

Abstract
Objective
Smooth muscle cell (SMC) phenotypic modulation, an important component of atherosclerosis progression, is critically regulated by the matrix, with normal components of the healthy SMC matrix limiting modulation and atherosclerosis-associated transitional matrix proteins promoting phenotypic modulation. We sought to determine how collagen IV (which comprises the healthy artery wall) and monomeric collagen I (which comprises atherosclerotic lesions) differentially affect SMC phenotype.
Methods and Results
Plating SMCs on collagen IV resulted in elevated expression of SMC contractility proteins compared to collagen I. Concurrent with enhanced contractile gene expression, collagen IV stimulates binding of SRF to CArG boxes in the promoters of smooth muscle actin and smooth muscle myosin heavy chain. Coll IV also stimulated the expression of myocardin, a critical SRF coactivator required to drive expression of SMC specific genes. In contrast to collagen IV, collagen I stimulated enhanced expression of the inflammatory protein vascular cell adhesion molecule (VCAM)-1. NF-κB and NFAT-binding sites in the VCAM-1 promoter are critical for collagen I–mediated expression of VCAM-1 promoter activity. However, only inhibitors of NFAT, not NF-κB, were able to reduce collagen I–associated VCAM expression, and collagen I but not collagen IV stimulated NFAT transcriptional activity.
Conclusion
These results show for the first time that collagen IV and collagen I differentially affect smooth muscle phenotypic modulation through multiple pathways.
Keywords: transcription, differentiation, inflammation, NFAT, matrix
Cardiovascular disease is the leading cause of death in developed countries and is showing increasing incidence in the developing world.1 More than 80% of the cardiovascular disease–associated mortality is attributable to atherosclerosis, a chronic inflammatory disease of the vessel wall. At early stages, atherogenesis is driven by endothelial cell dysfunction and monocyte recruitment, while at late stages progression is generated by smooth muscle cell (SMC) proliferation, migration, and fibrosis.2 SMCs regulate vascular tone through alterations in their contractile properties and as such express a unique subset of contractility-related genes, including SM α-actin (SMA), smooth muscle myosin heavy chain (SM-MHC), smoothelin, calponin, and desmin. SMC activation during atherosclerotic progression induces the loss of this “contractile phenotype” and the adoption of a “synthetic phenotype,” characterized by enhanced proliferation, migration, and expression of fibrotic and inflammatory proteins concomitant with decreased expression of contractility-associated proteins.3,4 This change, termed phenotypic modulation, is thought to play a critical role in the progression of atherosclerosis from a clinically silent fatty streak into an advanced atheroma, and stimulating SMCs to transition back to a contractile phenotype may induce plaque stabilization.4,5 While the function of cell cycle regulators and profibrotic genes in atherosclerotic progression are well described, the role of inflammatory gene expression in SMCs has puzzled scientists for more than a decade.6
The extracellular matrix regulates both tissue architecture and cell phenotype. Cells use matrix receptors, such as integrins, to detect the changes in matrix rigidity and composition that occur during tissue remodeling. The resulting intracellular signals regulate a number of cellular processes including cell proliferation, survival, differentiation, and gene expression.7 SMCs are normally surrounded by a basal laminae, but in regions of early atherosclerosis this matrix is degraded allowing SMC interactions with the interstitial matrix. Basal laminae proteins such as collagen IV (Coll IV), laminin, and perlecan can limit SMC growth, enhance contractile gene expression, reduce inflammatory gene expression, reduce LDL uptake in culture, and inhibit matrix calcification.8,9 In contrast, the interstitial matrix proteins, including collagen I (Coll I), collagen III, fibronectin, and osteopontin, enhance SMC growth concomitant with elevated extracellular signal regulated kinase (ERK) phosphorylation and expression of cell cycle regulators.8,9 Inhibiting the integrins that bind these interstitial matrix proteins is sufficient to block SMC proliferation in response to PDGF, EGF, and bFGF10 and to reduce migration and neointima formation in vivo.11 Whereas Coll IV and polymerized Coll I limit SMC growth and promote the contractile phenotype,12 monomeric Coll I reduces contractile gene expression.13 Monomeric Coll I may be relevant to atherosclerotic plaques, because enhanced Coll I degradation in the atherosclerotic plaque14 is thought to promote the release of monomeric Coll I. Despite 30 years of research in the area, the molecular mechanisms by which the matrix alters SMC gene expression in phenotypic modulation are unknown.
Smooth muscle–specific contractile gene promoters are active under physiological conditions but are temporally silenced in response to injury or atherosclerosis.15–17 Nearly all gene promoters for smooth muscle-specific contractile proteins contain at least 2 CArG cis regulatory elements.3,18 CArG boxes bind SRF (serum response factor), which requires myocardin or myocardin-related transcription factors (MRTFs).19–21 Many growth factors and cytokines implicated in SMC phenotypic modulation have been shown to regulate this process, including platelet-derived growth factor (PDGF), angiotensin-II, transforming growth factor beta (TGF-β), and recently oxidized phospholipids and sphingosine-1–phosphate.3,22,23 However, regulation by the extracellular matrix has not been addressed.
The current work examines the molecular mechanisms by which a “protective” matrix (Coll IV) or an “atherosclerotic” matrix (monomeric collagen I) differentially affect SMC gene expression. We now show that the extracellular matrix alters SMC-specific contractile gene expression through differential effects on SRF recruitment to CArG boxes in the promoters of SMA and SM-MHC and through differential regulation of myocardin expression. We also demonstrate for the first time that Coll I promotes the expression of the inflammatory protein VCAM-1, and we show that this induction is NFAT-dependent.
Methods
The current study uses rat aortic SMCs plated under serum-free conditions on either Coll I (20 μg/mL; Sigma) or Coll IV (20 μg/mL; Chemicon and Trevigen). Promoter-luciferase reporter constructs for SMA, SM-MHC, VCAM-1, and NFATc (Clonetech) were transfected using FuGENE6 (Roche) and used to measure promoter activity. Total mRNA was extracted using Trizol (Invitrogen), cDNA was synthesized using the iScript cDNA synthesis kit (BioRad), and quantitative real-time polymerase chain reaction (PCR) was used to measure SMA, SM-MHC, myocardin, and VCAM-1 mRNA expression as previously described.24,25 Western blotting was used to measure protein expression. Chromatin immunoprecipitation (ChIP) was used to measure SRF enrichment on the SMA and SM-MHC promoter as previously described.24,25 Please see the supplemental materials for expanded Materials and Methods section (available online at http://atvb.ahajournals.org).
Results
Coll IV and Coll I Differentially Regulate Smooth Muscle Cell Differentiation Marker Expression
Matrix composition affects SMC phenotype both in vitro and in vivo. Although the molecular mechanisms regulating matrix-specific SMC proliferation and migration are well established, little is known concerning how matrix-specific signals affect the changes in gene expression associated with smooth muscle phenotypic modulation. Therefore, we sought to determine how matrices associated with the contractile vessel (Coll IV) and matrices associated with atherosclerosis (Coll I) differentially affect smooth muscle gene expression. To test this, primary rat aortic SMCs were transfected with luciferase reporter constructs driven by the SMA and SM-MHC promoters and plated in serum free media onto Coll I or Coll IV matrices for 6 hours. Plating cells on Coll IV significantly increased luciferase activity compared to Coll I (Figure 1A), suggesting that Coll IV stimulates the SMA and SM-MHC promoter. The rapid effect of Coll IV on smooth muscle contractile gene expression suggests that the effect occurs because of interactions with Coll IV and not because of interactions with another cell-derived matrix. To verify these luciferase assays, SMCs were plated on Coll I or Coll IV in serum-free media for 3 days to induce smooth muscle cell quiescence and stabilize the expression of smooth muscle-specific contractility proteins. The absence of serum also prevented the contamination of the collagen matrices with serum-derived matrix proteins, such as fibronectin and vitronectin. Expression of SMA and SM-MHC was then determined by quantitative RT-PCR and Western blotting. Plating cells on Coll IV enhanced the expression of both SMA and SM-MHC mRNA (Figure 1B and C) and stimulated a significant increase in SMA protein expression (Figure 1D) compared to cells on Coll I.
Figure 1.

Regulation of smooth muscle–specific gene expression by collagen isotypes. A, Rat aortic SMCs expressing luciferase constructs driven by the SMA or SM-MHC promoter were plated on Coll I or Coll IV for 6 hour. Luciferase assays were performed on cell lysates, P<0.05. B, SMCs were plated on Coll I or Coll IV for 3 days, and mRNA was extracted. SMA (B) and SM-MHC (C) expression levels were measured using quantitative RT-PCR and shown as a ratio to 18S expression, P<0.05. D, SMCs plated on Coll isotypes for 3 days were lysed and SMA protein levels were quantified using Western blotting and normalized to total protein. A representative image is shown, P<0.0001. All experiments represent 3 biological replicates, each replicate run in triplicate.
To address the molecular mechanism of the matrix-specific contractility protein expression, we analyzed the role of SRF recruitment to the SMA and SM-MHC promoters by ChIP. Plating cells on Coll IV enhanced the recruitment of SRF to both the SMA (Figure 2A) and SM-MHC (Figure 2B) promoters compared to cells on Coll I and increased H4Ac (data not shown). Because SRF binds to CArG boxes in the promoters of SMA and SM-MHC, we next used luciferase constructs driven by the SMA promoter containing point mutations in both CArG boxes within the promoter region and in the intronic CArG box. Mutation of any of the CArG boxes reduced the enhanced promoter activity in cells plated on Coll IV (Figure 2C), suggesting that Coll IV enhances the expression of smooth muscle contractile genes by promoting the recruitment of SRF to the CArG boxes present in the promoters of the target genes. We then tested whether the SRF coactivator myocardin showed differential expression in response to different Coll matrices. Quantitative RT-PCR revealed that plating cells on Coll IV induced a significant increase in myocardin mRNA expression compared to cells on Coll I (Figure 2D), suggesting differential regulation of myocardin expression is an underlying cause of matrix-specific phenotypic modulation. SRF protein expression was not altered by Coll IV compared to Coll I (Figure 2E). These data suggest that Coll isoforms differentially affect SMC contractile gene expression by altering the formation of SRF-myocardin complexes on the CArG box regions in the promoters of SMC contractile genes.
Figure 2.

Coll IV promotes SRF/CArG-dependent regulation of SMA and SMMHC. SMCs were plated on either Coll I or Coll IV for 3 days in serum free media. SRF enrichment by ChIP was performed and associated DNA complexes were analyzed by quantitative RT-PCR. Primers specific to the CArG A and CArG B regions of the (A) SMA promoter (P<0.001, n=4) and the (B) SM-MHC promoter (P<0.002, n=3) were used. C, SMCs were transfected with luciferase reporters driven by the SMA promoter containing mutations in each of 3 CArG boxes. Cells were trypsinized and plated on Coll I– and Coll IV–coated plates in serum free media for 24 hours. Luciferase assays were performed. Biological replicates, n=3, in triplicate, P<0.05. D, SMCs were plated on either Coll I or Coll IV for 3 days, mRNA was extracted, and myocardin expression levels were determined using quantitative RT-PCR. Representative results are shown as a ratio of myocardin expression to 18S; n=3, in triplicate, P<0.05. E, SMCs were plated on either Coll I or Coll IV for 3 days. Cells were lysed, and SRF protein levels were determined by Western blotting. Representative blot is shown.
Coll IV and Coll I Differentially Regulate Smooth Muscle Cell VCAM-1 Expression
Although smooth muscle cell phenotypic modulation is classically described as a two-state model whereby cells are in the contractile or synthetic state, it is now clear that there are multiple SMC phenotypes, including an inflammatory phenotype, that depend on the specific gene expression profiles and transcriptomes. VCAM-1 is a cell surface adhesion molecule classically involved in tethering leukocytes to the endothelial cell surface. Although the role of VCAM-1 expression in smooth muscle physiology is unknown,26 it is postulated to modulate the local inflammatory response within the atherosclerotic plaque.6 To test whether matrix similarly affected the expression of inflammatory genes, SMCs were plated on either Coll I or Coll IV for 1, 2, or 5 days, and VCAM-1 expression was measured using quantitative RT-PCR and Western blotting. Consistent with Coll I–enhanced phenotypic modulation, SMCs on Coll I displayed a striking increase in both VCAM-1 mRNA (Figure 3A) and protein (Figure 3B) compared to cells plated on Coll IV. The enhanced expression of VCAM-1 in cells on Coll I could be seen by 24 hours both at the mRNA and the protein level, and the differential expression pattern was maintained for at least 5 days.
Figure 3.
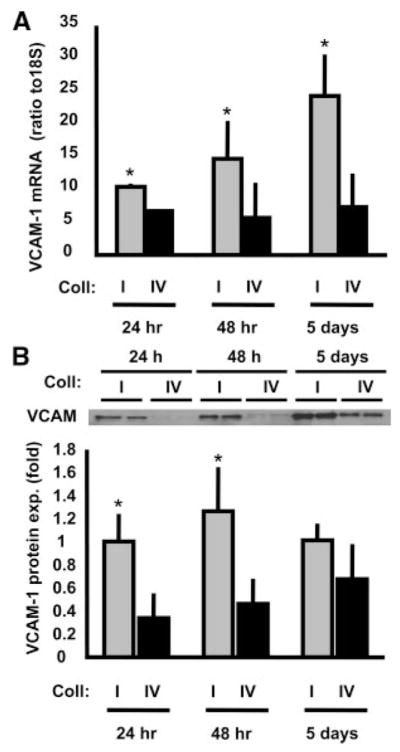
Coll I promotes VCAM-1 expression. SMC plating times on Coll I or Coll IV were varied at 24 hours, 48 hours, or 5 days, and VCAM-1 (A) mRNA expression (n=3, in triplicate, P<0.001) and (B) protein expression (n=3, in triplicate, P<0.001) were determined. A representative image is shown.
Calcineurin/NFAT Signaling Regulates Coll I–Induced Expression of VCAM-1
To determine the mechanism of matrix-specific VCAM-1 expression, we transfected SMCs with a luciferase construct driven by the VCAM-1 promoter, plated cells on Coll I or Coll IV, and analyzed whether point mutations in the VCAM-1 promoter affected its matrix-specific activation. Interestingly, we found that mutating the binding site for NF-κB, one of the classical regulators of VCAM-1 expression, was sufficient to abolish the matrix-specific effect on VCAM-1 expression (Figure 4). However, no difference could be detected in NF-κB activation between the two matrices by analyzing both nuclear translocation of NF-κB and its phosphorylation state (supplemental Figure I).
Figure 4.
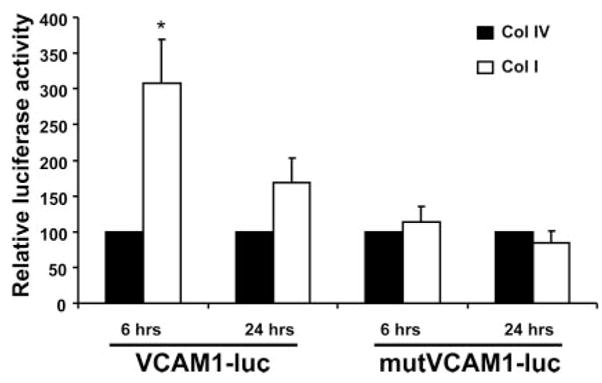
The NFAT/NF-κB binding site in the VCAM-1 promoter is required for Coll I–induced VCAM-1 expression. SMCs expressing luciferase constructs driven by the VCAM-1 promoter or a mutant VCAM-1 promoter lacking the NF-κB and NFAT cis elements were plated on Coll I or Coll IV for 6 or 24 hours. Luciferase assays were then performed on cell lysates. n=3, in triplicate, P<0.05.
The transcription factor NFAT also binds this NF-κB site in the VCAM-1 promoter.27 Therefore, we used pharmacological inhibitors of both NFAT and NF-κB to determine which transcription factor was required for matrix-specific activation of the VCAM-1 promoter. Treating SMCs plated on Coll I with the NF-κB inhibitor SN50, a cell-permeable inhibitory peptide, had no effect on VCAM-1 expression (Figure 5A), although SN50 was able to reduce interleukin (IL)-1β–induced VCAM-1 expression (supplemental Figure II). NFAT activation requires its dephosphorylation by the calcium-sensitive phosphatase calcineurin.28 Treating cells with the calcineurin inhibitor Cyclosporine A (CsA) resulted in a profound inhibition of VCAM-1 expression at both the protein (Figure 5A) and mRNA level (Figure 5B). The effect of CsA on VCAM-1 expression was not altered by simultaneous addition of SN50, suggesting that NFAT may be the primary regulator of matrix-specific VCAM-1 expression. In addition, both CsA and the NFATc inhibitor A-28522229 block Coll I-induced VCAM-1 promoter activity in the luciferase assay (Figure 5C). To verify that NFAT transcriptional activity was enhanced on Coll I, we transfected SMCs with an NFATc-luciferase reporter construct (3 NFATc cis elements, Clontech) and plated cells on Coll I or Coll IV in the absence or presence of CsA for 6 and 24 hours. Consistent with NFAT-driven VCAM-1 expression, NFAT activity was significantly enhanced on Coll I compared to Coll IV at the 6-hour time point (Figure 5D). CsA treatment abolished the increased luciferase activity seen on Coll I, confirming that the increased luciferase activity is NFATc-dependent.
Figure 5.

Coll I promotes NFAT-dependent VCAM-1 expression. A, SMCs were pretreated with SN50 (30 μg/mL), CsA (10 μmol/L), or a combination of the two for 1 hour. Cells were plated onto Coll I–coated dishes in the presence of the inhibitors for 24 hours. VCAM-1 protein expression was determined. A representative image is shown. n=3, in triplicate, P<0.01. B, SMCs were pretreated with CsA for 1 hour and plated onto Coll I–coated dishes in the presence of the inhibitors. After 24 hours, mRNA was extracted and VCAM-1 expression was determined by quantitative RT-PCR (representative results from a triplicate experiment; n=3, in triplicate, P<0.05) C, SMCs were transfected with VCAM-1 luciferase constructs. Cells were trypsinized and plated onto Coll I– and Coll IV–coated plates in the presence of CsA or A-285222 (10 μmol/L). Luciferase assays were performed after 6 hours of plating. Biological replicates in triplicate; n=3, in triplicate, P<0.05. D, SMCs expressing an NFATc-driven luciferase construct were trypsinized and plated Coll I– and Coll IV–coated plates in the presence of CsA for 6 and 24 hours. Luciferase assays were performed. n=3, in triplicate; *different from Coll IV, Coll IV+CsA, and Coll I+CsA, P<0.05.
Discussion
The work presented herein describes for the first time a mechanism by which different matrices regulate SMC contractile and inflammatory gene expression. Compared to Coll I, SMCs plated on the normal basement membrane component Coll IV have high levels of SMA and SM-MHC expression, enhanced SRF recruitment to CArG boxes in the SMA and SM-MHC promoters, and increased expression of the SRF coactivator myocardin (Figure 6). In contrast to the contractile genes, cells plated on Coll I have significantly enhanced VCAM-1 expression. Luciferase constructs driven by the VCAM-1 promoter illustrate that mutation of the NF-κB and NFAT-binding sites result in reduced promoter activity. Whereas NF-κB does not show differential activation on the matrices and NF-κB inhibitory peptides do not reduce VCAM-1 expression in cells on Coll I, NFAT transcriptional activity is enhanced in cells on Coll I and blocking NFAT activation significantly reduces Coll I–associated VCAM-1 expression (Figure 6). Although these data do not specifically exclude a role for matrix-specific effects on NF-κB activity, nor identify the specific NFATc isoform (future directions), the differential VCAM-1 expression when NFAT is inhibited but NF-κB activity is not suggests that any differential affects on NF-κB activity are insufficient to produce matrix-specific VCAM-1 expression.
Figure 6.
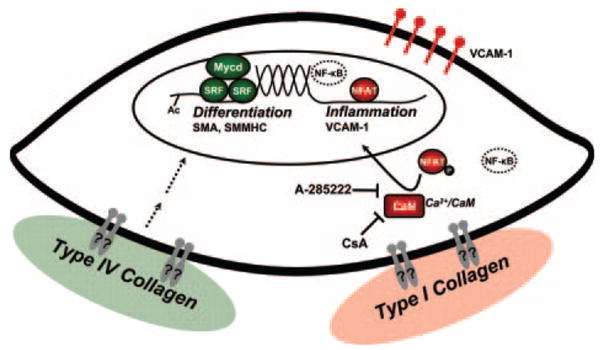
Molecular mechanisms of matrix-dependent SMC phenotypic modulation. Proteins colored in green are proposed to favor Coll IV–dependent activation of SMC differentiation marker expression (Myoc, SMMHC, SMA) and the contractile phenotype; dashed lines indicate that the signaling pathways to this molecular fingerprint are unknown. Proteins colored in red are proposed to favor Coll I–dependent activation of the inflammatory phenotype and VCAM-1 via calcineurin (CaN) and NFAT. NF-κB is depicted as a dashed transcription factor because although it was shown herein that Coll I does not mediate NF-κB–dependent regulation of VCAM-1, NF-κB has been shown to regulate VCAM-1 in response to other stimuli such at IL-1β. It is currently unknown which integrins/receptors mediate matrix-dependent regulation of SMC phenotype (gray receptors with ?). CsA indicates cyclosporine A; Ac, acetylation.
Collagen and the Regulation of Smooth Muscle–Specific Gene Expression
SRF regulates both “immediate early” genes and smooth muscle-specific contractile gene expression, the latter of which involves the presence of specific cofactors such as myocardin.19 During development, myocardin is required and sufficient to induce a smooth muscle–specific gene expression program. In contrast, expression of myocardin in SMCs is decreased during phenotypic modulation in vivo and in response to known regulators of phenotypic modulation in vitro.19,20 Regulation of myocardin expression involves an enhancer sequence 35 kb distal to the myocardin promoter containing binding sites for the transcription factors Mef2c, Foxo4, and TEAD.30 Mef2c can be activated by p38, which has been reported to be regulated by collagen in other systems.31 Recently, p38 signaling was shown to be critical for cardiac differentiation through its effects on Mef2c and myocardin.32 However, we could not identify any changes in the p38 activation profile between Coll I and Coll IV (supplemental Figure III). Posttranslational modifications including phosphorylation and acetylation regulate Foxo4 activity,33 whereas phosphatases and deacetylases can inhibit Foxo4. Calcineurin can dephosphorylate and inhibit Foxo4,34 and data presented herein suggest that Coll I enhances calcineurin-NFAT signaling to induce VCAM-1 expression. Therefore, enhanced calcineurin activation on Coll I could mediate its effects on both proinflammatory and smooth muscle-specific genes. Future studies will attempt to identify the transcription factors important for matrix-specific myocardin expression and to identify the molecular mechanisms involved in this regulation (dashed arrows in Figure 6).
NFAT and VCAM-1 Expression
While the function of cell cycle regulators and profibrotic genes in atherosclerotic progression are well described, the role of inflammatory gene expression in smooth muscle cells has puzzled scientists for more than a decade.6 Multiple models of atherosclerosis show enhanced SMC expression of VCAM-1 and P-selectin, proteins involved in leukocyte recruitment.35–37 Although the precise role remains unknown, the expression of VCAM-1 in neointimal SMCs may serve to limit the migration of inflammatory cells in the tissue6 or enhance the local activation of T cells.38,39 Surprisingly, recent work using VCAM-1 siRNA demonstrated a requirement for VCAM-1 expression for SMC migration and to a lesser extent proliferation, although the mechanisms behind this regulation are unknown.40 Activation of the proinflammatory transcription factor NF-κB in SMCs results in enhanced expression of matrix metalloproteinases41 and inhibitor of apoptosis protein-1,42 which has profound clinical implications because degradation of the basement membrane and SMC apoptosis are both associated with plaque vulnerability.43,44 Thus, inflammatory gene expression can be used as a marker of smooth muscle phenotypic modulation and may play an important role in the pathology of atherosclerosis.
The NF-κB family of transcription factors is a classical proinflammatory signaling pathway, and activation of NF-κB is required for VCAM-1 induction in several experimental systems.45 For those reasons, it was surprising to see no difference in NF-κB activation between the two matrices and no effect of the NF-κB inhibitor SN50 on Coll I–induced VCAM-1 expression. Although Coll I–induced VCAM-1 expression did require the NF-κB binding sites in the VCAM-1 promoter, these sites can also recruit NFAT to induce VCAM-1 expression.27 Although we could not detect reliable enhancement of NFATc1 or c4 binding to the VCAM-1 promoter by ChIP assay (data not shown), NFAT transcriptional activity was increased in SMCs on Coll I, and inhibiting CaN/NFAT signaling with the calcineurin inhibitor CsA or the NFATc inhibitor A-285222 completely blocked VCAM-1 expression in cells on Coll I. Underscoring the importance of this region in the VCAM-1 promoter, recent work comparing the atheroprone C57Bl/6J mouse and the atheroresistent C3H mouse demonstrated that C3H mice contain a single nucleotide polymorphism in the NF-κB binding site of the VCAM-1 promoter associated with reduced VCAM-1 expression and diminished atherosclerosis.46 Identifying the specific NFATc isoform(s) (1–4) will be paramount to further understanding the role of NFAT in SMC inflammatory gene responses.
Collagen Receptors in Phenotypic Modulation
One potentially interesting aspect of this work is the mechanism by which cells differentiate between Coll I and Coll IV. When denatured, both Coll I and Coll IV can bind to the RGD-binding integrin αvβ3 typically associated with phenotypic modulation.47,48 However, interactions with αvβ3 likely do not mediate these matrix-specific effects because the Coll was coated under native conditions and αvβ3 binds both denatured matrices. Both types of native collagen bind to similar integrins, although α1β1 preferentially binds Coll IV and α2β1 preferentially binds to Coll I.49 In addition, α1β1 shows strong expression in healthy SMCs, whereas α2β1 shows enhanced expression during phenotypic modulation both in vivo and in vitro.50 The role of α2β1-specific signaling in SMC phenotypic modulation is an interesting target for future studies. Another potential mechanism for collagen isotype-specific signaling is differential regulation of the discoidin domain family of receptors, including both DDR1 and DDR2. DDR1 knockout mice have impaired migration on Coll I and show reduced expression of matrix metalloproteinase (MMP)-2 and MMP-9.51 Whether differential signaling through these receptors could be mediating the matrix specific effects on smooth muscle phenotypic modulation is currently unknown and a future area of investigation (Figure 6). Although the relevance of monomeric Coll I to SMCs in vivo is unclear, monomeric Coll I provides a useful model system to identify the receptors and signaling mechanisms that mediate Coll-induced changes in SMC phenotype.
The current work shows for the first time a molecular mechanism for differential SMC contractile and inflammatory gene expression in response to protective basement membrane Coll IV and atherosclerotic monomeric Coll I (Figure 6). These data underscore the importance of the matrix in smooth muscle physiology and identify novel levels of regulation by the SMC matrix. Phenotypic modulation of SMCs is known to play an important role in atherosclerotic progression, with SMCs contributing to the clinical outcome of this pathological vessel remodeling. Furthermore, SMCs play a major role in neointimal formation in balloon-induced injury from angioplasty, in vein grafts, and in response to vascular stents. Through better understanding of how SMCs sense the protective versus atherosclerotic matrices, we hope to identify novel therapeutic approaches to limit SMC phenotypic modulation and to improve the design of vascular stents to limit pathological SMC growth.
Supplementary Material
Acknowledgments
We thank Patrick Albert for assistance with quantitative RT-PCR, Sean Garvey for assistance with the inhibitor experiments, and William Aird (Harvard Medical School, Boston, Mass) and Takashi Minami (The University of Tokyo) for the generous gift of the VCAM-1 luciferase constructs.
Sources of Funding
This work was supported by American Heart Association Scientist Development Grant (0735308N) to A.W.O. and by American Heart Association Scientist Development Grant and NIH HL081682 to B.R.W.
Footnotes
Disclosures
None.
References
- 1.Yusuf S, Reddy S, Ounpuu S, Anand S. Global burden of cardiovascular diseases: part I: general considerations, the epidemiologic transition, risk factors, and impact of urbanization. Circulation. 2001;104:2746–2753. doi: 10.1161/hc4601.099487. [DOI] [PubMed] [Google Scholar]
- 2.Ross R. Atherosclerosis–an inflammatory disease.[see comment] N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 4.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 5.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 6.Libby P, Li H. Vascular cell adhesion molecule-1 and smooth muscle cell activation during atherogenesis. J Clin Invest. 1993;92:538–539. doi: 10.1172/JCI116620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz MA. Integrin signaling revisited. Trends in Cell Biology. 2001;11:466–470. doi: 10.1016/s0962-8924(01)02152-3. [DOI] [PubMed] [Google Scholar]
- 8.Barnes MJ, Farndale RW. Collagens and atherosclerosis. Exp Gerontol. 1999;34:513–525. doi: 10.1016/s0531-5565(99)00038-8. [DOI] [PubMed] [Google Scholar]
- 9.Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52:372–386. doi: 10.1016/s0008-6363(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 10.Mawatari K, Liu B, Kent KC. Activation of integrin receptors is required for growth factor-induced smooth muscle cell dysfunction. J Vasc Surg. 2000;31:375–381. doi: 10.1016/s0741-5214(00)90167-8. [DOI] [PubMed] [Google Scholar]
- 11.Kappert K, Blaschke F, Meehan WP, Kawano H, Grill M, Fleck E, Hsueh WA, Law RE, Graf K. Integrins alphavbeta3 and alphavbeta5 mediate VSMC migration and are elevated during neointima formation in the rat aorta. Basic Res Cardiol. 2001;96:42–49. doi: 10.1007/s003950170076. [DOI] [PubMed] [Google Scholar]
- 12.Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto M, Yamamoto K, Noumura T. Type I collagen promotes modulation of cultured rabbit arterial smooth muscle cells from a contractile to a synthetic phenotype. Exp Cell Res. 1993;204:121–129. doi: 10.1006/excr.1993.1016. [DOI] [PubMed] [Google Scholar]
- 14.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000;106:1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wamhoff BR, Hoofnagle MH, Burns A, Sinha S, McDonald OG, Owens GK. A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ Res. 2004;95:981–988. doi: 10.1161/01.RES.0000147961.09840.fb. [DOI] [PubMed] [Google Scholar]
- 17.McDonald OG, Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100:1428–1441. doi: 10.1161/01.RES.0000266448.30370.a0. [DOI] [PubMed] [Google Scholar]
- 18.Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577–593. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–864. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 21.Wang DZ, Olson EN. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr Opin Genet Dev. 2004;14:558–566. doi: 10.1016/j.gde.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N, Owens GK. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801. doi: 10.1161/CIRCRESAHA.107.152736. [DOI] [PubMed] [Google Scholar]
- 23.Wamhoff BR, Macdonald T, Lynch KR, Owens GK. Differential regulation of smooth muscle phenotype by sphingosine-1-phosphate receptors in vitro and in response to vascular injury. Arterioscler Thromb Vasc Biol. doi: 10.1161/ATVBAHA.107.159392. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116:36–48. doi: 10.1172/JCI26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ Res. 2004;95:406–414. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 26.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–819. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minami T, Miura M, Aird WC, Kodama T. Thrombin-induced autoinhibitory factor, Down syndrome critical region-1, attenuates NFAT-dependent vascular cell adhesion molecule-1 expression and inflammation in the endothelium. J Biol Chem. 2006;281:20503–20520. doi: 10.1074/jbc.M513112200. [DOI] [PubMed] [Google Scholar]
- 28.Lee M, Park J. Regulation of NFAT activation: a potential therapeutic target for immunosuppression. Mol Cells. 2006;22:1–7. [PubMed] [Google Scholar]
- 29.Nilsson LM, Sun ZW, Nilsson J, Nordstrom I, Chen YW, Molkentin JD, Wide-Swensson D, Hellstrand P, Lydrup ML, Gomez MF. Novel blocker of NFAT activation inhibits IL-6 production in human myometrial arteries and reduces vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C1167–C1178. doi: 10.1152/ajpcell.00590.2005. [DOI] [PubMed] [Google Scholar]
- 30.Creemers EE, Sutherland LB, McAnally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development. 2006;133:4245–4256. doi: 10.1242/dev.02610. [DOI] [PubMed] [Google Scholar]
- 31.Klekotka PA, Santoro SA, Zutter MM. alpha 2 integrin subunit cytoplasmic domain-dependent cellular migration requires p38 MAPK. J Biol Chem. 2001;276:9503–9511. doi: 10.1074/jbc.M006286200. [DOI] [PubMed] [Google Scholar]
- 32.Hernandez-Torres F, Martinez-Fernandez S, Zuluaga S, Nebreda A, Porras A, Aranega AE, Navarro F. A role for p38alpha mitogen-activated protein kinase in embryonic cardiac differentiation. FEBS Lett. 2008;582:1025–1031. doi: 10.1016/j.febslet.2008.02.050. [DOI] [PubMed] [Google Scholar]
- 33.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 34.Lara-Pezzi E, Winn N, Paul A, McCullagh K, Slominsky E, Santini MP, Mourkioti F, Sarathchandra P, Fukushima S, Suzuki K, Rosenthal N. A naturally occurring calcineurin variant inhibits FoxO activity and enhances skeletal muscle regeneration. J Cell Biol. 2007;179:1205–1218. doi: 10.1083/jcb.200704179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landry DB, Couper LL, Bryant SR, Lindner V. Activation of the NF-kappa B and I kappa B system in smooth muscle cells after rat arterial injury. Induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am J Pathol. 1997;151:1085–1095. [PMC free article] [PubMed] [Google Scholar]
- 36.Breuss JM, Cejna M, Bergmeister H, Kadl A, Baumgartl G, Steurer S, Xu Z, Koshelnick Y, Lipp J, De Martin R, Losert U, Lammer J, Binder BR. Activation of nuclear factor-kappa B significantly contributes to lumen loss in a rabbit iliac artery balloon angioplasty model. Circulation. 2002;105:633–638. doi: 10.1161/hc0502.102966. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. Inducible expression of vascular cell adhesion molecule-1 by vascular smooth muscle cells in vitro and within rabbit atheroma. Am J Pathol. 1993;143:1551–1559. [PMC free article] [PubMed] [Google Scholar]
- 38.van Seventer GA, Newman W, Shimizu Y, Nutman TB, Tanaka Y, Horgan KJ, Gopal TV, Ennis E, O’Sullivan D, Grey H, et al. Analysis of T cell stimulation by superantigen plus major histocompatibility complex class II molecules or by CD3 monoclonal antibody: costimulation by purified adhesion ligands VCAM-1, ICAM-1, but not ELAM-1. J Exp Med. 1991;174:901–913. doi: 10.1084/jem.174.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Damle NK, Klussman K, Linsley PS, Aruffo A. Differential costimulatory effects of adhesion molecules B7, ICAM-1, LFA-3, and VCAM-1 on resting and antigen-primed CD4+ T lymphocytes. J Immunol. 1992;148:1985–1992. [PubMed] [Google Scholar]
- 40.Petersen EJ, Miyoshi T, Yuan Z, Hirohata S, Li JZ, Shi W, Angle JF. siRNA silencing reveals role of vascular cell adhesion molecule-1 in vascular smooth muscle cell migration. Atherosclerosis. 2008;98:301–306. doi: 10.1016/j.atherosclerosis.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bond M, Chase AJ, Baker AH, Newby AC. Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc Res. 2001;50:556–565. doi: 10.1016/s0008-6363(01)00220-6. [DOI] [PubMed] [Google Scholar]
- 42.Erl W, Hansson GK, de Martin R, Draude G, Weber KS, Weber C. Nuclear factor-kappa B regulates induction of apoptosis and inhibitor of apoptosis protein-1 expression in vascular smooth muscle cells. Circ Res. 1999;84:668–677. doi: 10.1161/01.res.84.6.668. [DOI] [PubMed] [Google Scholar]
- 43.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 44.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pei H, Wang Y, Miyoshi T, Zhang Z, Matsumoto AH, Helm GA, Tellides G, Shi W. Direct evidence for a crucial role of the arterial wall in control of atherosclerosis susceptibility. Circulation. 2006;114:2382–2389. doi: 10.1161/CIRCULATIONAHA.106.640185. [DOI] [PubMed] [Google Scholar]
- 47.Davis GE. Affinity of integrins for damaged extracellular matrix: alpha v beta 3 binds to denatured collagen type I through RGD sites. Biochem Biophys Res Commun. 1992;182:1025–1031. doi: 10.1016/0006-291x(92)91834-d. [DOI] [PubMed] [Google Scholar]
- 48.Pedchenko V, Zent R, Hudson BG. Alpha(v)beta3 and alpha(v)beta5 integrins bind both the proximal RGD site and non-RGD motifs within noncollagenous (NC1) domain of the alpha3 chain of type IV collagen: implication for the mechanism of endothelia cell adhesion. J Biol Chem. 2004;279:2772–2780. doi: 10.1074/jbc.M311901200. [DOI] [PubMed] [Google Scholar]
- 49.Heino J. The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biology. 2000;19:319–323. doi: 10.1016/s0945-053x(00)00076-7. [DOI] [PubMed] [Google Scholar]
- 50.Skinner MP, Raines EW, Ross R. Dynamic expression of alpha 1 beta 1 and alpha 2 beta 1 integrin receptors by human vascular smooth muscle cells. Alpha 2 beta 1 integrin is required for chemotaxis across type I collagen-coated membranes. Am J Pathol. 1994;145:1070–1081. [PMC free article] [PubMed] [Google Scholar]
- 51.Hou G, Vogel WF, Bendeck MP. Tyrosine kinase activity of discoidin domain receptor 1 is necessary for smooth muscle cell migration and matrix metalloproteinase expression. Circ Res. 2002;90:1147–1149. doi: 10.1161/01.res.0000022166.74073.f8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.