

Abstract
Most GIST patients develop clinical resistance to KIT/PDGFRA tyrosine kinase inhibitors (TKI). However, it is unclear whether clinical resistance results from single or multiple molecular mechanisms in each patient. KIT and PDGFRA mutations were evaluated in 53 GIST metastases obtained from 14 patients who underwent surgical debulking after progression on imatinib or sunitinib. To interrogate possible resistance mechanisms across a broad biological spectrum of GISTs, inter- and intra-lesional heterogeneity of molecular drug-resistance mechanisms were evaluated in the following: conventional KIT (CD117)-positive GISTs with KIT mutations in exon 9, 11 or 13; KIT-negative GISTs; GISTs with unusual morphology; and KIT/PDGFRA wild-type GISTs. Genomic KIT and PDGFRA mutations were characterized systematically, using complementary techniques including D-HPLC for KIT exons 9, 11–18 and PDGFRA exons 12, 14, 18, and mutation-specific PCR (V654A, D820G, N822K, Y823D). Primary KIT oncogenic mutations were found in 11/14 patients (79%). Of these, 9/11 (83%), had secondary drug-resistant KIT mutations, including six (67%) with two to five different secondary mutations in separate metastases, and three (34%) with two secondary KIT mutations in the same metastasis. The secondary mutations clustered in the KIT ATP binding pocket and kinase catalytic regions. FISH analyses revealed KIT amplicons in 2/10 metastases lacking secondary KIT mutations. This study demonstrates extensive intra- and inter-lesional heterogeneity of resistance mutations and gene amplification in patients with clinically progressing GIST. KIT kinase resistance mutations were not found in KIT/PDGFRA wild-type GISTs or in KIT-mutant GISTs showing unusual morphology and/or loss of KIT expression by IHC, indicating that resistance mechanisms are fundamentally different in these tumours. Our observations underscore the heterogeneity of clinical TKI resistance, and highlight the therapeutic challenges involved in salvaging patients after clinical progression on TKI monotherapies.
Keywords: imatinib, sunitinib, drug resistance mechanisms, GIST, heterogeneity
Introduction
Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours of the gastrointestinal (GI) tract and are refractory to radiation and conventional chemotherapy. 85–90% of GISTs have activating mutations of the KIT or PDGFRA receptor tyrosine kinase genes [1–3], resulting in oncoproteins that are crucial diagnostic and therapeutic targets in GIST [2,3]. Indeed, therapeutic inhibition of KIT/PDGFRA kinase activity by imatinib has emerged as the first-line treatment option in patients with inoperable GIST [4–6]. Notably, imatinib response depends on KIT/PDGFRA mutational status [7,8]. Patients whose GISTs have KIT exon 11 mutations have a higher response rate and longer median survival compared to those with wild-type KIT/PDGFRA or with KIT exon 9 mutations [1]. Complete responses to imatinib in metastatic GIST are rare (≤5%) and most responding patients develop secondary resistance [6].
The most common secondary resistance mechanism appears to be mutation of the KIT kinase domain; however, additional resistance mechanisms include KIT/PDGFRA genomic amplifications and activation of alternative oncogenes [9,10]. Therapeutic options for patients whose GISTs progress on imatinib include dose escalation or treatment with sunitinib malate (SUTENT Pfizer, New York, USA), a Federal Drug Administration (FDA)-approved drug with demonstrated efficacy, acceptable tolerability and safety in a double-blind placebo-controlled Phase III trial [11]. Previous studies have focused on individual imatinib resistance mechanisms in GIST lesions progressing on imatinib therapy, but the heterogeneity of these mutations, in a given patient, remains unclear. Therefore, the aim of this study was to characterize intra- and inter-lesional drug resistance mechanisms in GIST tumour samples obtained during debulking procedures performed on patients with imatinib or sunitinib resistance.
Material and methods
Tumour selection
Fifty-three GIST metastases from 14 patients (12 male and 2 female, age range 50–75 years, median age 62 years) were studied. All patients progressed clinically on imatinib or sunitinib, according to the conventional Southwest Oncology Group/Response Evaluation Criteria in Solid Tumours [12]. All patients underwent resection during 2001–2004 at the Brigham and Women's Hospital, Boston, MA, USA. Imatinib or sunitinib was discontinued within 1 week prior to debulking surgeries. Surgery was performed in five patients progressing after imatinib alone and in nine patients progressing after imatinib and sunitinib treatment. To interrogate possible resistance mechanisms in a broad spectrum of GISTs, we examined not only tumours with typical morphology, but tumours that were KIT (CD117) negative, had unusual morphology or were KIT/PDGFRA wild-type. This study was approved by the Institutional Review Board of Brigham and Women's Hospital.
Haematoxylin and eosin-stained sections from 276 paraffin blocks were reviewed to confirm the diagnoses prior to inclusion in the study. Tumour regions from different metastases or different areas within metastases were selected from each patient, with an emphasis on variation in tumour cytology, KIT expression (KIT-positive or KIT-negative) and mitotic activity. In total, 57 tumour areas from 53 metastases were selected. The morphological appearance (spindle cell, epithelioid cell, mixed cell type, unusual morphology), tumour size, location, treatment effects (necrosis, hyalinosis, pseudo-chondroid changes, haemorrhage) were evaluated, as well as mitotic rate [expressed as the number of mitotic figures per 50 high power fields (HPFs) in the most mitotic area, using a ×40 objective and a ×10 ocular, field size 0.25 mm2] (Table 1). Histological treatment response was scored in each metastasis, using a previously proposed grading scheme [8]: 1, minimal (0–10% response); 2, low (>10% and <50% responses); 3, moderate (>50% and <90% response); and 4, high (>90% response).
Table 1.
Summary of clinicopathological and molecular findings in 14 patients undergoing debulking procedures for progressing GIST during TKI treatment
| Patient no. | Treatment | Age/sex | Location | Size (cm) | Morphology | Mitoses/ 50HPF |
Histological treatment response |
KIT/IHC | Primary KIT mutation | Secondary KIT mutation
|
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| DHPLC | |||||||||||
| 1 | IM | 57M | Serosal nodule (1) | 1.8 | Spindle | 20 | 1 | + | Exon 9 (insertion AY502–503) |
NMF | NMF |
| Serosal nodule (2) | 2.7 | Spindle (perivascular growth) | 7 | 1 | + | NMF | |||||
| Serosal nodule (3) | 4.0 | Spindle (perivascular growth) | 18 | 1 | + | NMF | |||||
| 2 | IM,SU | 66M | Serosal nodule right lower quadrant | 1.7 | Mixed (clear CP) | 4 | 3 | + | Exon 9 (insertion AY502–503) |
NMF | NMF |
| Omental nodule | 0.7 | Spindle | 10 | 1 | + | NMF | |||||
| Sigmoid mass (1) | 5.0 | Epithelioid | 3 | 3 | + | NMF | |||||
| Sigmoid mass (2) | 5.0 | Epithelioid | 6 | 3 | − | NMF | |||||
| Periumbilical tumour | 12.0 | Mixed | 9 | 2 | + | Exon 17 | N822Y | ||||
| Serosal nodule right upper quadrant (1) | 2.0 | Epithelioid | 34 | 1 | + | N822K | |||||
| Serosal nodule right upper quadrant (2) | 4.0 | Epithelioid | 33 | 2 | + | N822K | |||||
| 3 | IM,SU | 62M | Stomach (1) | 24.0 | Spindle (palisading) | 100 | 2 | + | Exon 11 (deletion EVQWKV554–559) |
Exon 17 | D8I6H |
| Stomach (2) | 24.0 | Spindle (palisading) | 40 | 2 | NMF | NMF | |||||
| 4 | IM | 50M | Liver metastasis | 9.5 | Mixed | 19 | 1 | + | Exon 11 (deletion KPMYEVQWK550–558) |
Exon 18 | A829P |
| Stomach (1) | 12.3 | Mixed | 54 | 2 | Exon 13 | V654A | |||||
| Stomach (2) | 12.3 | Epithelioid (giant cells) | 26 | 1 | V654A | ||||||
| 5 | IM,SU | 59F | Liver metastasis | 6.5 | Spindle | 7 | 1 | + | Exon 1 1 (deletion PYD577–579) |
Exon 13 | V654A |
| Mesenteric nodule (1) | 0.9 | Spindle | 22 | 1 | Exon 17 | N822Y | |||||
| Mesenteric nodule (2) | 2.9 | Spindle | 34 | 1 | N822Y | ||||||
| Nodule attached to gallbladder | 3.0 | Spindle (giant cells) | 20 | 1 | D820G | ||||||
| 6 | IM | 75 M | Intra-abdominal 1 | 6.0 | Spindle | 36 | 1 | + | Exon 11 (deletion VEEINGNNYVYIDPTQL560–576) |
Exon 13 | V654A |
| Intra-abdominal 2 | 5.0 | Spindle | 88 | 1 | V654A | ||||||
| Intra-abdominal 2 | 5.0 | Spindle | 88 | 1 | V654A | ||||||
| 7 | IM,SU | 67F | Stomach | 35.0 | Epithelioid (intracytoplasmic inclusions) |
2 | 1 | − | Exon 11 (deletion WKV557-559(F)) |
NMF | NMF |
| Liver metastasis | 2.7 | Epithelioid (intracytoplasmic inclusions and atypia) |
13 | 2 | NMF | ||||||
| Serosal nodule | 3.0 | Epithelioid (intracytoplasmic inclusions and atypia) |
14 | 2 | NMF | ||||||
| 8 | IM | 54M | Abdominal tumour (1) | 3.0 | Spindle | 1 | 3 | + | Exon 11 (deletion WKV557-559C) |
NMF | NMF |
| Tumour Stomach wall | 15.4 | Mixed | 26 | 2 | Exon 17 | Y823D | |||||
| Abdominal tumour (2) | 2.5 | Epithelioid | 8 | 3 | D820Y | ||||||
| Abdominal tumour (3) | 2.5 | Epithelioid | 8 | 3 | D820Y | ||||||
| 9 | IM,SU | 75M | Mesenteric nodule 1 | 1.9 | Spindle | 16 | 2 | + | Exon 11 (point mutation L576P) |
Exon 13 | V654A |
| Skin metastasis | 3.5 | Spindle | 57 | 1 | V654A | ||||||
| Mesenteric nodule 2 | 3.3 | Spindle | 72 | 2 | V654A | ||||||
| Mesenteric nodule 4 | 1.8 | Spindle | 24 | 1 | V654A | ||||||
| Mesenteric nodule 3 | 2.9 | Spindle | 44 | 1 | Exon 13 and 17 | V654A, D820G | |||||
| 10 | IM | 74M | Stomach wall (1) | 13.0 | Epithelioid (intracytoplasmic inclusions) |
15 | 1 | + | Exon 13 (point mutation K642E) |
NMF | NMF |
| Stomach wall (2) | Epithelioid | 52 | 1 | + | NMF | ||||||
| Stomach wall (3) | Epithelioid | 61 | 2 | + | NMF | ||||||
| 11 | IM,SU | 63M | Tumour mid-abdomen | 3.8 | Mixed | 1 | 2 | − | Exon 13 (point mutation K642E) |
NMF | NMF |
| Serosal nodule left upper quadrant (1) | 3.6 | Spindle | 1 | 4 | + | Exon 14 | T670I | ||||
| Serosal nodule ileum (1) | 0.5 | Spindle | 7 | 1 | + | Exon 17 | D8I6H | ||||
| Serosal nodule ileum (2) | 1.0 | Epithelioid | 34 | 1 | + | D8I6H | |||||
| Serosal nodule terminal ileum | 0.7 | Spindle | 1 | 1 | + | D8I6H | |||||
| Serosal nodule left upper quadrant (2) | 2.8 | Mixed | 3 | 2 | + | D8I6H | |||||
| 12 | IM,SU | 60M | Omental nodule | 4.5 | Epithelioid | 36 | 1 | + | NMF | NMF | |
| Liver metastasis (1) | 2.8 | Epithelioid (huge eosinophilic CP) |
18 | 1 | + | ||||||
| Tumour sigmoid wall | 14.0 | Epithelioid | 15 | 1 | + | ||||||
| Liver metastasis (2) | 2.1 | Epithelioid (huge eosinophilic CP) |
16 | 1 | + | ||||||
| 13, NF Type 1 | IM,SU | 44M | Serosal nodule adjacent to bowel | 3.5 | Spindle (storiform) | 1 | 2 | + | NMF | NMF | |
| Nodule lig. of Tritz | 3.0 | Spindle | 1 | 3 | + | ||||||
| Nodule duodenum | 2.5 | Spindle (storiform) | 2 | 2 | + | ||||||
| Mesenteric nodule | 15.0 | Spindle (storiform) | 6 | 2 | + | ||||||
| 14 | IM,SU | 65M | Perigastric mass (1) | 1.5 | Spindle with atypia | 48 | 1 | − | NMF | NMF | |
| Perigastric mass (2) | 2.5 | Epithelioid | 4 | 1 | + | ||||||
| Perigastric mass (3) | 2.4 | Epithelioid | 14 | 2 | + | ||||||
| Liver metastasis | 19.8 | Epithelioid | 58 | 1 | + | ||||||
| Large nodule lesser curvature (1) | 3.5 | Epithelioid with mild atypia | 19 | 1 | + | ||||||
| Large nodule lesser curvature (2) | 3.5 | Spindle with atypia | 60 | 1 | − | ||||||
pt, patient; IM, imatinib; SU, sunitinib; CP, cytoplasm; +, positive; −, negative; NMF, no mutation found.
All listed samples were examinated for KIT mutations in exons 9, 11, 12, 13, 14, 15, 16, 17, 18 and PDGFRA mutations in exons 12, 14, 18. Exons not specifically listed are wild-type.
Immunohistochemistry
Immunohistochemical studies for KIT (CD117) (Dako, Carpinteria, CA, USA; polyclonal A4502, 1 : 250) were performed in all cases without epitope retrieval, as previously described [13]. In cases lacking KIT expression, additional immunohistochemical stains using antibodies against SMA (Sigma, St. Louis, MO, USA; 1A4, 1 : 20.000); Desmin (Dako; D33, 1 : 500); Caldesmon (Dako; h-CD, 1 : 300; heat-induced epitope retrieval) and MYF4 (Novocastra, Burlingame, CA, USA; LO26,1 : 600, heat-induced epitope retrieval) were performed. The Envision Plus detection system (Dako) was used for all antibodies. Appropriate positive and negative controls were included.
DNA Extraction and initial mutation screening
The 57 tumour areas of interest were marked and collected from unstained sections by manual tumour tissue dissection. Tumour tissue was deparaffinized as previously reported [14]. Mutational analyses were performed on the extracted genomic DNA, using a combination of polymerase chain reaction (PCR) amplification, denaturing high-performance liquid chromatography (D-HPLC) screening and automated sequencing, as described previously [1,2,15]. KIT exons 9, 11, 12, 13, 14, 15, 16, 17, 18 and PDGFRA exons 12, 14, 18 were evaluated.
Mutation screening by allele-specific PCR
Five known hotspots for KIT secondary resistance mutations [V654A, D820G, N822K (T → A and T → G), Y823D] were screened by novel allele-specific PCR assays. Details on the development of these assays will be the subject of another report (Kepten et al, manuscript in preparation). In brief, mutation-specific forward primers were designed such that the nucleotide substitution of interest was matched by a locked nucleic acid at the 3′ end. Amplicons were detected by hydrolysable, dual-labelled probes to exon 13 or exon 17, depending on the site of mutation. PCR conditions were established such that dilutions of GIST DNA samples with known mutations (estimated to be 50% by direct sequencing) were routinely positive down to the level of 1 : 100 (estimated 0.5% mutant allele). DNA from formalin-fixed, paraffin-embedded normal tissue was either negative or had Ct > 5 cycles beyond that of the 1 : 100 dilution. Dilution controls and normal DNA controls were included in each assay. All abnormal allele-specific assays were repeated at least once.
Fluorescence in situ hybridization (FISH)
FISH analyses of KIT copy number were performed on 4 μm tissue sections that were pre-baked for 2 h at 60 °C. The sections were deparaffinized in xylene three times (each 15 min) and dehydrated twice in 100% ethanol for 2 min. The slides were then immersed in TRIS–EDTA (100 mm Tris base, 50 mm EDTA, pH 7.0) for 45 min at 95–99 °C and rinsed in ×1 PBS for 5 min. Proteolytic digestion of the sections was performed using Digest–ALL 3 (Invitrogen, Carlbad, CA, USA) at 37 °C for 20 min, twice. The sections were then sequentially dehydrated in alcohol (70%, 85%, 95% and 100%) for 2 min each and air-dried. The KIT probe comprised two overlapping BAC clones, C00-84L10 and RP11-586A2, labelled by random priming with digoxigenin and detected with FITC anti-digoxigenin, and co-hybridized with a spectrum orange-labelled chromosome 4 centromeric probe (CEP4; Vysis); 100 interphase nuclei were evaluated from each specimen. The cytogenetic patterns were classified as FISH-negative (no or low genomic gain: ≤ four copies of KIT in >40% of cells), FISH-positive (high level of polysomy: ≥ four copies of KIT in ≥40% of cells), or gene amplification (presence of tight KIT gene clusters and a ratio of KIT and chromosome 4 cen ≥2 per cell, or ≥15 copies of KIT per cell in ≥10% of analyzed cells) [16].
Results
Morphological correlates of resistance
To direct the genomic studies and to address morphological correlates for TKI resistance heterogeneity, we sampled a broad spectrum of tumour areas in each patient by evaluating several morphological parameters. Among the 57 GIST samples, the cellular morphology ranged from typical spindle cell (n = 26), to epithelioid (n = 15), to mixed (n = 7) to lesions with unusual (n = 9) features (Figure 1A–H). The spindle cell GISTs were composed of cells with palely eosinophilic fibrillary cytoplasm, ovoid nuclei and ill-defined cell borders, often with syncytial appearance (Figure 1A). GISTs with epithelioid cell morphology were composed of round cells with eosinophilic to clear cytoplasm, arranged in sheets and nests (Figure 1B). One or more samples from patients 1, 3 and 13 had a prominent perivascular, palisading (Figure 1A) or storiform growth pattern. Two tumours obtained from patient 14 showed epithelioid morphology with abrupt transformation to a pleomorphic spindle cell sarcoma (Table 1, Figure 1C and 1D). Other unusual morphologies included huge epithelioid cells with nuclear atypia (Figure 1E) and focal intracytoplasmic inclusions, as well as pleomorphic spindle cell areas. In total, nine samples with unusual morphology were collected from patients 7, 10, 12 and 14 (Table 1). Multinucleated giant cells were present in two samples obtained from patients 4 and 5 (Table 1).
Figure 1.
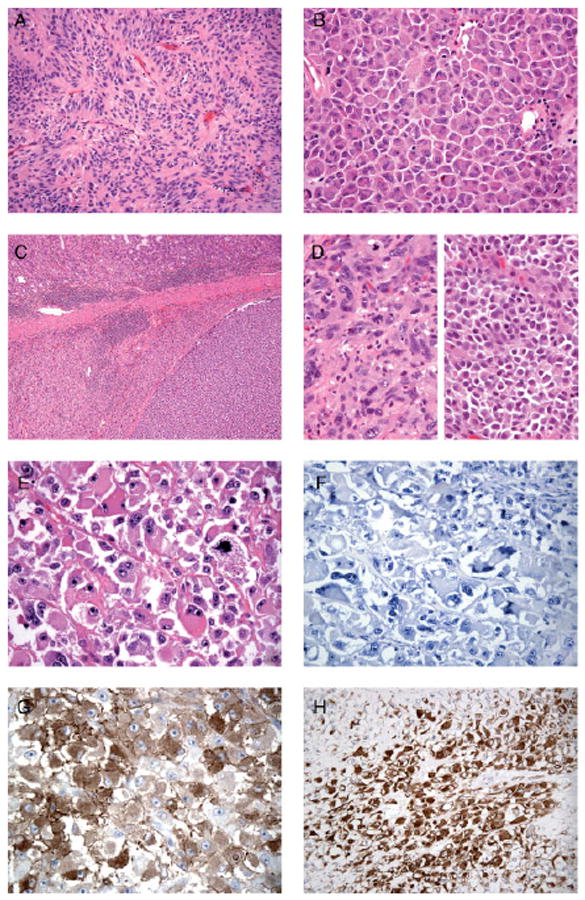
(A) GIST with spindle cell morphology composed of cells with a pale eosinophilic fibrillary cytoplasm, ovoid nuclei and ill-defined cell borders, with syncytial appearance and palisading (patient 3). (B) GIST with epithelioid cell morphology composed of round cells with eosinophilic cytoplasm arranged in sheets (patient 2). (C) Low-power view of a GIST in the stomach wall, with abrupt transition between two morphologically different tumour components (patient 14; KIT and PDGFR wild-type). Higher-power images of this tumour are demonstrated in (D), showing epithelioid morphology (right) and pleomorphic spindle cell morphology (left). (E) GIST showing unusual morphology, with huge epithelioid cells, vesicular nuclei and prominent nucleoli (patient 7; primary KIT exon 11 mutation). (F) KIT-negative GIST (patient 7). (G) KIT-negative GIST showing strong cytoplasmic staining for caldesmon (patient 7). (H) KIT-negative GIST showing strong cytoplasmic staining for desmin (patient 7)
Histological treatment response was heterogeneous within and between metastases selected from a given patient. Thirty-one (54.4%) samples showed minimal, 18 (31.6%) low, 7 (12.3%) moderate and only 1 (1.7%) sample showed high treatment response (Table 1). Of the samples from patients studied after progression on sunitinib, 24 (54.5%) tumours showed minimal, 15 (34.1%) low, four (9.1%) moderate and only one (2.3%) tumour showed high treatment response, respectively.
Mitotic activity was in the range 1–100 mitoses/50 HPFs. Metastases with moderate and high treatment response (eight samples) had a median mitotic activity of 4/50 HPF (range 1/50–8/50 HPFs), whereas metastases with minimal and low response (49 samples) had a median mitotic activity of 29/50 HPFs (range 1/50–100/50 HPFs). The sizes of the individual tumour nodules were in the range 0.5–35 cm (median 6.1 cm). Tumour size did not correlate with treatment effect or the frequency of detected secondary mutations (Tables 1, 2). All post-treatment samples showed robust blood vessels surrounded by smooth muscle cells/pericytes, arguing against an antiangiogenic effect of therapy. Treatment effects were noted only in the tumour parenchyma.
Table 2.
Summary of secondary imatinib resistant KIT mutations evaluated by D-HPLC and AS–PCR
| Patient no. | Treatment | Location | Primary KIT mutations | Secondary KIT mutation frequencies
|
|||
|---|---|---|---|---|---|---|---|
| DHPLC | AS–PCR* | AS–PCR* | |||||
| ∼15% | >5% | 5–0.5% | |||||
| 1 | IM | Serosal nodule (1) | Exon 9 | NMF | NMF | NMF | NMF |
| 2 | IM, SU | Serosal nodule right lower quadrant | Exon 9 | NMF | NMF | NMF | NMF |
| Omental nodule | Exon 9 | NMF | NMF | NMF | V654A | ||
| Periumbilical tumour | Exon 9 | Exon 17 | N822Y | NMF | D820G | ||
| Serosal nodule right upper quadrant (1) | Exon 9 | Exon 17 | N822K | N822K (T → G) | V654A | ||
| Serosal nodule right upper quadrant (2) | Exon 9 | Exon 17 | N822K | N822K (T → G) | NMF | ||
| 3 | IM, SU | Stomach (1) | Exon 11 | Exon 17 | D816H | NMF | NMF |
| 4 | IM | Liver metastasis | Exon 11 | Exon 18 | A829P | NMF | NMF |
| Stomach (1) | Exon 11 | Exon 13 | V654A | V654A | NMF | ||
| 5 | IM, SU | Liver metastasis | Exon 11 | Exon 13 | V654A | V654A | D820G |
| Mesenteric nodule (1) | Exon 11 | Exon 17 | N822Y | NMF | N822K (T → A) | ||
| Mesenteric nodule (2) | Exon 11 | Exon 17 | N822Y | NMF | N822K (T → G) | ||
| Nodule attached to gallbladder | Exon 11 | Exon 17 | D820G | D820G | NMF | ||
| 6 | IM | Intra-abdominal 1 | Exon 11 | Exon 13 | V654A | V654A | NMF |
| Intra-abdominal 2 | Exon 11 | Exon 13 | V654A | V654A | NMF | ||
| 7 | IM, SU | Stomach | Exon 11 | NMF | NMF | NMF | NMF |
| Liver metastasis | Exon 11 | NMF | NMF | NMF | NMF | ||
| Serosal nodule | Exon 11 | NMF | NMF | NMF | NMF | ||
| 8 | IM | Abdominal tumour (1) | Exon 11 | NMF | NMF | NMF | NMF |
| Abdominal tumour (2) | Exon 11 | Exon 17 | D820Y | NMF | NMF | ||
| Abdominal tumour (3) | Exon 11 | Exon 17 | D820Y | NMF | NMF | ||
| 9 | IM, SU | Mesenteric nodule 1 | Exon 11 | Exon 13 | V654A | V654A | NMF |
| Skin metastasis | Exon 11 | Exon 13 | V654A | V654A | NMF | ||
| Mesenteric nodule 4 | Exon 11 | Exon 13 | V654A | V654A | N822K (T → A) | ||
| 10 | IM | Stomach wall (3) | Exon 13 | NMF | NMF | NMF | D820G |
| 11 | IM, SU | Tumour mid-abdomen | Exon 13 | NMF | NMF | NMF | NMF |
| Serosal nodule left upper quadrant (1) | Exon 13 | Exon 14 | T670I | NMF | NMF | ||
| Serosal nodule ileum (1) | Exon 13 | Exon 17 | D816H | NMF | NMF | ||
| Serosal nodule ileum (2) | Exon 13 | Exon 17 | D816H | NMF | NMF | ||
| Serosal nodule terminal ileum | Exon 13 | Exon 17 | D816H | NMF | NMF | ||
| Serosal nodule left upper quadrant (2) | Exon 13 | Exon 17 | D816H | NMF | NMF | ||
| 12 | IM, SU | Omental nodule | NMF | NMF | NMF | NMF | |
| Liver metastasis (1) | |||||||
| Tumour sigmoid wall | |||||||
| Liver metastasis (2) | |||||||
| 13, NF Type I | IM, SU | Serosal nodule adjacent to bowel | NMF | NMF | NMF | NMF | |
| Nodule ligamentum of Tritz | |||||||
| Nodule duodenum | |||||||
| Mesenteric nodule | |||||||
| 14 | IM, SU | Perigastric mass (1) | NMF | NMF | NMF | NMF | |
| Perigastric mass (2) | |||||||
| Perigastric mass (3) | |||||||
| Liver metastasis | |||||||
| Large nodule lesser curvature (1) | |||||||
| Large nodule lesser curvature (2) | |||||||
AS–PCR detects: exon 13, V654A, exon 17, D820G, N822K (T → A), N822K (T → G), Y823D.
pt, patient; NMF, no mutation found.
Heterogeneity in the immunohistochemical staining profile
KIT (CD117) staining was scored as positive in 50/57 tumour samples. All samples from patient 7 (Figure 1E) were KIT-negative (Figure 1F) but showed multifocal strong positivity for SMA, caldesmon (Figure 1G) and desmin (Figure 1H), whereas MYF4 staining was negative. In three patients (2, 11 and 14) a mixture of KIT-positive and -negative regions was observed. KIT-negative samples from patients 11 and 14 showed immunoreactivity with caldesmon and SMA, respectively. By contrast, the KIT-negative sample from patient 2 did not stain for muscle markers.
Mutational heterogeneity
Primary KIT mutations were detected by D-HPLC and sequencing in 11 patients (nos 1–11) (Table 1), whereas all GIST samples from the remaining three patients (nos 12–14) were KIT and PDGFRA wild-type, including four samples obtained from a patient (no. 13) with neurofibromatosis type 1 (Table 1). In all tumour samples collected from a given patient, the same primary mutation was detected. Seven patients showed a primary KIT mutation in exon 11 (nos 3–9) and in two patients a primary mutation in exon 9 [nos 2 and 17] or exon 13 (nos 10 and 11) was detected (Table 1).
All 57 tumour samples were screened for secondary imatinib-resistance mutations in KIT by D-HPLC (Table 1, Figure 2A), which has a sensitivity of ∼15% mutant alleles. In addition, 46/57 tumour samples were screened by allele-specific PCR (AS–PCR; Table 2, Figure 2B), which has a sensitivity of 0.5% mutant alleles. By using these complimentary techniques, secondary imatinib-resistance mutations in KIT were found in 9/11 patients (82%), irrespective of the primary (exon 9, 11 or 13) KIT mutation. The secondary imatinib-resistant KIT mutations were clustered in two regions, the ATP binding pocket of the KIT kinase (exons 13 and 14) and in the kinase activation loop (exon 17) (Figure 2A, B). Furthermore, multiple different secondary imatinib-resistance mutations (between two and five) were found in 6/9 patients (67%) (nos 2, 4, 5, 8, 9, 11; Tables 1, 2). In seven samples obtained from three patients (nos 2, 5, 9), two secondary imatinib-resistance KIT mutations were detected in one or more individual tumour samples (Tables 1, 2).
Figure 2.
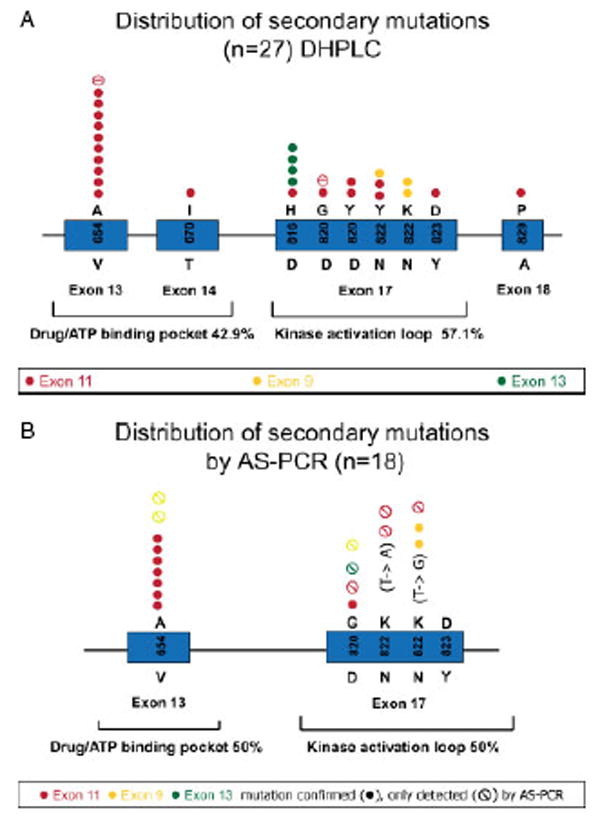
(A) Summary of secondary KIT resistance mutations detected by D-HPLC in 27/57 tumour samples from 14 patients with progressing GISTs. Associated primary KIT mutations are indicated in red (exon 11 mutations), yellow (exon 9 mutations) and green (exon 13 mutations). (B) Summary of secondary KIT resistance mutations detected by AS–PCR only (
 ) or detected by both AS–PCR and D-HPLC (●) in 18 tumour samples. Associated primary KIT mutations are indicated in red (exon 11 mutations), yellow (exon 9 mutations) and green (exon 13 mutations)
) or detected by both AS–PCR and D-HPLC (●) in 18 tumour samples. Associated primary KIT mutations are indicated in red (exon 11 mutations), yellow (exon 9 mutations) and green (exon 13 mutations)
AS–PCR assays were performed for five resistance mutation hotspots [V654A, D820G, N822K (T → A and T → G) and Y823D]. In all cases where one of these mutations was detected by D-HPLC, there was a strong signal (>5%) by AS–PCR (Table 2). AS–PCR identified secondary KIT resistance mutations that were not demonstrable by D-HPLC in one or more tumour samples from four patients (nos 2, 5, 9, 10; Table 2). These additional resistance mutations were present at low abundance (0.5–5%) in the background of more abundant (D-HPLC-detectable) secondary mutations, or as the only anomaly. Interestingly, all samples collected from patient 7 lacked KIT expression and neither D-HPLC nor AS–PCR revealed secondary resistance mutations in these samples (Tables 1, 2). Furthermore, D-HPLC and AS–PCR did not detect secondary KIT resistance mutations in tumour samples showing unusual morphological features, in KIT/PDGFRA wild-type GISTs or in KIT-negative GISTs (Tables 1, 2). A metastasis showing a T670I resistance mutation was the only tumour in this study with high morphological treatment response (Table 1).
Fluorescence in situ analysis
FISH was performed in 10/14 GIST samples showing a primary KIT mutation in exon 9 (nos 1, 2), exon 11 (nos 3, 7, 8) and exon 13 (nos 10,11) but lacking a secondary KIT mutation. Eight of the 10 samples were FISH-negative (Figure 3A), whereas two samples, both from patient 10, had KIT amplification (Figure 3B).
Figure 3.
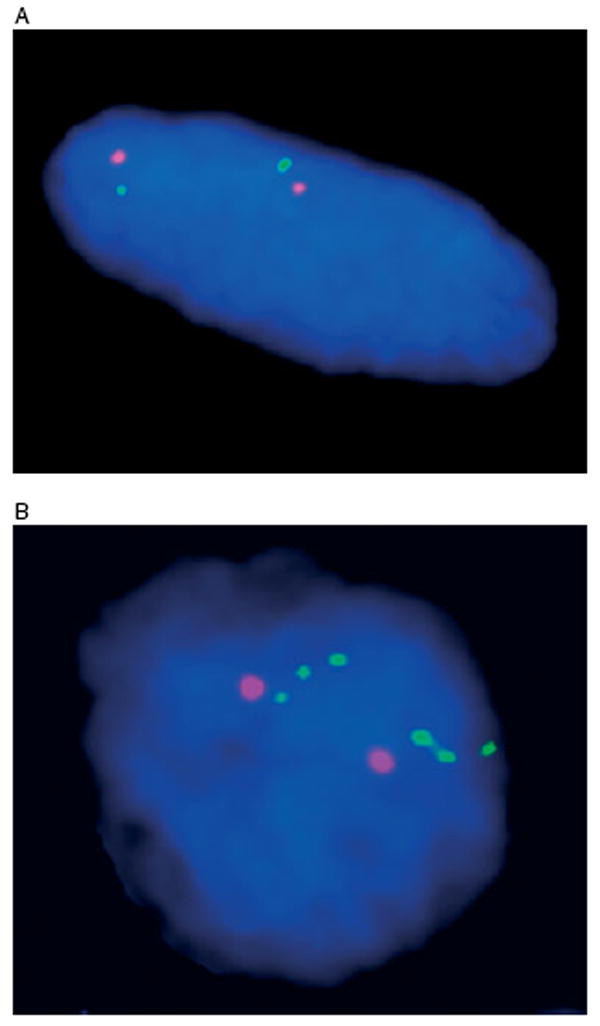
(A) FISH analysis of abdominal tumour 1 (spindle cell morphology) from patient 8, with disomic (FISH-negative) pattern. The chromosome 4 centromere probe is shown in orange and the KIT probe in green. (B) FISH analysis of stomach wall (2) tumour (epithelioid morphology) from patient 10, with KIT amplification. Chromosome 4 centromere probe is shown in orange and the KIT probe in green
Discussion
The aim of this study was to determine the heterogeneity of tyrosine kinase inhibitor (TKI) resistance mutations within and between different metastatic GIST lesions and to examine the relationship between drug resistance and morphological variability, mitotic activity and immunophenotype. Secondary KIT mutations were interrogated using two complementary techniques, D-HPLC and AS–PCR. D-HPLC provided a broad unbiased screening approach to detect secondary mutations in various KIT and PDGFRA exons, with a sensitivity of ∼15%. By contrast, the novel AS–PCR approach was designed to detect five hotspot secondary imatinib-resistant KIT mutations, with a sensitivity of ∼0.5%. Secondary KIT kinase domain mutations were found in 82% of patients after imatinib or sunitinib therapy, irrespective of whether the primary mutation was in KIT exon 9, 11 or 13. The secondary mutations were clustered in the ATP binding pocket (D-HPLC 42.9% and AS–PCR 50%) and in the activation loop (D-HPLC 57.1% and AS–PCR 50%) of the KIT kinase domain. D-HPLC demonstrated two alternative secondary KIT resistance mutations (involving exons 13 and 17) in only 1/56 tumour samples, whereas the more sensitive AS–PCR method demonstrated more than one resistance mutation in each of six metastases. These findings underscore the complexity of clinically important TKI resistance mechanisms. Whereas previous reports have demonstrated resistance mutations in 44–67% of GISTs progressing after imatinib therapy [6,18–20], the combination of D-HPLC and AS–PCR revealed secondary KIT TKI-resistance mutations in 9/10 patients (90%) whose GIST had a primary KIT oncogenic mutation and whose tumour expressed KIT protein. In addition, we demonstrated two or more (between two and five) TKI-resistance mutations in most patients in this series, whereas previous studies have shown multiple resistance mutations in ∼10–30% of GIST patients at the time of clinical progression [6,20]. Contributing factors to the higher level of demonstrable TKI-resistance mutations might include the more extensive sampling of multiple metastases, incorporation of the highly sensitive AS–PCR detection method, and clonal selection for additional resistance mutations in patients receiving sunitinib. Patients receiving sunitinib treatment showed substantially more (one to five) secondary imatinib-resistance mutations compared to patients only treated with imatinib (at most two). However, the mutation types seen after sunitinib therapy, in this study, were similar to those reported after progression on imatinib therapy alone [6,8,10]. Secondary resistance mutations in the KIT activation loop (exon 17) were seen in ∼60% (Table 2) of sunitinib-treated metastases, which is in accord with in vitro evidence that mutations in this region are sunitinib-resistant [21]. Although multiple KIT exons (9,11–18) were screened for secondary mutations outside the ATP binding pocket and the kinase activation loop, D-HPLC did not reveal mutations in these regions.
KIT V654A is the most frequent secondary mutation in patients whose GISTs have primary KIT exon 11 mutations and who eventually progress during imatinib treatment [6,8]. Interestingly, the V654A mutation, which is sunitinib-sensitive based on in vitro studies [21], was found in ∼27% (Table 2) of samples after clinical progression on sunitinib. In addition, these same samples showed minimal to low morphological evidence of treatment response. Such observations suggest that sunitinib might be cytostatic rather than cytocidal in GISTs with secondary V654A mutations. This is in keeping with the clinical evidence from a randomized, placebo-controlled, multi-centre trial demonstrating that stable GIST was the best overall tumour response on sunitinib treatment [11]. In addition, the presence of low-level TKI resistance mutations on the same KIT alleles encoding the V654A might also account for the persistence of V654A alleles during sunitinib therapy.
In theory, sunitinib therapeutic activity could result from either antiangiogenic or direct antitumoral activity, as this is a multikinase inhibitor that inhibits KIT, PDGFR, VEGFR, FLT3 and RET [22–26]. However, there is no compelling evidence for sunitinib antiangiogenic activity in GIST patients [27]. In the present study, moderate-to-high morphological evidence of sunitinib treatment response was found in 11.4% of metastases, even at time of clinical progression on sunitinib. Viable tumour cells from these samples were often clustered around blood vessels, and a reduction of overall tumour vasculature was not detected, suggesting that the treatment response in these cases resulted largely from direct inhibition of crucial KIT-mediated survival pathways in the GIST cells, rather than anti-angiogenic effects. As expected, GIST metastases with moderate and high treatment response had lower mitotic activity overall (median 4/50HPF) compared to those with minimal or low treatment response (median 29/50HPF). However, there was substantial variability in the mitotic activity amongst samples with minimal and low treatment response, such that mitotic activity alone is not a reliable marker for clinical progression. Interruption of TKI therapy, as might occur pre-operatively, is known to augment KIT phosphorylation and the biochemical activity of downstream signalling proteins in GIST, and to cause PET scan ‘flares’ [18]. Therefore, evaluation of treatment effects based on more fixed morphological correlates, such as necrosis and fibrosis, might be a more reliable indicator than mitotic activity in distinguishing clinically stable from progressing GIST lesions. In our study, tumour size did not correlate with the presence of treatment effect, and this has been noted in a previous report [28]. Furthermore, there was no correlation between tumour size and frequency of detectable secondary KIT kinase mutations.
Using complementary D-HPLC and AS–PCR mutational screening approaches, we demonstrated secondary resistance mutations as the overriding resistance mechanism in 9/14 patients. By contrast, all patients whose GISTs had wild-type KIT/PDGFRA lacked kinase domain resistance mutations, indicating that mechanisms of resistance are fundamentally different in these GISTs. KIT wild-type GISTs feature levels of KIT activation similar to KIT-mutant GISTs [29,30], suggesting that KIT has a key oncogenic role in the pathogenesis of these tumours. However, imatinib is a less potent inhibitor of wild-type KIT compared with exon 11-mutant KIT [37, Heinrich et al, submitted], possibly accounting for the lower clinical imatinib responsiveness for GISTs expressing wild-type KIT.
Unusual morphologies were seen in four patients, including two whose GISTs were KIT/PDGFRA wild-type and two with metastases that lacked KIT expression. The distinctive morphological features included large epithelioid cells with vesicular chromatin, prominent nucleoli and focal intracytoplasmic eosinophilic inclusions, and abrupt transformation/dedifferentiation in a high-grade spindle cell tumour. Mutational analyses of samples entirely comprised of these unusual morphological features did not reveal novel KIT mutations as the potential molecular mechanism accounting for these bizarre histological evolutions. However, the observed loss of KIT expression in some of these samples suggests that the histological changes resulted from activation of novel KIT-independent oncogenic pathways. Interestingly, smooth muscle markers were expressed strongly in six of the seven samples featuring loss of KIT expression. These observations fit with developmental biology evidence that smooth muscle cells and interstitial cells of Cajal (ICC) arise from a common progenitor cell, with KIT signalling committing these progenitor cells to ICC differentiation, and KIT inhibition resulting in a switch to smooth muscle differentiation [31]. Therefore, unusual morphological and immunohistochemical features are found in GISTs after TKI treatment, and might present diagnostic challenges, particularly if coupled with loss of KIT expression, as detected by IHC.
Our studies also confirm a role, albeit limited [32], for KIT amplification as a mechanism of drug resistance in GIST. Analyses in 10 GISTs lacking demonstrable secondary KIT mutations revealed localized KIT amplicons in both samples analysed from patient 10, whose GIST had a primary KIT exon 13 mutation. These findings suggest that increased gene dosage can contribute to clinical progression in some GISTs.
Overall, the molecular, FISH and histological assessments described here underscore a striking inter- and intra-lesional heterogeneity in TKI resistance mechanisms. With regard to treatment approaches, newer generations of broad-spectrum KIT and PDGFRA kinase inhibitors, as well as combination therapies with various such inhibitors, could prolong GIST remissions in a manner similar to treatment approaches used in HIV, by suppressing a broader spectrum of tumour clones from the outset of therapy. Novel treatment options include inhibition of the KIT chaperone HSP90 [33], which may result in KIT oncoprotein degradation, irrespective of the TKI-resistance mutations present. Other broadly relevant therapeutic strategies include blockage of crucial KIT-mediated signalling pathways, as might be accomplished via PI3-K inhibition [34].
In summary, our study underscores the overriding contribution of secondary KIT mutations to TKI resistance in GISTs and demonstrates substantial heterogeneity of resistance mutations within and between metastases from individual patients. However, the highly sensitive and complementary D-HPLC and AS–PCR methods did not show TKI-resistance mutations in tumours wild-type for KIT/PDGFRA, indicating that resistance mechanisms are fundamentally different in these GISTs. Likewise, secondary KIT TKI-resistance mutations were not found in clinically progressing GISTs that lacked KIT expression, suggesting that activation of novel oncogenic pathways accounts for TKI resistance in such cases. Our observations on the complexity of TKI resistance underscore the challenges in achieving long-term disease control with kinase inhibitor monotherapies, and raise concern over the ultimate effectiveness of second- and third-line TKI drugs in GIST patients resistant to imatinib.
Acknowledgments
This work was supported by grants from an anonymous donor, GI SPORE 1P50CA127003-01, the Life Raft Group, the Cesarini Team for the Pan-Massachusetts Challenge, the Virginia and Daniel K. Ludwig Trust for Cancer Research, the Ronald O. Perelman Fund for Cancer Research, the Stutman GIST Cancer Research Fund, the BP Lester Foundation, the Rubenstein Foundation, Leslie's Links, a VA Merit Review Grant and the FWF Austrian Science Fund.
Footnotes
No conflicts of interest were declared.
References
- 1.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 2.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 3.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 4.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 5.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 6.Heinrich M, Corless C, Blanke C, et al. Molecular correlates of imatinib resistence in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–4774. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 7.Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40(5):689–695. doi: 10.1016/j.ejca.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 8.Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher J, Corless C, Dimitrijevic S, et al. Mechanisms of resistance to imatinib mesylate (IM) in advanced gastrointestinal stromal tumors (GIST) Proc Am Soc Clin Oncol. 2003;22:3275–3277. [Google Scholar]
- 10.Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128(2):270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 11.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 12.Therasse P, Le Cesne A, Van Glabbeke M, Verweij J, Judson I. RECIST vs. WHO: prospective comparison of response criteria in an EORTC phase II clinical trial investigating ET-743 in advanced soft tissue sarcoma. Eur J Cancer. 2005;41(10):1426–1430. doi: 10.1016/j.ejca.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Hornick JL, Fletcher CD. Immunohistochemical staining for KIT (CD117) in soft tissue sarcomas is very limited in distribution. Am J Clin Pathol. 2002;117(2):188–193. doi: 10.1309/LX9U-F7P0-UWDH-8Y6R. [DOI] [PubMed] [Google Scholar]
- 14.Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002;160(5):1567–1572. doi: 10.1016/S0002-9440(10)61103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–5364. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- 16.Cappuzzo F, Varella-Garcia M, Shigematsu H, Domenichini I, Bartolini S, Ceresoli Gl, et al. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. J Clin Oncol. 2005;23(22):5007–5018. doi: 10.1200/JCO.2005.09.111. [DOI] [PubMed] [Google Scholar]
- 17.Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition TS1 of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002;20(6):1692–1703. doi: 10.1200/JCO.2002.20.6.1692. [DOI] [PubMed] [Google Scholar]
- 18.Agaram NP, Besmer P, Wong GC, Guo T, Socci ND, Maki RG, et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(1):170–181. doi: 10.1158/1078-0432.CCR-06-1508. [DOI] [PubMed] [Google Scholar]
- 19.Prenen H, Cools J, Mentens N, Folens C, Sciot R, Schoffski P, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12(8):2622–2627. doi: 10.1158/1078-0432.CCR-05-2275. [DOI] [PubMed] [Google Scholar]
- 20.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12(6):1743–1749. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- 21.Heinrich MC, Corless CL, Liegl B, Fletcher CD, Raut CP, Donsky R, et al. Mechanisms of sunitinib malate (SU) resistance in gastrointestinal stromal tumors (GISTs) J Clin Oncol. 2007;25(I No 18S June 20 suppl) Abstract No: 10006. [Google Scholar]
- 22.Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis. 2004;7(3):225–233. doi: 10.1007/s10456-004-3149-y. [DOI] [PubMed] [Google Scholar]
- 23.Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2(5):471–478. [PubMed] [Google Scholar]
- 24.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. [PubMed] [Google Scholar]
- 25.O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101(9):3597–3605. doi: 10.1182/blood-2002-07-2307. [DOI] [PubMed] [Google Scholar]
- 26.Schueneman AJ, Himmelfarb E, Geng L, Tan J, Donnelly E, Mendel D, et al. SU11248 maintenance therapy prevents tumor regrowth after fractionated irradiation of murine tumor models. Cancer Res. 2003;63(14):4009–4016. [PubMed] [Google Scholar]
- 27.Seandel M, Shia J, Linkov I, Maki RG, Antonescu CR, Dupont J. The activity of sunitinib against gastrointestinal stromal tumor seems to be distinct from its antiangiogenic effects. Clin Cancer Res. 2006;12(20):6203–6204. doi: 10.1158/1078-0432.CCR-06-1292. part 1. [DOI] [PubMed] [Google Scholar]
- 28.Agaram NP, Baren A, Arkun K, Dematteo RP, Besmer P, Antonescu CR. Comparative ultrastructural analysis and KIT/PDGFRA genotype in 125 gastrointestinal stromal tumors. Ultrastruct Pathol. 2006;30(6):443–452. doi: 10.1080/01913120600854186. [DOI] [PubMed] [Google Scholar]
- 29.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61(22):8118–8121. [PubMed] [Google Scholar]
- 30.Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67(19):9084–9088. doi: 10.1158/0008-5472.CAN-07-1938. [DOI] [PubMed] [Google Scholar]
- 31.Torihashi S, Nishi K, Tokutomi Y. Blockade of KIT signaling induces transdifferentiation in interstitiall cells of Cajal to smooth muscle phenotype. Gastroenterology. 1999;117:140–148. doi: 10.1016/s0016-5085(99)70560-3. [DOI] [PubMed] [Google Scholar]
- 32.Miselli F, Casieri P, Negri T, Orsenigo M, Lagonigro MS, Gronchi A, et al. c-KIT/PDGFRA gene status alterations possibly related to primary imatinib resistance in gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(8):2369–2377. doi: 10.1158/1078-0432.CCR-06-1745. [DOI] [PubMed] [Google Scholar]
- 33.Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66(18):9153–9161. doi: 10.1158/0008-5472.CAN-06-0165. [DOI] [PubMed] [Google Scholar]
- 34.Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26(54):7560–7568. doi: 10.1038/sj.onc.1210558. [DOI] [PubMed] [Google Scholar]