

Abstract
A practical, functional group tolerant method for the Rh-catalyzed direct arylation of a variety of pharmaceutically important azoles with aryl bromides is described. Many of the successful azole and aryl bromide coupling partners are not compatible with methods for the direct arylation of heterocycles using Pd(0) or Cu(I) catalysts. The readily prepared, low molecular weight ligand, Z-1-tert-butyl-2,3,6,7-tetrahydrophosphepine, which coordinates to Rh in a bidentate P-olefin fashion to provide a highly active yet thermally stable arylation catalyst, is essential to the success of this method. By using the tetrafluoroborate salt of the corresponding phosphonium, the reactions can be assembled outside of a glove box without purification of reagents or solvent. The reactions are also conducted in THF or dioxane, which greatly simplifies product isolation relative to most other methods for direct arylation of azoles employing high-boiling amide solvents. The reactions are performed with heating in a microwave reactor to obtain excellent product yields in two hours.
Keywords: Rhodium catalysis, Direct arylation, C-H activation, Heterocycles
Introduction
The direct arylation of privileged heteroarenes1 provides a highly efficient means to synthesize functional biaryl compounds utilized extensively throughout the pharmaceutical and materials industries.2 This approach eliminates the need for the organometallic starting materials required in traditional cross-coupling methods,3 and over the past several years a number of reactions that exploit directing groups,4 repulsive steric interactions,5 electron-rich substrates,6 or C-H bond acidity7 to selectively activate and functionalize a specific C-H bond with a transition metal catalyst have been developed. When applicable to a substrate class, these methods reduce reaction byproducts, increase the number of available substrates, and decrease the synthetic effort required for formation of the desired C-C bond.
Our group recently described a Rh-catalyzed arylation method in which mixtures of endo- and exo-9-cyclohexylbicyclo-[4.2.1]-9-phosphanonane (cyclohexylphobane) served as highly active ligands for the direct arylation of a variety of heterocycles using aryl bromides (eq 1).8 Experimental and computational studies on the heterocycle activation step of this reaction provided evidence that coordination of the heterocycle to the catalytically active Rh-phosphine fragment precedes an intramolecular C-H activation step, which provides a Rh-H intermediate that ultimately tautomerizes to an N-heterocyclic carbene complex.9 This mechanism for C-H bond activation leads to unique selectivity and has enabled the arylation of wide variety of heterocycles using Rh catalysis, 10 many of which are incompatible with electrophilic Pd-catalyzed methods or Cu-catalyzed methods7e that require strong bases and presumably proceed via heterocycle deprotonation. In particular, unprotected N-H azoles and non-aromatic heterocycles were viable arylation substrates, and a wide array of functionalized aryl bromides were compatible with the arylation conditions. However, a number of important problems regarding substrate scope, functional group tolerance, and practicality still remained.
 |
Herein, we report the solution to many of these problems through the synthesis and evaluation of a set of 2,3,6,7-tetrahydrophosphepine P-olefin ligands previously unexplored in transition metal catalysis. The development of these ligands was based on an investigation into the reaction of [RhCl(coe)2]2 with 1a that revealed dehydrogenation of 1a to form a P-olefin Rh complex (vide infra). The method developed using these new ligands significantly expands the scope of heterocycle and aryl bromide coupling partners that may be used over that observed for 1a/b. Moreover, the method was further modified to allow assembly of the reactions without the need for a glove box or reagent purification.
Investigation of Rh-Phosphine Complexes
Aryl halide hydrodehalogenation11 was identified as a key side reaction in early studies on Rh-catalyzed arylation employing PCy3 as a ligand.12 While greatly reduced through the use of 1a/b and aryl bromides, this deleterious side reaction was still observed to a significant extent in sluggish arylations between poorly reactive coupling partners. Dehydrogenation of 1a/b, in a manner similar to that previously observed for PCy3, was suspected as the “H2” source for aryl bromide hydrodehalogenation, though the site(s) of dehydrogenation in 1a/b was not known.13 Eliminating this reaction was thus deemed essential to further optimization efforts. Our work toward this goal commenced by heating d8-THF solutions of 1a and [RhCl(coe)2]2 and monitoring the 1H and 31P NMR spectra of this mixture in situ (eq 2).14 Conversion to a complex assigned as bis-phosphine 2 based on 31P chemical shifts was initially observed when four equivalents of 1a were utilized with respect to [RhCl(coe)2]2. Subsequently, complete conversion of 2 to complex 3 was observed. Importantly, complex 3 was also present in the crude arylation reaction mixtures utilizing 1a/b under optimized arylation conditions.
 |
Complex 3 was prepared in good yield by heating a THF solution of 1a and [RhCl(coe)2]2 at 125 °C for 2 h, concentrating the mixture, and then washing the resulting yellow solid with pentane. This material was crystallized from pentane/THF, and the structure of 3 was determined using single crystal X-ray analysis (Figure 1). The structure clearly showed that one of the phoban ligands had been selectively dehydrogenated to generate a P-olefin binding motif with Rh1-P1 and Rh1-P2 bond distances of 2.2816(18) Å and 2.379(2) Å, respectively, and Rh1-C16 and Rh1-C17 bond distances of 2.123(9) Å and 2.123(7) Å, respectively.15 The second phoban ligand was not dehydrogenated as evidenced by the sp3 hybridization of the carbon atoms in the 4-carbon bridge of the ligand. The stability of this complex even under extended heating at 125 °C indicated tighter chelation of the rhodium center relative to the analogous complex formed in situ from Rh/PCy3, which we assume underwent multiple rounds of cyclometallation/β-hydride elimination due to the observed pseudo catalytic hydrodehalogenation of PhI and complete decomposition of the Rh species.12
Figure 1.

a) ORTEP diagram of 3. b) Line illustration of 3.
The activity of 3 as a catalyst for the arylation of benzimidazole using PhBr was next explored in order to determine the effects of the introduced unsaturation (eq 3, Figure 2). As shown in Figure 2, complex 3 catalyzed the arylation of benzimidazole to provide a final yield of product similar to that obtained with the use of [RhCl(coe)2]2/1a/b. Fairly similar kinetic behavior between the different catalysts was observed, with the apparent zero order kinetic behavior being a notable feature of the reactions. In general, these data indicate that complex 3 formed under the reaction conditions employing phosphines 1a/b and exhibits surprising stability, presumably due to bidentate P-olefin Rh chelation. This, in turn, suggests that conversion of [RhCl(coe)2]2/1a/b to the stable complex 3, rather than solely ligand sterics and electronics, is largely responsible for the superior activity of arylation catalysts derived from phosphines 1a/b.
Figure 2.

Plot of conversion versus time for arylation of benzimidazole using PhBr in the presence of various Rh catalysts.
 |
While complex 3 was an active arylation catalyst with interesting kinetic behavior and attractively low levels of aryl bromide hydrodehalogenation, it was also a step back in terms of catalyst simplicity and ligand modularity. We therefore sought to simplify the phosphine ligand while maintaining the P-olefin binding motif that conferred the unique activity of 3 and correspondingly [RhCl(coe)2]2/1a/b, which undergoes in situ ligand dehydrogenation to 3. Specifically, it was envisioned that a ligand lacking the two-carbon bridge present in the phoban skeleton, such as a (Z)-2,3,6,7-tetrahydrophosphepine, might fulfill these design specifications (Figure 3).
Figure 3.

Conceptual evolution of ligands used for heterocycle arylation leading to the (Z)-2,3,6,7-tetrahydrophosphepine skeleton.
Synthesis and Evaluation of (Z)-2,3,6,7-Tetrahydrophosphepines
Substituted (Z)-2,3,6,7-tetrahydrophosphepines have been reported in the literature16 but were completely unexplored as ligands for transition metal catalysis, despite recent advances in the use of other P-olefin ligands for a variety of transformations.17 Recently, Lammertsma and coworkers published the synthesis of (Z)-1-phenyl-2,3,6,7-tetrahydrophosphepine, but used the material in situ to generate a variety of interesting Mo-phosphepine complexes and did not isolate the free ligand.18 We utilized a modified version of the Lammertsma procedure in order to prepare a small set of phosphepine ligands (Scheme 1).19 Specifically, a three step procedure, involving addition of two equivalents of 3-butenylmagnesium bromide to the appropriate phosphine chloride followed by an alcohol quench, peroxide oxidation, aqueous workup, and olefin metathesis was developed to construct the substituted 2,3,6,7-tetrahydrophosphepine ring with only a single purification after the final step. The desired phosphines were then obtained by heating toluene solutions of the phosphine oxides and phenylsilane to reflux, concentrating the reaction mixture in vacuo, and purifying the products by Kügelrohr distillation.
Scheme 1.

Synthesis of (Z)-1-Substituted-2,3,6,7-tetrahydrophosphepine Ligands.aStarting from PhP(O)Cl2; no oxidation conducted.
In order to verify the structure of the Rh catalysts generated from these ligands, we next prepared a d8-THF solution of 5c and [RhCl(coe)2]2 and monitored the mixture using 1H NMR spectroscopy. Again, we initially observed formation of a bisphosphine species, followed by complete conversion to P-olefin complex 7 (eq 4). The structure of this material was confirmed by single crystal X-ray analysis (Figure 4a). The ligand is coordinated to Rh in a bidentate P-olefin fashion with a Rh1-P1 bond distance of 2.190(1) Å, and Rh1-C4 and Rh1-C5 bond distances of 2.130(6) Å and 2.105(5) Å, respectively. The Rh-binding motifs found in 3 and 7 exhibited a great deal of similarity indicating that removing the two-carbon bridge from 1a did not significantly alter the desired coordination geometry (Figure 4c).
Figure 4.

a) X-ray structure of 7. b) Line illustration of 7. c) Overlay of common Rh-binding motifs of 3 and 7. d) Diagram deconstructing overlay shown in Figure 4-c.
 |
Having established the Rh coordination chemistry of 5c, we next investigated the activity of ligands 5a-e in the arylation of benzimidazole with PhBr under reaction conditions utilizing conventional heating (eq 5, Figure 5). While all of the ligands provided active arylation catalysts when mixed with [RhCl(coe)2]2 in an optimized ratio of 1.5:1 (P:Rh), phosphepine 5b with a tert-butyl substituent was by far the most effective and resulted in quantitative reaction conversion. Phosphepine 5a with a phenyl substituent was also interesting due to the rapid initial reaction rate but gave only moderate conversion due to catalyst inactivation. A number of aryl phosphepine derivatives were prepared and evaluated, including 2,6-disubstituted phenyl phosphepines designed to prevent orthometallation, but improvements in catalyst stability were not observed (data not shown)
Figure 5.

Plot of conversion versus time for arylation using 5a-e.
 |
Having observed quantitative product formation and essentially no hydrodehalogenation of PhBr during the course of the arylation reaction with tert-butyl-substituted phosphepine 5b, we next re-evaluated this ligand using microwave heating in order to shorten the reaction time. Conditions previously optimized using ligands 1a/b were first investigated for the arylation of benzimidazole with PhBr (eq 6). A 74% yield of the desired 2-phenylbenzimidazole was observed under these conditions (Table 1, entry 1), but functionalized aryl bromides were poorly tolerated. These results utilizing microwave heating differed greatly from those observed in d8-THF at 150 °C under conventional heating, under which broad functional group tolerance was observed by 1H NMR spectroscopy over long reaction times (data not shown). Product decomposition was suspected as a cause for the low yields obtained in some of these cases; however, catalyst decomposition, indicated by the presence of significant Rh-black precipitate in the reaction vessels, was also cause for concern.
Table 1.
Optimization of Reaction Conditions Using Microwave Heating.
| Entry | Solvent | Time (min) | Temp (°C) | Yield (%)a |
|---|---|---|---|---|
| 1 | DCB | 40 | 250 | 74 |
| 2 | DCB/THF (1:1) | 40 | 250 | 77 |
| 3 | DCB/THF (1:1) | 120 | 200 | 88 |
| 4 | DCB/THF (1:1) | 240 | 200 | 84 |
| 5 | THF | 120 | 200 | 83 |
Determined by 1H NMR spectroscopy relative to 2,6-dimethoxytoluene internal standard.
Both of these difficulties most probably arose from either the high temperature or the solvent, 1,2-dichlorobenzene (DCB), employed in our previously published conditions.8 Investigation of reaction solvent, time, and temperature for reactions conducted in a microwave reactor revealed that the reaction temperature could be reduced to 200 °C from 250 °C by extending the reaction time to 2 h from 40 min in a DCB/THF solvent mixture (Table 1, entries 1-4). Furthermore, at this reduced temperature, THF could be used without a high-boiling co-solvent to provide the desired product in good yield and with no Rh black precipitation (entry 5).20 The elimination of DCB, a halogenated and toxic solvent, has significant practical implications for reaction workup, waste disposal, and product purification.
 |
Substrate Scope
The optimized reaction conditions were then utilized to arylate benzimidazole with a variety of aryl bromides in good to excellent yield (eq 7, Table 2). Very high functional group tolerance was observed, and many substrates not compatible with our previously published methods, and not demonstrated using Pd catalysis, are now viable. In particular, sulfinyl, chloro, acetamide, free hydroxy, and free amine groups were all tolerated (entries 1-12). However, while para- and meta-substitution were tolerated, ortho substitution was not. Electron rich heteroaryl bromides, including 5-bromo-1-methylindole, 5-bromobenzoxazole, 5-bromobenzothiazole, and 3-bromothiophene, also coupled in excellent yields (entries 13-16). These results are particularly notable given that these electron-rich heterocycles undergo electrophilic metallation by Pd catalysts, which complicates their use in palladium-mediated direct arylation chemistry.1a
Table 2.
Scope of Aryl Bromides Compatible with Arylation Reaction.
| Entry | ArBr | Product | Yield (%)a | |
|---|---|---|---|---|
| 1 | CF3 | 11 | 93 | |
| 2 | S(O)Me | 12 | 60 | |
| 3 | Cl | 13 | 86 | |
| 4 | C(O)Et | 14 | 91 | |
| 5 |  |
CO2Et | 15 | 96 |
| 6 | C(O)NH2 | 16 | 85 | |
| 7 | H | 17 | 98 | |
| 8 | NHAc | 18 | 88 | |
| 9 | OMe | 19 | 69 | |
| 10 | OH | 20 | 66 | |
| 11 |  |
H | 21 | 67 |
| 12 | NH2 | 22 | 56 | |
| 13 |  |
NMe | 23 | 63 |
| 14 | O | 24 | 81 | |
| 15 | S | 25 | 100 | |
| 16 | 26 | 64 | ||
Isolated yield of pure product.
 |
A variety of additional heterocycles were also compatible with the Rh-catalyzed arylation conditions, and conveniently, optimal yields with these substrates were obtained at an increased reaction concentration of 0.3 M (eq 8, Table 3). N-Methylbenzimidazole and benzoxazole coupled in good yield (entries 1 and 2, respectively), and for the first time using Rh-catalysis, benzothiazole was a viable substrate (entry 3). Pharmaceutically important bisarylimidazoles were also excellent arylation substrates, and both indolyl- and pyridyl-substitution, common to a number of known drug candidates, could be present on the imidazole ring (entries 4-7), although pyridine substitution did result in reduced yield (entry 7).21 Finally, arylation of 4,5-dimethylthiazole and the non-aromatic 4,4-dimethyloxazoline provided moderate yields of the corresponding arylated products (entries 8 and 9).
Table 3.
Scope of Heterocycles Compatible with Arylation Reaction.
| Entry | Heterocycle | Product | Yield (%)a | |
|---|---|---|---|---|
| 1 | 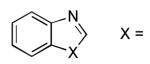 |
NMe | 27 | 79 |
| 2 | O | 28 | 66 | |
| 3 | S | 29 | 38b | |
| 4 | 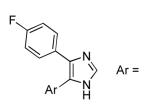 |
phenyl | 30 | 91 |
| 5 | 3-indolyl | 31 | 80 | |
| 6 | 4-pyridyl | 32 | 28 | |
| 7 | 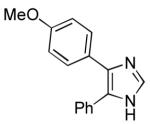 |
33 | 92 | |
| 8 | 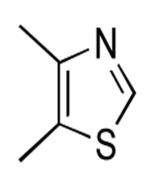 |
34 | 47 | |
| 9 | 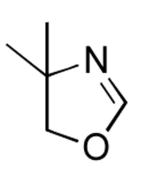 |
35 | 52 | |
Isolated yield of pure product.
6 h reaction time.
 |
Direct arylation using [5bH]BF4 under convenient conditions without use of a glove box
The success observed for the arylation of heterocycles using 5b next led us to pursue strategies designed to simplify the experimental procedure by avoiding the use of a glove box and purified reagents. We therefore sought to protect the air sensitive ligand 5b as its corresponding HBF4 salt, as previously described by Fu and coworkers for the protection of PtBu3.22 The excess base present under our reaction conditions could then be exploited to generate the active phosphine in situ. HBF4 salt formation was readily accomplished by briefly stirring 5b with excess aqueous HBF4, extracting the salt with CH2Cl2, and concentrating the organic extracts to yield [5bH]BF4 as an air-stable white solid. Importantly, this material provided results identical to those obtained using the free phosphine (5b) in the coupling of benzimidazole and PhBr (eq 9, Table 4, entries 1 and 2).
Table 4.
Comparison of Reaction Conditions.
| Entry | Phosphine | [Rh] | Base | Set-up | Yield (%)a |
|---|---|---|---|---|---|
| 1 | 5b | [RhCl(coe)2]2 | i-Pr2i-BuN | glove box | 98 |
| 2 | [5bH]BF4 | [RhCl(coe)2]2 | i-Pr2i-BuN | glove box | 98 |
| 3 | 5b | [RhCl(cod)]2 | i-Pr2i-BuN | glove box | 100 |
| 4 | [5bH]BF4 | [RhCl(cod)]2 | i-Pr2i-BuN | N2 line | 90 |
Isolated yield of pure product.
 |
Significantly, [RhCl(coe)2]2, an expensive and air sensitive catalyst, could be replaced with [RhCl(cod)]2 (cod = cyclooctadiene), a much cheaper and air-stable catalyst, with essentially no change in product yield (Table 4, entry 3). Previous attempts to use this Rh source in both the arylation and alkylation reactions developed in our group had resulted in decreased product yields, presumably due to the greater difficulty of displacing a bidentate olefin ligand with the monodentate, sterically hindered phosphines employed in those methods. In contrast, the bidentate P-olefin ligand 5b readily displaces the cod ligand to provide the active arylation catalyst. Most importantly, the aforementioned phosphine and Rh substitutions enabled easy assembly of the arylation reactions outside a glove box, without purification of any reagents, using only a N2 line to provide an inert atmosphere in the microwave vessel (entry 4). A small set of arylation reactions were repeated under the modified conditions in order to establish the generality of this protocol (eq 10, Table 5). Good to high yields of the desired products were obtained for a range of aryl bromides and heterocycles. Only modest decreases in product yield were observed relative to the original protocol, so it is anticipated that these conditions will be appropriate for most applications. Due to the higher boiling point of dioxane relative to THF, a subset of reactions were also performed with dioxane at a reduced 2.5 % precatalyst loading, and good yields were also observed under these reaction conditions (entries 2, 4, 6, and 8).
Table 5.
Arylation of Azoles using [5bH]BF4.
| Entry | Heterocycle | ArBr | Solvent | Conc.a | [RhCl(cod)]2 (%) | Product | Yield (%)b |
|---|---|---|---|---|---|---|---|
| 1 | 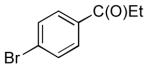 |
THF | 0.1 | 5 | 15 | 74 | |
| 2 | dioxane | 0.3 | 2.5 | 15 | 58 | ||
| 3 |  |
THF | 0.1 | 5 | 17 | 89 | |
| 4 | 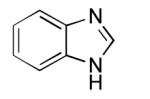 |
dioxane | 0.3 | 2.5 | 17 | 99 | |
| 5 |  |
THF | 0.1 | 5 | 18 | 85 | |
| 6 | dioxane | 0.3 | 2.5 | 18 | 67 | ||
| 7 |  |
THF | 0.1 | 5 | 25 | 93 | |
| 8 | dioxane | 0.3 | 2.5 | 25 | 87 | ||
| 9 |  |
THF | 0.3 | 5 | 27 | 70 | |
| 10 |  |
 |
THF | 0.3 | 5 | 28 | 55 |
| 11 |  |
THF | 0.3 | 5 | 31 | 75 |
Concentration of heterocycle.
Isolated yield of pure product.
 |
Catalyst loading
Preliminary investigation of catalyst loading, which becomes increasingly important for large scale reactions, was also performed (eq 11, Table 6). For this study conventional heating was used because microwave heating is not typically performed with large scale reactions. At 1% loading of the [RhCl(coe)2]2 precatalyst, high conversion with high isolated yields could be achieved by performing the reaction in dioxane with benzimidazole at 0.3 M (entries 3-5). By performing the reaction at 175 °C, a high yield of arylation product was obtained within 24 h (entry 5). Lower conversions and/or extended reaction times were required when the reactions were performed in THF (entry 1) or at a lower benzimidazole concentration (entry 2).
Table 6.
Determination of catalyst loading.
| Entry | Solvent | Conc.a | Temp (°C) | Time (h) | Yield (%)b |
|---|---|---|---|---|---|
| 1 | THF | 0.1 | 165 | 120 | 62 |
| 2 | dioxane | 0.1 | 165 | 120 | 82 |
| 3 | dioxane | 0.3 | 165 | 72 | 100 |
| 4 | dioxane | 0.3 | 165 | 48 | 90c |
| 5 | dioxane | 0.3 | 175 | 24 | 90c |
Concentration of benzimidazole.
Yield determined by GC with dodecane as an internal standard unless otherwise noted.
Isolated yield of pure product.
 |
Proposed Mechanism
A possible mechanism that accounts for heterocycle arylation utilizing these new ligands is presented in Figure 6. Combination of 5b and [RhCl(coe)2]2 provides the dimer complex 7 (vide supra). Dissociation of this complex and coordination of the heterocycle would provide the heterocycle complex 36, which is very similar to that observed in the very well characterized, related reactions of N-methylbenzimidazole, 3,4-dihydroquinazoline, and other heterocycles with RhCl(PCy3)2.9. The formation of carbene complex 37 is presumed to occur via a C-H activation/tautomerization process that was also previously documented in the study of carbene-rhodium complex formation with 3,4-dihydroquinoline.9 This low-valent, electron rich Rh complex is ideally suited for oxidative addition of aryl halides to generate the (aryl)(carbene)rhodium complex 38. Elimination of HBr from this complex in either an intramolecular fashion or assisted by the added amine base would then lead to complex 39. Finally, reductive elimination of the desired biaryl product would regenerate the Rh catalyst.
Figure 6.

Proposed Mechanism for Heterocycle Arylation Employing Rh/5b.
The high product yields and long catalyst lifetimes observed in heterocycle arylation reactions using ligand 5b in combination with [RhCl(L)2]2 (L = coe or cod) pre-catalysts are believed to result from stabilization of the Rh center by hemilabile olefin coordination. On the other hand, a variety of bidentate phosphines and P-N ligands were also screened as ligands for the Rh-catalyzed arylation of benzimidazole with bromobenzene, but little-or-no product was observed in each case. The (Z)-2,3,6,7-tetrahydrophosphepine scaffold must therefore provide a unique coordination environment well-suited for our heterocycle arylation chemistry and may prove useful in other transition metal catalyzed reactions.
Conclusions
In summary, we have developed a practical, functional group tolerant method for the Rh-catalyzed direct arylation of a variety of pharmaceutically important heterocycles with aryl bromides. Essential to the success of this method was the use of Z-1-tert-butyl-2,3,6,7-tetrahydrophosphepine, 5b, as a ligand which coordinates to Rh in a bidentate P-olefin fashion to provide a highly active yet thermally stable catalyst. Furthermore, the initial reaction mixtures could be assembled without the use of a glove box or purified reagents when the tetrafluoroborate salt of the phosphine was employed. The arylation reaction likely occurs via a novel C-H bond activation pathway involving a substrate-based NHC intermediate, which provides substrate scope orthogonal to that reported for Pd-catalyzed reactions. In particular, unprotected N-H azoles and a wide array of functionalized aryl bromides and electron rich heteroaryl bromides are compatible with the Rh-phosphepine catalyst. The reactions can be conducted in THF, which greatly simplifies product isolation relative to the procedure required under our previously published conditions, which require 1,2-dichlorobenzene as solvent, and Pd-based heterocycle arylation methods carried out with high-boiling amide solvents. By heating the reaction solutions in a microwave reactor, excellent product yields were achieved with two hour reaction times.
Experimental
(Z)-1-tert-Butyl-2,3,6,7-tetrahydro-1H-phosphepine (5b)
To an oven-dried round bottom flask under N2 was added tert-butyldichlorophosphine (2.1 g, 13.2 mmol) and THF (45 mL). The solution was cooled to 0 °C, and a solution of 3-butenylmagnesium bromide (0.76 M, 37.5 mL, 27.7 mmol) was added dropwise via syringe. The reaction mixture was allowed to warm to room temperature overnight (ca. 12 h) and quenched by slow addition of i-PrOH (10 mL) and water (10 mL). Aqueous hydrogen peroxide (3 mL, 30% wt/wt) was then added slowly via syringe (caution, exothermic reaction), and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was further diluted with water (100 mL), and the volatile materials were removed by rotary evaporation. The reaction mixture was then extracted with CH2Cl2 (3 × 100 mL), and the combined CH2Cl2 extracts were dried, filtered, and concentrated to give the crude product (dibut-3-enyl(tert-butyl)phosphine oxide, 3.6 g) as a viscous oil in good purity as judged by 31P NMR spectroscopy. This residue was degassed using three cycles of evacuation and N2 purging, dissolved in CH2Cl2, and added to a round bottom flask fitted with a condenser and septum under N2. A CH2Cl2 solution of Grubbs’ first generation metathesis catalyst (0.50 g, 0.046 mmol) was added through the condenser, and the resulting solution (275 mL total volume) was heated at reflux for 24 h to affect nearly complete conversion to the desire phosphepine oxide as judged by 31P NMR spectroscopic analysis of an aliquot taken from the reaction mixture. The solution was concentrated, loaded onto a Biotage samplet using a minimal amount of CH2Cl2 (ca. 1-3 mL), and purified using a MeOH/CH2Cl2 gradient (Rf = 0.3 in 4% MeOH/CH2Cl2) to provide 2.33 g (95%) of 4b as a brown oil that partially solidified on standing. 1H NMR (400 MHz, CDCl3): δ 5.80 (m, 2H), 2.63 (m, 2H), 2.17 (m, 2H), 1.79 (m, 2H), 1.42 (t, J = 13.6 Hz, 2H), 1.04 (d, J = 13.9 Hz, 9H). 13C NMR (100 MHz, CDCl3): δ 132.2 (s), 31.3 (d, J = 67.5 Hz), 24.1 (s), 22.7 (d, J = 59.4 Hz), 18.5 (d, J = 5.1 Hz). 31P NMR (162 MHz, CDCl3): δ 58.0. IR (ZnSe, thin film) νmax (cm-1): 3020, 2947, 2907, 2862, 1647, 1463, 1394, 1365, 1190, 1151 (P=O). HRMS-EI (m/z): [M]+ calcd for C10H19OP, 186.117354; found, 186.116842.
A vial containing phosphepine oxide 4b (2.33 g, 12.5 mmol) was fitted with a septum and degassed using three cycles of evacuation and N2 purging. This material was dissolved in toluene (10 mL) and added to a round bottom flask fitted with a reflux condenser and a septum under N2. Phenylsilane (6.0 mL, 50 mmol) was added, and the reaction mixture was heated at reflux until complete conversion of the starting material had occurred (16 h). The volatile materials were removed under high vacuum and the residue was distilled under reduced pressure (0.15 mm Hg, 50 °C) using a Kügelrohr apparatus to provide 1.187 g (56%) of 5b as a colorless liquid. 1H NMR (400 MHz, C6D6): δ 5.72 (m, 2H), 2.31 (m, 4H), 1.50 (m, 2H), 1.24 (m, 2H), 0.95 (d, J = 10.9 Hz, 9H). 13C NMR (100 MHz, C6D6): δ 131.6 (s), 27.3 (d, J = 11.7 Hz), 26.9 (d, J = 13.2 Hz), 24.3 (d, J = 13.2 Hz), 20.0 (d, J = 19.0 Hz). 31P NMR (162 MHz, C6D6): δ 7.0. HRMS-EI (m/z): [M]+ calcd for C10H19P, 170.122439; found, 170.122376.
(Z)-1-tert-Butyl-2,3,6,7-tetrahydro-1H-phosphonium tetrafluoroborate ([5bH]BF4)
In an inert atmosphere box, 5b (0.101 g, 0.587 mmol) and a stir bar were added to a vial. The vial was sealed with a septum and removed from the box. CH2Cl2 (6 mL) and HBF4 (0.50 mL, 2.94 mmol, 48% wt. aqueous solution) were added and the mixture was vigorously stirred for 15 min. The mixture was extracted with 3 × 25 mL of CH2Cl2, and the combined organic extracts were dried with MgSO4, filtered, and concentrated to provide 0.0992 g (65%) of [5bH]BF4 as a white solid. 1H NMR (400 MHz, CDCl3): δ 6.39 (dm, J = 488.7 Hz, 1H), 5.94 (m, 2H), 2.72 (m, 6H), 1.94 (m, 2H), 1.41 (d, J = 17.2 Hz, 9H). 13C NMR (100 MHz, CDCl3): δ 131.0 (s), 28.6 (d, J = 43.3 Hz), 24.9 (s), 19.9 (d, J = 6.6 Hz), 12.7 (d, J = 43.3 Hz). 31P NMR (162 MHz, C6D6): δ 34.4 ppm. IR (ZnSe, thin film) νmax (cm-1): 2954 (br, w), 1472 (w), 1409 (w), 1378 (w), 1199 (w), 1093 (m), 1045 (s). HRMS-FAB (m/z): [M]+ calcd for C10H20P, 171.130264; found, 171.130860.
Typical Procedure for Cross-Coupling Azoles and Aryl Bromides (Microwave Protocol), 2-phenyl-1H-benzo[d]imidazole (17)
In an inert atmosphere box, a stir bar, the appropriate heterocycle, e.g., benzimidazole (0.0481 g, 0.4 mmol), and 1 mL of THF were added to a 5 mL glass microwave vial. Into a separate vial were weighed 5b (0.102 g, 0.06 mmol) and [RhCl(coe)2]2 (0.0143 g, 0.02 mmol), and the catalyst was transferred to the microwave vial using 2 mL of THF. The appropriate aryl bromide, e.g., bromobenzene (0.1261 g, 0.8 mmol) was transferred to the microwave vial using 1 mL of THF, and i-Pr2i-BuN (0.245 mL, 1.2 mmol) was added directly to the vial via syringe. The vial was sealed, removed from the inert atmosphere box, and heated for 2 h at 200 °C. The reaction mixture was then cooled, quenched with excess Et3N (0.5 mL), and concentrated under reduced pressure. The residue was dissolved in a minimal amount of methanol/methyelene chloride (ca. 1-2 mL), loaded onto a silica gel samplet (Biotage No. SAM-1107-16016), and purified using flash chromatography. A suitable gradient was calculated by the Biotage SP (10-80% ethyl acetate/hexanes) given a product Rf of 0.32 in 40% ethyl acetate/hexanes. The desired product was obtained as 0.0775 g (98% yield, Table 2, entry 7) of a white solid. For other heterocycles, the concentration of the reactants was increased to 0.3 M. The amount of heterocycle (mmol) and the concentration (M) of the reaction is therefore provided for each entry. mp 284-285 °C (lit. 285-286 °C).23 1H NMR (400 MHz, CD3OD): δ 8.10 (m, 2H), 7.61 (br s, 2H), 7.54 (m, 2H), 7.27 (m, 2H).
Supplementary Material
Acknowledgment
This work was supported by the NIH GM069559 to J.A.E. and by the Director and Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DE-AC03-76SF00098 to R.G.B. NIH postdoctoral fellowship support to A.M.B. is also gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures, analytical and spectral characterization data for all new compounds, and crystallographic information files (CIF) for compounds 3 and 7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1 (a).This area has enjoyed explosive growth, so only references from the past five years are listed. For a nice method utilizing aryl chlorides, see: Chion HA, Daugulis O. Org. Lett. 2007;9:1449–1451. doi: 10.1021/ol0702324.Direct arylation of electron-rich heterocycles is described, but unprotected N-H azoles were not tolerated, and over-arylation was observed in some cases. See references therein for previous scattered examples utilizing aryl chlorides. For examples using aryl bromides, see: Bellina F, Calandri C, Cauteruccio S, Rossi R. Tetrahedron. 2007;63:1970–1980.(three examples, two equivalents CuX required).Cerna I, Pohl R, Kepetarova B, Hocek M. Org. Lett. 2006;8:5389. doi: 10.1021/ol062324j. (one example).Pariseien M, Valette D, Fagnou K. J. Org. Chem. 2005;70:7578–7584. doi: 10.1021/jo051039v. (six examples).Alagille D, Baldwin RM, Tamagnan GD. Tetrahedron Lett. 2005;46:1349–1351. (electron rich aryl bromides were coupled with benzothiazole and benzoxazole). Rieth RD, Mankad NP, Calimano E, Sadighi JP. Org. Lett. 2004;6:3981. doi: 10.1021/ol048367m. (stoichiometric metalation of pyrrole required). Park C-H, Ryabova R, Seregin IV, Sromek AW, Gevorgyan V. Org. Lett. 2004;6:1159–1162. doi: 10.1021/ol049866q. (only indolizine arylation reported). Mori A, Sekiguchi A, Masui K, Shimada T, Horie M, Osakada K, Kawamoto M, Ikeda T. J. Am. Chem. Soc. 2003;125:1700–1701. doi: 10.1021/ja0289189. (one example). Yokooji A, Okazawa T, Satoh T, Miura M, Nomura M. Tetrahedron. 2003;59:5685–5689. (thiazole arylation). Li W, Nelson DP, Jensen MS, Hoerrner RS, Javadi GJ, Cai D, Larsen RD. Org. Lett. 2003;5:4835–4837. doi: 10.1021/ol035878k. (imidazo[1,2-a]pyridine arylation). Glover B, Harvey KA, Liu B, Sharp MJ, Tymoschenko MF. Org. Lett. 2003;5:310–304. doi: 10.1021/ol027266q. (furan and thiophene arylation). Okazawa T, Satoh T, Miura M, Nomura M. J. Am. Chem. Soc. 2002;124:5286–5287. doi: 10.1021/ja0259279. (thiazole multiple arylation). McClure SM, Glover B, McSorley E, Millar A, Osterhout MH, Roschangar F. Org. Lett. 2001;3:1677. doi: 10.1021/ol0158866. (two examples of furan arylation).
- 2 (a).Carey JS, Laffan D, Thomson C, Williams MT. Org. Biomol. Chem. 2006;4:2337. doi: 10.1039/b602413k.Hashimoto H, Maeda K, Ozawa K, Haruta J, Wakitani K. Bioorg. Med. Chem. Lett. 2002;12:65–68. doi: 10.1016/s0960-894x(01)00670-9.Sisko J, Kassick AJ, Mellinger M, Filan JJ, Allen A, Olsen MA. J. Org. Chem. 2000;65:1516–1524. doi: 10.1021/jo991782l. and references therein.Roncali J. Chem. Rev. 1997;97:173–206. doi: 10.1021/cr950257t.
- 3 (a).The importance of modern cross-coupling methods, particularly the Suzuki reaction, for the formation of aryl-aryl bonds cannot be overstated. For recent developments, see: Billingsley K, Buchwald SL. J. Am. Chem. Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j.Littke AF, Dai C, Fu GC. J. Am. Chem. Soc. 2000;122:4020–4028.
- 4 (a).Daugulis O, Zaitsev VG. Angew. Chem., Int. Ed. 2005;44:4046. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]; (b) Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; (c) Thalji RK, Ahrendt KA, Bergman RG, Ellman JA. J. Org. Chem. 2005;70:6775–6781. doi: 10.1021/jo050757e. [DOI] [PubMed] [Google Scholar]; (d) Kakiuchi F, Murai S. Acc. Chem. Res. 2002;35:826–834. doi: 10.1021/ar960318p. [DOI] [PubMed] [Google Scholar]
- 5 (a).Chen H, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]; (b) Cho J-Y, Iverson CN, Smith MR. J. Am. Chem. Soc. 2000;122:12868. [Google Scholar]; (c) Ishiyama T, Takagi J, Kousaku I, Miyaura N, Anastasi NR, Hartwig JF. J. Am. Chem. Soc. 2001;124:390. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- 6 (a).The vast majority of methods for heterocycle arylation fall in to this category.1 In addition, see: Tunge JA, Foresee LN. Organometallics. 2006;24:6440–6444.Yanagisawa S, Sudo T, Noyori R, Itami K. J. Am. Chem. Soc. 2006;128:11748–11749. doi: 10.1021/ja064500p.Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2003;125:9578–9579. doi: 10.1021/ja035054y.Jia CG, Piao DG, Oyamada JZ, Lu WJ, Kitamura T, Fujiwara Y. Science. 2000;287:1992–1995. doi: 10.1126/science.287.5460.1992.
- 7 (a).Garcia-Cuadrado D, Braga AAC, Maseras F, Echavarren AM, Echevarran J. Am. Chem. Soc. 2006;128:1066–1067. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]; (b) Lafrance M, Fagnou KJ. Am. Chem. Soc. 2006;128:16496–16497. doi: 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]; (c) Lafrance M, Shore D, Fagnou K. Org. Lett. 2006;8:5097–5100. doi: 10.1021/ol0619967. [DOI] [PubMed] [Google Scholar]; (d) Lafrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754–8756. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]; (e) Stuart DR, Fagnou K. Science. 2007;316:1172–1175. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]; (f) Do H-Q, Daugulis OJ. Am. Chem. Soc. 2007;129:12404–12405. doi: 10.1021/ja075802+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis JC, Wu JY, Bergman RG, Ellman JA. Angew. Chem. Int. Ed. 2006;118:1619–1621. doi: 10.1002/anie.200504289. [DOI] [PubMed] [Google Scholar]
- 9 (a).Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2002;124:3202–3203. doi: 10.1021/ja017351d. [DOI] [PubMed] [Google Scholar]; (b) Wiedemann SH, Lewis JC, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2006;128:2452. doi: 10.1021/ja0576684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10 (a).The corresponding heterocycle alkylation reactions were developed and extensively studied in our group prior to our work on heterocycle arylation. Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2001;123:2685. doi: 10.1021/ja0058738.Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2002;124:13964. doi: 10.1021/ja0281129.Tan KL, Park S, Ellman JA, Bergman RG. J. Org. Chem. 2004;69:7329. doi: 10.1021/jo048666p.Wiedemann SH, Bergman RG, Ellman JA. Org. Lett. 2004;6:1685. doi: 10.1021/ol049417q.Wiedemann SH, Ellman JA, Bergman RG. J. Org. Chem. 2006;71:1969. doi: 10.1021/jo052345b.
- 11.For an extensive review on hydrodehalogenation see: Alonso F, Beletskaya IP, Yus M. Chem. Rev. 2002;102:4009–4091. doi: 10.1021/cr0102967.
- 12 (a).Lewis JC, Wiedemann SH, Bergman RG, Ellman JA. Org. Lett. 2004;6:35–38. doi: 10.1021/ol035985e.See also: Christ ML, Sabo-Etienne S, Chaudret B. Organometallics. 1995;14:1082–1084., and references therein.
- 13 (a).For other recent studies on phoban-based catalyst systems, see: Klingler RJ, Chen MJ, Rathke JW, Kramarz KW. Organometallics. 2007;26:352–357.Dwyer CL, Kirk MM, Meyer WH, van Rensburg W.Janse, Forman GS. Organometallics. 2006;25:3806–3812.Konya D, Lenero K. Q. Almeida, Drent E. Organometallics. 2006;25:3166–3174.
- 14.We previously demonstrated that 1b is converted to 1a upon heating, so 1a was utilized in our stoichiometric studies in order to simplify analysis of reaction mixtures (see reference 8).
- 15.This transformation is analogous to that described by Frost, Weller, and coworkers for the dehydrogenation of tricyclopentyl phosphine: Douglas TM, Le Nôtre J, Brayshaw SK, Frost CG, Weller AS. Chem. Commun. 2006:3408–3410. doi: 10.1039/b608129k.
- 16 (a).Van Reijendam JW, Baardman F. Tetrahedron Lett. 1972;51:5181–5182. [Google Scholar]; (b) Shima Takanori, Bauer Eike B., Hampel Frank, Gladysz JA. J. Chem. Soc., Dalton Trans. 2004;7:1012–1028. doi: 10.1039/b400156g. [DOI] [PubMed] [Google Scholar]
- 17 (a).Park Y, Meek DW. Bull. Kor. Chem. Soc. 1995;16:524–528.Thomaier J, Boulmaaz S, Schönberg H, Rüegger H, Currao A, Grützmacher H, Hillebrecht H, Pritzkow H. New. J. Chem. 1998:947–958. Deblon S, Liesum L, Harmer J, Schönberg H, Grützmacher H. Chem. Eur. J. 2004;10:4198.Maire P, Deblon S, Breher F, Geier J, Böhler C, Rüegger H, Schönberg H, Grützmacher H. Chem. Eur. J. 2004;10:4198. doi: 10.1002/chem.200400441. Shintani R, Duan W-L, Nagano T, Okamoto A, Hayashi T. Angew. Chem. Int. Ed. 2005;44:4611. doi: 10.1002/anie.200501305. Shintani R, Duan W-L, Okamoto K, Hayashi T. Tetrahedron Asym. 2005;16:3400–3405. Thoumazet C, Richard L, Grützmacher H, Le Floch P. Chem. Commun. 2005:1592–1594. doi: 10.1039/b417716a. See reference 15. Piras E, Läng F, Rüegger H, Stein D, Wörle M, Grützmacher H. Chem. Eur. J. 2006;12:5849–5858. doi: 10.1002/chem.200501470. Mora G, van Zuphen S, Thoumazet C, Le Goff XF, Ricard L, Grützmacher H. Organometallics. 2006;25:5528–5532. Kasák P, Arion VB, Widhalm M. Tetrahedron: Asymmetry. 2006;17:3084–3090.
- 18.van Assema SGA, Ehlers AW, de Kanter FJJ, Schakel M, Spek AL, Lutz M, Lammertsma K. Chem. Eur. J. 2006;12:4333–4340. doi: 10.1002/chem.200501531. [DOI] [PubMed] [Google Scholar]
- 19 (a).We also explored a borane protection route as described by Gouverneur and coworkers: Schuman M, Trevitt M, Redd A, Gouverneur V. Angew. Chem. Int. Engl. 2000;39:2491–2493.Slinn CA, Redgrave AJ, Hind SL, Edlin C, Nolan SP, Gouverneur V. Org. Biomol. Chem. 2003;1:3820–3825. doi: 10.1039/b306940k.This ultimately failed, perhaps due to olefin hydroboration as describd in: Scheideman M, Shapland P, Vedejs E. J. Am. Chem. Soc. 2003;125:10502–10503. doi: 10.1021/ja034655m. And references therein.
- 20.The reaction pressures obtained under these conditions can reach the limits of commercial microwave reactors, so care must be taken to leave enough head space in the reaction vessel to avoid over-pressure errors and vial bursts (3 mL of THF can be heated to 200 °C in a 5 mL Biotage microwave vial without incident).
- 21 (a).Chang LL, Sidler KL, Cascieri MA, de Laszlo S, Koch G, Li B, MacCoss M, Mantlo N, O’Keefe S, Pang M, Rolando A, Hagmann WK. Bioorg. Med. Chem. Lett. 2001;11:2549. doi: 10.1016/s0960-894x(01)00498-x.de Laszlo S, Hacker C, Li B, Kim D, MacCoss M, Mantlo N, Pivnichny JV, Colwell L, Koch GE, Cascieri MA, Hagmann WK. Bioorg. Med. Chem. Lett. 1999;9:641. doi: 10.1016/s0960-894x(99)00081-5.. See also references 2a-c.
- 22.Netherton MR, Fu GC. Org. Lett. 2001;3:4295–4298. doi: 10.1021/ol016971g. [DOI] [PubMed] [Google Scholar]
- 23.Perry RJ, Wilson BD. J. Org. Chem. 1993;58:7016–7021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.