. Structure of the flavivirus helicase: implications for catalytic activity, protein interactions, and proteolytic processing. J Virol. 2005;79:10268–10277. doi: 10.1128/JVI.79.16.10268-10277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo D, Xu T, Watson RP, Scherer-Becker D, Sampath A, Jahnke W, Yeong SS, Wang CH, Lim SP, Strongin A, Vasudevan SG, Lescar J. Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 2008;27:3209–3219. doi: 10.1038/emboj.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wardell AD, Errington W, Ciaramella G, Merson J, McGarvey MJ. Characterization and mutational analysis of the helicase and NTPase activities of hepatitis C virus full-length NS3 protein. J Gen Virol. 1999;80:701–709. doi: 10.1099/0022-1317-80-3-701. [DOI] [PubMed] [Google Scholar]
- 27.Lam AM, Keeney D, Eckert PQ, Frick DN. Hepatitis C virus NS3 ATPases/helicases from different genotypes exhibit variations in enzymatic properties. J Virol. 2003;77:3950–3961. doi: 10.1128/JVI.77.7.3950-3961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kyono K, Miyashiro M, Taguchi I. Expression and purification of a hepatitis C virus NS3/4A complex, and characterization of its helicase activity with the Scintillation Proximity Assay system. J Biochem (Tokyo) 2004;135:245–252. doi: 10.1093/jb/mvh029. [DOI] [PubMed] [Google Scholar]
- 29.Neumann-Haefelin C, Frick DN, Wang JJ, Pybus OG, Salloum S, Narula GS, Eckart A, Biezynski A, Eiermann T, Klenerman P, Viazov S, Roggendorf M, Thimme R, Reiser M, Timm J. Analysis of the evolutionary forces in an immunodominant CD8 epitope in hepatitis C virus at a population level. J Virol. 2008;82:3438–3451. doi: 10.1128/JVI.01700-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heck JA, Lam AM, Narayanan N, Frick DN. Effects of mutagenic and chain terminating nucleotide analogs on enzymes isolated from various hepatitis C virus genotypes. Antimicrob Agents Chemother. 2008;52:1901–1991. doi: 10.1128/AAC.01496-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howe AY, Chase R, Taremi SS, Risano C, Beyer B, Malcolm B, Lau JY. A novel recombinant single-chain hepatitis C virus NS3–NS4A protein with improved helicase activity. Protein Sci. 1999;8:1332–1341. doi: 10.1110/ps.8.6.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sigel H. Isomeric equilibria in complexes of adenosine 5′-triphosphate with divalent metal ions. Solution structures of M(ATP)2- complexes. Eur J Biochem. 1987;165:65–72. doi: 10.1111/j.1432-1033.1987.tb11194.x. [DOI] [PubMed] [Google Scholar]
- 33.Porter DJ, Short SA, Hanlon MH, Preugschat F, Wilson JE, Willard DHJ, Consler TG. Product release is the major contributor to kcat for the hepatitis C virus helicase-catalyzed strand separation of short duplex DNA. J Biol Chem. 1998;273:18906–18914. doi: 10.1074/jbc.273.30.18906. [DOI] [PubMed] [Google Scholar]
- 34.Ali JA, Lohman TM. Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science. 1997;275:377–380. doi: 10.1126/science.275.5298.377. [DOI] [PubMed] [Google Scholar]
- 35.Levin MK, Patel SS. The helicase from hepatitis C virus is active as an oligomer. J Biol Chem. 1999;274:31839–31846. doi: 10.1074/jbc.274.45.31839. [DOI] [PubMed] [Google Scholar]
- 36.Jennings TA, Mackintosh SG, Harrison MK, Sikora D, Sikora B, Dave B, Tackett AJ, Cameron CE, Raney KD. NS3 helicase from the hepatitis C Virus can function as a monomer or oligomer depending on enzyme and substrate concentrations. J Biol Chem. 2009;284:4806–4814. doi: 10.1074/jbc.M805540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll SS, Tomassini JE, Bosserman M, Getty K, Stahlhut MW, Eldrup AB, Bhat B, Hall D, Simcoe AL, LaFemina R, Rutkowski CA, Wolanski B, Yang Z, Migliaccio G, De Francesco R, Kuo LC, MacCoss M, Olsen DB. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J Biol Chem. 2003;278:11979–11984. doi: 10.1074/jbc.M210914200. [DOI] [PubMed] [Google Scholar]
- 38.Murakami E, Bao H, Ramesh M, McBrayer TR, Whitaker T, Micolochick Steuer HM, Schinazi RF, Stuyver LJ, Obikhod A, Otto MJ, Furman PA. Mechanism of activation of β-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob Agents Chemother. 2006 doi: 10.1128/AAC.00400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boguszewska-Chachulska AM, Krawczyk M, Stankiewicz A, Gozdek A, Haenni AL, Strokovskaya L. Direct fluorometric measurement of hepatitis C virus helicase activity. FEBS Lett. 2004;567:253–258. doi: 10.1016/j.febslet.2004.04.072. [DOI] [PubMed] [Google Scholar]
- 40.Levin MK, Patel SS. Helicase from hepatitis C virus, energetics of DNA binding. J Biol Chem. 2002;277:29377–29385. doi: 10.1074/jbc.M112315200. [DOI] [PubMed] [Google Scholar]
- 41.Lam AM, Rypma RS, Frick DN. Enhanced nucleic acid binding to ATP-bound hepatitis C virus NS3 helicase at low pH activates RNA unwinding. Nucleic Acids Res. 2004;32:4060–4070. doi: 10.1093/nar/gkh743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandelkow E, Johnson KA. The structural and mechanochemical cycle of kinesin. Trends Biochem Sci. 1998;23:429–433. doi: 10.1016/s0968-0004(98)01278-x. [DOI] [PubMed] [Google Scholar]
- 43.Quinkert D, Bartenschlager R, Lohmann V. Quantitative analysis of the hepatitis C virus replication complex. J Virol. 2005;79:13594–13605. doi: 10.1128/JVI.79.21.13594-13605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho HS, Ha NC, Kang LW, Chung KM, Back SH, Jang SK, Oh BH. Crystal structure of RNA helicase from genotype 1b hepatitis C virus. A feasible mechanism of unwinding duplex RNA. J Biol Chem. 1998;273:15045–15052. doi: 10.1074/jbc.273.24.15045. [DOI] [PubMed] [Google Scholar]
- 45.Delagoutte E, von Hippel PH. Helicase mechanisms and the coupling of helicases within macromolecular machines. Part I: Structures and properties of isolated helicases. Q Rev Biophys. 2002;35:431–478. doi: 10.1017/s0033583502003852. [DOI] [PubMed] [Google Scholar]
- 46.Vamvakopoulos NC, Vournakis JN, Marcus SL. The effect of magnesium and manganese ions on the structure and template activity for reverse transcriptase of polyribocytidylate and its 2′-0-methyl derivative. Nucleic Acids Res. 1977;4:3589–3597. doi: 10.1093/nar/4.10.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matson SW, Tabor S, Richardson CC. The gene 4 protein of bacteriophage T7. Characterization of helicase activity. J Biol Chem. 1983;258:14017–14024. [PubMed] [Google Scholar]
- 48.Frick DN, Lam AM. Understanding helicases as a means of virus control. Curr Pharm Des. 2006;12:1315–1338. doi: 10.2174/138161206776361147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]