

Abstract
A growing body of evidence indicates a close relationship between tyrosine kinase receptor trafficking and signaling. Biochemical and molecular analyses of the expression, fate, and kinetics of membrane trafficking of the nerve growth factor (NGF) receptor TrkA were performed in PC12 cells. Pulse-chase experiments indicate that TrkA is synthesized as a 110-kDa N-glycosylated precursor that leads to the mature 140-kDa form of the receptor with a half-life of conversion of ∼24 ± 0.5 min. Neuraminidase digestion shows that modification of the carbohydrate moiety of the receptor by sialylation occurs during maturation. The 140-kDa form is rapidly translocated to the cell surface as assessed by cell surface biotinylation performed on intact PC12 cells. Mature receptor half-life is ∼138 ± 4 min and is shortened to 86 ± 8 min by NGF treatment. Flow cytometric analysis indicates that NGF induces clearing of this receptor from the cell surface within minutes of treatment. The addition of NGF decreases the half-life of cell surface gp140TrkA from 100 to 35 min and leads to enhanced lysosomal degradation of the receptor. The process of NGF-induced TrkA internalization is clearly affected by interfering with ligand binding to p75NTR. An analysis of receptor activation kinetics also shows that receptor signaling primarily takes place from an intracellular location. Together, these data show that the primary effect of NGF treatment is a p75NTR-modulated decrease in TrkA transit time at the cell surface.
NGF1 is the prototypic member of the neurotrophin family of ligands that includes brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5) (1, 2). Each neurotrophin interacts specifically with a member of the Trk receptor tyrosine kinase family; NGF binds to TrkA, BDNF and NT-4 bind to TrkB, and NT-3 binds to TrkC (1, 2). NT-3 can also activate the other Trk receptors with lower efficacy. Neurotrophins can also associate with the neurotrophin receptor p75NTR, which lacks intrinsic catalytic activity. NGF is necessary for differentiation and survival of certain sensory and sympathetic neurons (3, 4). PC12 cells express both TrkA and p75NTR (5). This cell line has been extensively studied as a model for NGF-induced signal transduction events because it can mimic NGF-induced survival or differentiation observed in neuronal cells (6). Binding of NGF to TrkA induces autophosphorylation of the receptor on specific tyrosine residues (7, 8). This initiates a cascade of events leading to the activation of phosphatidylinositol 3-kinase (PI-3K), mitogen-activated protein kinase (MAPK), and phospholipase C-γ (PLC-γ) (9-11).
Signaling events mediated by TrkA appear to be mediated by trafficking of the receptor. It has been shown, for example, that NGF treatment results in the accumulation of PC12 cells in the G1 phase of the cell cycle (12, 13). During early G1, there is an apparent enhanced expression of the TrkA receptor at the PC12 cell surface (14). Moreover, upon NGF binding TrkA has been reported (15) to exit from raft fractions where it is initially located. It has also been shown that, following ligand binding, TrkA undergoes a dynamin-dependent internalization probably via the coated pit pathway (16, 17). Interfering with this process inhibits neurotrophic activity of the growth factor (17, 18). Thus, molecular processes regulating both TrkA targeting to the cell surface and internalization from this location may play a role in modulating signaling via this receptor. In mature neurons, additional spatial constraints come into play because NGF is restricted to the synaptic area located far from the cell body where the growth factor is believed to exert its effect (19). Numerous studies support the idea that signaling from the growth cone to the cell body is mediated by signaling vesicles containing the activated NGF-TrkA complex (16, 20-22). Maturation events offer further means for the potential regulation of signaling by NGF. Two protein forms of TrkA predominate in the cell extracts, a 110 kDa N-glycosylated form called gp110TrkA and a 140-kDa fully matured form, gp140TrkA (23). gp110TrkA is proposed to be the precursor for the mature gp140TrkA. It has been shown that levels of gp110TrkA and gp140TrkA are not affected in the same manner by NGF treatment (24). Moreover, maturation of the receptor appears to depend on the cellular background in which TrkA is expressed. Indeed, depending on the cellular model used for the study of receptor maturation, the receptor form activated by NGF can be gp110TrkA or gp140TrkA (25, 26).
From synthesis to degradation, transmembrane proteins are directed to several different locations within the cell. Along the biosynthetic pathway they are inserted into the membrane bilayer at the level of the endoplasmic reticulum where some co-translational modifications may occur (27). Further maturation of the proteins occurs in the Golgi network where additional modifications of the lumenal domain are believed to take place (28). After this step, proteins may be directly translocated to the cell surface or to intracellular membrane compartments (29). Once at the plasma membrane, transmembrane proteins may enter the endocytic pathway, which brings them inside the cell within endosomes (30). From this location, proteins can return to the cell surface (recycling) or be targeted to the lysosome (degradation). In certain cases, extra-lysosomal proteolytic cleavage of the cytoplasmic portion of a receptor can occur, releasing a fragment that can go into other compartments such as the nucleus (31).
TrkA trafficking along both biosynthetic and endocytic pathways has been studied. Results presented herein offer an approximation of the kinetic parameters for the translocation of TrkA to and from the cell surface and the effect of NGF thereon. This was achieved by a complementary approach of cell surface biotinylation and flow cytometric analysis of cell surface receptor expression. These experiments allow us not only to measure the kinetics of receptor maturation and turnover but also to analyze how these kinetics correlate with variations in, and the activation state of, the total TrkA cellular pools found within different cellular compartments. The ability of NGF to modify receptor internalization from the cell surface has also been tested. Finally, the contribution of p75NTR to NGF-induced TrkA internalization has been evaluated.
MATERIALS AND METHODS
Reagents
Sulfo-NHS-biotin and streptavidin-agarose were purchased from Pierce. Anti-TrkA extracellular domain (RTA) and anti-p75NTR extracellular domain (REX) antibodies were prepared as previously described (32). Anti-transferrin receptor (HTR68-4) was kindly provided by I. Trowbridge (The Salk Institute, La Jolla, CA). Anti-p75 carboxyl-terminal sera was a kind gift of Dr. Moses V. Chao (New York University Medical Center). Anti-Trk (C-14), anti-phosphotyrosine (PY99), and anti-phospho 490 Trk (E6) were from Santa Cruz Biotechnology. 3NGF, a generous gift of Dr. C. Ibanez, was produced as described previously (33). NGF from mouse submaxillary glands was from Quality Controlled Biologicals. Protein A-Sepharose 4 Fast Flow and wheat germ agglutinin (WGA) were from Amersham Biosciences. R-phycoerythrin-conjugated Affinipure F(ab’)2 fragment donkey anti-rabbit IgG and rhodamine-conjugated anti-mouse IgG were from Jackson ImmunoResearch Laboratories. Neuraminidase and the anti-p75NTR extracellular domain monoclonal antibody MC192 were from Roche Molecular Biochemicals. Chloroquin, ammonium chloride, tunicamycin, and concanavalin A were from Sigma.
Metabolic Labeling, Cell Surface Biotinylation, and Immunoprecipitation
In pulse-chase experiments, cells were incubated for 2 h in methionine- and cysteine-free media. Cells (3-4 million/time point) were then exposed to 100 μCi/ml [35S]methionine and cysteine for 30 min. Cells were then washed twice with complete medium and chased for different periods of time. In some experiments, cells were incubated in the presence of 1 μg/ml tunicamycin before and during pulse-chase experiments. At the end of the chase period, cell surface biotinylation was performed. Intact cells were incubated for 45 min in ice-cold PBS containing 0.5 mg/ml sulfo-NHS-biotin. Cells were then washed four times in PBS with 2 mm lysine to remove unbound reactive biotin. After metabolic labeling and cell surface biotinylation, proteins were extracted using ice-cold lysis buffer (20 mm Tris-HCl, pH 8, 137 mm NaCl, 2 mm EDTA, 10% glycerol, 1% Nonidet P-40, 20 μm leupeptin, 1 mm sodium vanadate, 1 mm Pefabloc, 0.15 units/ml aprotinin, 1 mm β-glycerophosphate, and 6 mm sodium fluoride). The extracts were clarified by centrifugation at 12,000 × g for 10 min. Cleared lysates were then immunoprecipitated using the Trk C-14 antibody directed against the human TrkA carboxyl terminus. Immunocomplexes were collected using protein A-Sepharose beads and eluted by boiling for 10 min in 10% SDS. The resulting immunoprecipitated protein was divided in two parts. One part was kept as the “total TrkA” sample and the second part was subjected to a second precipitation step with streptavidin-agarose beads to obtain the “surface TrkA” fraction. Proteins were then subjected to SDS-PAGE, dried, and exposed to a phosphorimaging screen for quantification with a Storm PhosphorImager (Amersham Biosciences).
Neuraminidase Digestion
Proteins were labeled and immunoprecipitated as described in the previous paragraph. Immunocomplexes were resuspended in acetate buffer (20 mm sodium acetate, pH 5.0, 5 mm CaCl2). Digestion was then performed for 3 h at 37 °C with 0.75 milliunits/ml neuraminidase. The digested proteins were then analyzed by SDS-PAGE and autoradiography.
Flow Cytometric Analysis
Cells were collected in warm PBS supplemented with 0.1 mm CaCl2 and 1 mm MgCl2. After centrifugation, cells were resuspended in culture medium and subjected to NGF treatment (concentration ranging from 0 to 50 ng/ml) at 37 °C on a rotating wheel. In some experiments, cells were also treated with 8 μg/ml MC192 antibody. Cells were then washed twice in ice-cold PBS and used as live intact cells for immunolabeling. All the subsequent labeling steps were performed on ice. Cells were washed twice in blocking buffer (PBS with 0.5% bovine serum albumin and 0.02% sodium azide) and incubated for 30 min in the same buffer containing an antibody directed against the RTA. After two washes in blocking buffer, cells were exposed for 30 min to phycoerythrin-labeled secondary antibody and washed again three times. Cells were then analyzed using a FACScan flow cytometer (BD Biosciences Immunocytometry Systems) equipped with an argon ion laser tuned to 488 nm. Emission fluorescence was measured with a DF 585-42 filter. Data acquisition and analysis were performed with CellQuest software (BD PharMingen).
Cell Culture
PC12 cells (obtained from Dr. G. Guroff, National Institutes of Health) and PC12 6-24 overexpressing human neuronal TrkA (provided by Dr. D. Martin-Zanca, Instituto de Microbiologia Bioquemica, Universida de Salamanca, Spain) were grown as described previously (34).
Microscopy
PC12 cells were transfected with rat TrkA-EGFP vector using the calcium phosphate procedure. 24 h post-transfection, cells were spread on collagen poly-l-lysine-coated cover slips (14). 48 h post-transfection, cells were fixed for 10 min in PBS with 3.7% formaldehyde. Labeling of cell surface TrkA was then carried out by incubation with RTA serum (1:2000) followed by incubation with a rhodamine-labeled anti-rabbit IgG. Cells were then permeabilized for 1 min in PBS with 0.5% Triton X-100. After washing with PBS, cells were blocked with PBS with 0.5% bovine serum albumin for 30 min and incubated with anti-phospho TrkA mouse monoclonal antibody (E6; 1:200) or anti-transferrin receptor mouse monoclonal antibody (1 μg/ml) for 30 min. Cells were washed three times in PBS and incubated an additional 30 min with Cy5-conjugated anti-mouse IgG. After washing, cells were mounted in Moviol. Images were acquired using the MRC1000 confocal laser unit (Bio-Rad, Hercules, CA) coupled to a Zeiss Axioplan Microscope equipped with a Zeiss 40×, C-apo, 1.3 numerical aperture oil immersion objective.
RESULTS
Synthesis and Maturation of TrkA
Mechanisms by which TrkA is transported to the plasma membrane were investigated. Analyses of the synthesis and cell surface targeting of this receptor were performed by metabolic labeling. Although detectable, the levels of endogenous TrkA expression in PC12 cells were too low to allow proper monitoring of changes in turnover using metabolic labeling. The PC12 6-24 line stably expressing higher levels of TrkA was therefore used for this type of analysis and certain others in the course of these studies. Preliminary experiments indicated that the synthesis and maturation of endogenous and exogenous TrkA were similar for those parameters where comparison was possible (apparent molecular weight of the different forms, sensitivity to neuraminidase, surface targeting, surface to lysosome trafficking) (data not shown). Pulse-chase experiments presented in Fig. 1A indicate that TrkA is synthesized as a 110-kDa glycoprotein, which is processed to the 140-kDa mature form with a half-life of 24 ± 0.5 min. Tunicamycin treatment suggests that gp110TrkA has a sugar component that modifies the overall apparent molecular weight by approximately 30 kDa, because the inhibition of N-glycosylation by this drug leads to the appearance of the 80-kDa form of the receptor (Fig. 2A). Moreover, the inhibition of N-glycosylation abolished receptor maturation as shown by the absence of the higher molecular weight form of the receptor after 2 h of chase in the presence of tunicamycin. This lack of receptor processing correlates with the inability of the unglycosylated receptor to reach the cell surface (Fig. 2A, right panel). Experiments presented in Fig. 2B (top panel) indicate that gp140TrkA is the only form of TrkA that is sensitive to neuraminidase digestion. It would appear therefore, that the final step of maturation requires sialylation of the sugar moiety of the receptor. Moreover, the precursor and mature forms of TrkA are differentially precipitated with concanavalin A and WGA lectin as are those for p75NTR (Fig. 2B, bottom). These data argue again in favor of the modification of glycosylation during the processing of TrkA from the precursor to the mature form. WGA has greater affinity for glycosyl groups enriched in N-acetyl-glucosamine residues (35). These latter residues are transferred to glycoprotein in the Golgi (27). Preferential precipitation of gp140TrkA by this lectin suggests that the later steps of TrkA maturation occur at the level of the Golgi network.
Fig. 1. Processing and cell surface expression of TrkA.
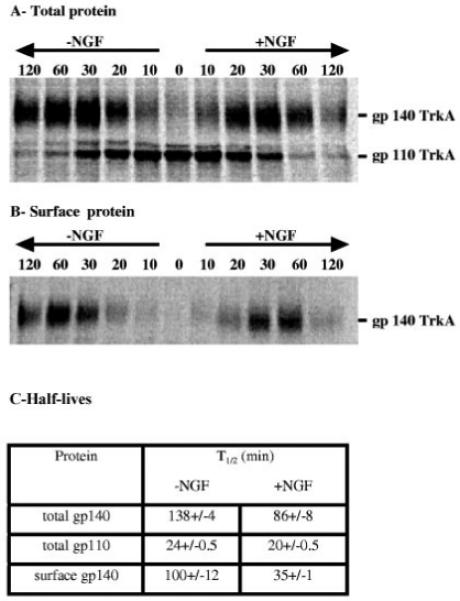
PC12 6-24 cells were used in pulse-chase experiments. Panels A and B are autoradiograms of total and cell surface TrkA (see “Materials and Methods” for details). When indicated, NGF (50 ng/ml) was added after the 30 min pulse period. Panel C indicates the half-lives of different TrkA pools as calculated from the quantification of autoradiograms. gp140TrkA half-lives are calculated using time points after which 90% of labeled gp110TrkA has disappeared. Values are means ± S.E. of three independent experiments.
Fig. 2. Maturation of TrkA.
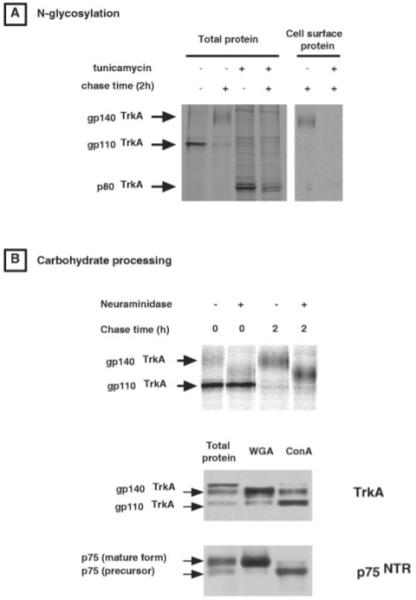
The lower panels of A and B are autoradiograms obtained from pulse-chase experiments in PC12 6-24 cells (see “Materials and methods” for details). A, the experiment has been performed either in the presence or in the absence of 1 μg/ml of the N-glycosylation inhibitor tunicamycin. B, the lower panel is an autoradiogram of TrkA proteins that have been subjected to neuraminidase digestion after immunoprecipitation. The upper panel presents the results of Western blot analysis performed on extracts precipitated with the lectins concanavalin A (ConA) or WGA. Membranes were probed with the RTA antibody directed against the TrkA extracellular domain or with the anti-p75NTR carboxyl-terminal antibody.
Translocation of TrkA to the Cell Surface
Cell surface protein biotinylation allows the monitoring of the cell surface appearance of the receptor during the time course of pulse-chase experiments. The mature gp140TrkA form of the receptor is detectable both in the total cell extract and at the cell surface after a 10 min chase (Fig. 1, A and B). These kinetics suggest that, as soon as it is mature, TrkA is translocated to the cell surface. The half-life of the mature receptor is 138 ± 4 min (Fig. 1C). Cell surface biotinylation allows calculation of the half-life of gp140TrkA at the cell surface as being 100 ± 12 min, which is ∼70% of the half-life of total gp140TrkA.
Effect of NGF on Receptor Half-life
Pulse-chase experiments followed by the quantification of total or cell surface TrkA show a differential impact of NGF treatment on the steps of TrkA processing and trafficking analyzed previously. Fig. 1C shows that the presence of 50 ng/ml of NGF induces only a slight decrease in the half-life of gp110TrkA from 24 to 20 ± 0.5 min. By contrast, the half-life of total gp140TrkA appears to be decreased by 40% in the presence of the growth factor (from 138 ± 4 to 86 ± 8 min). Moreover, there is a 65% reduction in the cell surface receptor half-life (from 100 ± 12 to 35 ± 1 min) following NGF treatment. These data indicate that receptor maturation is not affected by NGF, whereas the duration of gp140TrkA on the cell surface and the time needed for targeting of this protein to its sites of degradation are significantly decreased in the presence of NGF.
NGF Treatment Induces Modifications of the Cell Surface and Total gp140TrkA Cellular Pools
In PC12 cells, Western blot analyses of total cellular TrkA protein indicate that the ratio of gp140TrkA to gp110TrkA is decreased during NGF treatment (Fig. 3A). This is the result of a decrease in the cellular content of gp140TrkA, which is detectable within 30 min of incubation with NGF. Less than 10% of the initial level of this protein is detected after 5 h of growth factor treatment, whereas cellular pools of gp110TrkA and p75NTR are not affected (as ascertained from quantitation derived from Western blot data). Biotinylation experiments indicate that gp140TrkA is cleared from the cell surface within 5 min of NGF treatment, whereas the cell surface level of p75NTR remains constant (Fig. 3B).
Fig. 3. Effect of NGF on gp110TrkA and gp140TrkA pools.
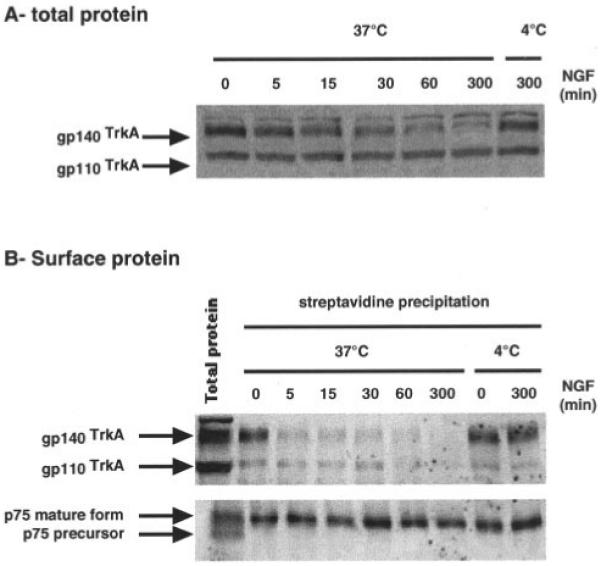
NGF kinetics (50 ng/ml) were performed in PC12 cells. At the end of NGF treatment, cells were submitted to cell surface biotinylation prior to protein extraction. Western blot analyses were then performed on total protein samples (A) or on cell surface proteins obtained by streptavidin precipitation (B). TrkA was probed with the RTA antibody, whereas p75NTR was detected using an antibody directed against the p75NTR carboxyl terminus.
gp140TrkA Is Degraded in Lysosomes
The presence of NGF does not appear to result in a decrease in TrkA synthesis, because total cellular gp110TrkA is not altered by growth factor treatment (Fig. 3A). Moreover, receptor maturation is not affected by NGF treatment as assessed by pulse-chase experiments (Fig. 1A). The gp140TrkA down-regulation observed in Fig. 3A is therefore most likely the result of enhanced receptor degradation rather than a decrease in the production of this protein. NGF-induced down-regulation of total gp140TrkA has been analyzed in the presence of two inhibitors of lysosomal enzymes. Data presented in Fig. 4 show that the two drugs tested lead to a decrease in receptor degradation with NH4Cl being the most efficient inhibitor. Treatment with NH4Cl also inhibits EGF-induced degradation of the EGF receptor. These results suggest that NGF-induced degradation of gp140TrkA occurs in lysosomes.
Fig. 4. Ligand-induced receptor degradation.
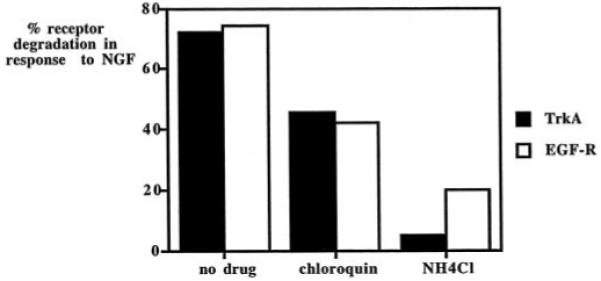
PC12 cells were incubated for 4 h with or without NGF or EGF at 50 ng/ml together with the drugs chloroquin (20 μm) or NH4Cl (10 mm). After treatment, proteins were extracted and analyzed by Western blot with the RTA antibody or the anti-EGF-R antibody. The graph presents the percentage of mature receptor degradation between growth factor-treated and -untreated cells calculated from the quantification of Western blot data obtained from two independent experiments.
A modified cell surface biotinylation experiment allows the monitoring of the time course of gp140TrkA targeting to the lysosome. In the experiment depicted in Fig. 5, cell surface biotinylation was performed prior to NGF treatment. The fates of biotinylated proteins were then followed upon additional incubation with or without NGF. An analysis of biotinylated protein shows that the cell surface gp140TrkA pool labeled at the beginning of the experiment begins to be degraded in the lysosome within 1 h of culture. The cell surface gp140TrkA pool half-life can be estimated from this type of experiment, indicating that the speed of receptor movement from the cell surface to the lysosome is increased 2-3-fold by the presence of NGF.
Fig. 5. Time course of gp140TrkA targeting to lysosomes.
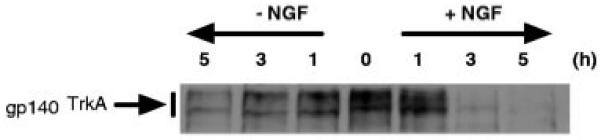
PC12 6-24 cells were collected, and cell surface biotinylation was performed. After cell surface protein labeling, cells were incubated at 37 °C for the indicated period of time with or without 50 ng/ml NGF. At the end of the incubation, proteins were extracted and subjected to streptavidin precipitation to recover biotinylated protein. Western blot analysis was then performed using RTA antibody to detect TrkA. The experiment was repeated three times.
NGF Binding to p75NTR Modulates TrkA Internalization
To monitor the cell surface levels of a receptor more easily, flow cytometric analysis was used to detect cell surface TrkA. This approach offers sensitivity comparable with that of cell surface biotinylation to detect TrkA clearing from the plasma membrane (data not shown). Using both of these techniques, we have analyzed NGF-induced TrkA internalization under conditions wherein NGF binding to p75NTR has been altered.
Modification of NGF binding to p75NTR was first obtained by incubating PC12 cells in the presence of the MC192 monoclonal antibody that is specific for the extracellular domain of p75NTR. This antibody increases the affinity of p75NTR for NGF by ∼2.5-fold (36). gp140TrkA internalization was evaluated by flow cytometry using concentrations of 1-50 ng/ml NGF. Fig. 6A shows that the use of this anti-p75NTR antibody almost completely abolishes NGF-induced TrkA internalization.
Fig. 6. Inhibition of NGF induced gp140TrkA internalization with anti-p75NTR antibodies.

A, PC12 cells were incubated prior to (30 min) and during NGF treatment with 8 μg/ml of the antibody MC192 directed against the p75NTR extracellular domain. NGF treatment was carried out for 30 min with concentrations ([NGF]) ranging from 0 to 50 ng/ml. At the end of the incubation time, the TrkA cell surface level was evaluated by flow cytometric analysis (see “Materials and Methods” for details). Values are means ± S.E. of four independent experiments. B, PC12 cells were incubated prior to (30 min) and during NGF treatment with a 1:100 dilution of REX serum directed against the extracellular domain of p75NTR. NGF treatment was carried out for 30 min with concentrations ([NGF]) ranging from 0 to 50 ng/ml. At the end of the incubation time, cell surface level of TrkA was evaluated by cell surface biotinylation. Values are means ± S.E. of three independent experiments.
The REX anti-p75NTR antibody inhibits binding of NGF through direct competition (32, 37). In the presence of REX, the internalization of TrkA, which is monitored by biotinylation of the cell surface receptor, is inhibited at 5 ng/ml NGF, whereas it is unaffected at 50 ng/ml (Fig. 6B).
Use of 3NGF, a mutated form of NGF with a reduced affinity for p75NTR, extends the observations made with the two anti-bodies. Results depicted in Fig. 7 using flow cytometric analysis show that a small decrease in the internalization efficiency was detectable when low levels of mutant 3NGF were used as compared with wild type growth factor (1 or 5 ng/ml). By contrast, at 50 ng/ml there was no significant difference between the wild type and the mutant neurotrophin. Comparable results were observed using biotinylation of the cell surface receptor (results not shown).
Fig. 7. 3NGF is less efficient than wild type NGF in promoting gp140TrkA internalization.
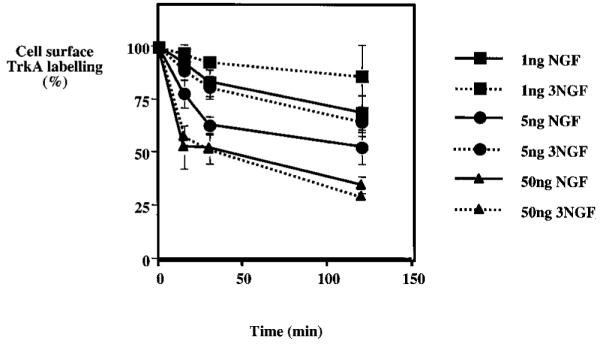
PC12 cells were treated for the indicated periods of time with either wild type NGF (NGF) or a mutant form of NGF (3NGF) with reduced affinity for p75NTR. Average cell surface TrkA was quantitated by flow cytometry as described under “Materials and Methods.” Each value is the mean ± S.E. of four independent experiments. Internalization in the presence of 1 and 5 ng/ml NGF differed significantly from internalization in the presence of the same concentrations of the mutant NGF (p < 0.01 by paired t test).
TrkA Activation through the Endocytotic Pathway
A large increase in total phosphorylated TrkA was detected via Western blot within 15 min of NGF treatment (Fig. 8A, gels at top). The subsequent decrease in phosphorylation of TrkA followed the decrease of total TrkA resulting from prolonged exposure to growth factor, as described above. By contrast, NGF treatment led only to a slight increase in cell surface phosphorylated TrkA as compared with the total phospho-TrkA in the cell (Fig. 8A). Cell surface phosphorylated TrkA appeared to be maintained at a constant low level from 15 to 300 min after NGF treatment. Such a disparity between the cell surface and the total phosphorylated receptor suggests that the bulk of the activated receptor is localized intracellularly.
Fig. 8. Localization of activated TrkA receptor.
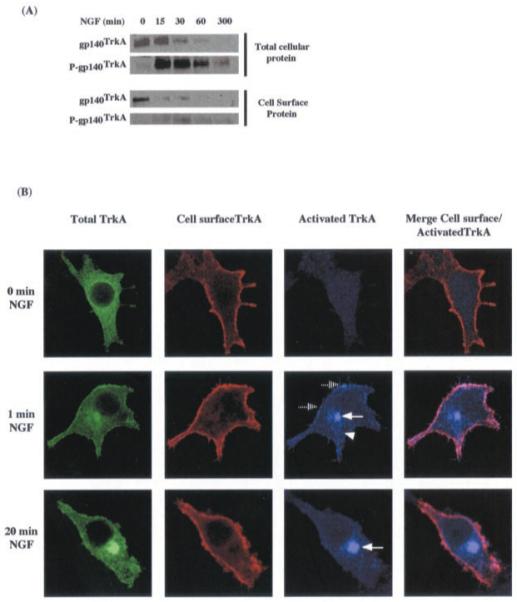
A, total cellular gp140TrkA and total phosphorylated gp140TrkA were detected by Western blot analysis performed on a total protein sample or on an anti-phosphotyrosine precipitated sample and revealed with the RTA antibody. Cell surface gp140TrkA was prepared as described under “Material and Methods.” Cell surface gp140TrkA and cell surface-phosphorylated gp140TrkA were analyzed by Western blot with RTA and anti-phosphotyrosine antibody respectively. B, triple labeling of total TrkA (green), cell surface TrkA (red), and phospho-TrkA (blue) in PC12 nnr5 cells transiently transfected with TrkA-EGFP vector and treated with NGF (50 ng/ml) for different periods of time. Co-localization of RTA and phospho-TrkA yields a pink color.
The precise intracellular localization of the activated receptor was monitored using confocal microscopy (Fig. 8B). Transfection of the chimeric TrkA-EGFP receptor in PC12 nnr5 cells allows simultaneous monitoring of three different pools of cellular TrkA in a single cell.2 (i) Total TrkA is reflected in GFP fluorescence, which corresponds to all of the cellular localizations of TrkA. (ii) Cell surface TrkA was identified by labeling unpermeabilized cells with the RTA antibody. (iii) Activated TrkA corresponds to immunolabeling of permeabilized cells using the anti-phospho-TrkA-specific antibody.
A phospho-TrkA signal was detected only in cells treated with NGF (Fig. 8B). Phospho-TrkA was located at the cell surface (dashed arrows) and in a perinuclear compartment (solid arrows). From 1 to 20 min after NGF treatment, activated TrkA at the cell surface decreased with a concomitant enhancement in the signal of perinuclear activated TrkA. After 1 min of treatment, activated TrkA is also observed in numerous cytoplasmic vesicles (triangle). It can also be noted that, even in cells treated for less than 1 min with NGF, a strong phospho-TrkA labeling is observed in the perinuclear compartment. This intracellular location probably corresponds to an endosomal compartment, because it appears to co-localize with the endosomal marker Tnf-R.2 These data corroborate the conclusions drawn from Western blot analyses and point to a central role for the endosome in TrkA signaling.
DISCUSSION
The expression, maturation, and fate of the NGF receptors TrkA and p75NTR were studied in an attempt to get a better understanding of the regulation of their trafficking. This study presents evidence for a very specific effect of NGF on TrkA trafficking that appears to be limited to a decrease in the transit time of the mature receptor on the cell surface. NGF treatment has only a minor impact on the synthesis and maturation of the TrkA receptor. The half-life for receptor degradation is essentially unmodified by this treatment. The only parameter affected is the cell surface transit time, which is significantly decreased in the presence of NGF. Analysis of the subcellular localization of the activated receptor suggests that signaling occurs in endosomes. Finally, initiation of TrkA internalization from the cell surface by NGF appears to depend on growth factor binding to the neurotrophin receptor p75NTR.
TrkA Maturation
Metabolic studies allow estimation of the kinetics of TrkA processing and stability. Maturation of the receptor along the biosynthetic pathway leads to a mature receptor with a half-life of about 24 min. This value is similar to those described for the maturation of other receptors such as the PDGF receptor and the neurotrophin receptor p75NTR (38, 39). The half-lives of mature receptors are much more diverse than those of precursor proteins. For example, the EGF receptor and p75NTR exhibit half-lives of >10 h in their mature forms (39, 40). By contrast, the PDGF receptor, the Neu receptor, the Epo receptor, and c-ret have half-lives of 2-3 h (38,41-44). TrkA has a similar half-life of 2 h and 20 min. These relatively short receptor half-lives may permit dynamic modulation of receptor levels. Under these conditions, even slight modifications in either the rate of synthesis or degradation of a receptor will lead to rapid changes in the amounts of the receptor. This is in contrast with TrkA for which the overall half-life is only shortened by 1.6-fold. The half-lives of Neu and the PDGF and EGF receptors have been reported to be shortened 4-6-fold in the presence of their corresponding growth factors (40, 42, 45). The disparity between the half-lives of the mature forms of TrkA and p75NTR may also account for the difference in cellular distribution of these two receptors at the steady state. Indeed, immunofluorescence studies in PC12 cells indicate that p75NTR is mostly present at the cell surface, whereas TrkA is mainly localized intracellularly (14). This may reflect the need for a continuous production of the TrkA precursor in the biosynthetic pathway to maintain a constant level of the mature receptor at the cell surface.
Tunicamycin treatment shows that TrkA transport to the surface requires N-glycosylation of the receptor. Studies by Watson et al. (46) indicate that 9 of the 13 potential N-glycosylation sites are used in human TrkA. The 30-kDa reduction in the molecular mass of the TrkA precursor in tunicamycin-treated cells may be explained by the removal of N-linked sugars of the native or partially matured forms (3 and 6 kDa, respectively) from these nine locations on the TrkA precursor (27). Nevertheless, it can not be excluded that additional co-translational maturation steps may account for some modifications of the TrkA core protein. Pulse-chase analysis and cell surface biotinylation clearly show that N-glycosylation is required for receptor targeting to the cell surface. This observation may explain the loss of NGF-induced differentiation of PC12 cells treated with tunicamycin. The inability of the receptor to reach the cell surface in the absence of N-glycosylation can be the result of receptor misfolding and endoplasmic reticulum retention (47, 48). However, the disruption of N-glycosylation is not a general requirement for the targeting of N-glycosylated receptors to the cell surface, because p75NTR is still targeted to the plasma membrane in the absence of its N- and O-linked sugar moieties (49).
NGF treatment seems to differentially affect the turnover rates of the different forms of TrkA. Maturation of TrkA from the gp110 to the gp140 form is only slightly increased in the presence of NGF (17% reduction of immature receptor half-life). By contrast, the stability of total and cell surface gp140TrkA is more strongly affected because these receptor populations show decreased half-lives of 40 and 60%, respectively. The difference between total receptor half-life and cell surface receptor half-life is comparable in NGF-treated and -untreated cells (40-50 min). The half-life of receptor trafficking from the cell surface to lysosomes can therefore be extrapolated from this data to be 40-50 min (i.e. the difference indicated above). This suggests that TrkA trafficking from the cell surface to the lysosome is not affected by NGF treatment. The main effect of NGF treatment is, therefore, to decrease the time the receptor resides at the cell surface.
Kinetics of Surface Expression and Degradation
Cell surface biotinylation followed by a cell culture in the absence or presence of a ligand indicates a time course of receptor degradation comparable with the previously mentioned value for lysosomal degradation. Indeed, degradation of the cell surface receptor pool was detectable within 1 h after receptor biotinylation at the cell surface. The half-lives of Neu and the PDGF- and EGF receptors have been reported to be shortened 4-6-fold in the presence of their corresponding growth factors (40, 42, 45).
The fate of endogenous TrkA in PC12 cells was also followed. gp140TrkA is the only form of TrkA readily detectable at the PC12 cell surface. Treatment by NGF produces clearing of more than 70% of the receptor from the cell surface within 15 min of treatment. This value correlates well with the observation of Grimes et al. (16), who have shown that 66% of the receptor initially labeled at the PC12 cell surface is found inside the cells after 20 min of NGF treatment. By contrast, the extent of TrkA internalization appears to be much higher in PC12 cells than in cell lines overexpressing the receptor; cell surface TrkA levels are reduced by 20% in the presence of NGF in a B104 neuroblastoma cell having equal numbers of TrkA and p75NTR (50). This probably reflects saturation of the trafficking pathway by high levels of the receptor. The present study indicates that cell surface TrkA is maintained at a low level even after prolonged NGF exposure (up to 1 day). Analysis of the total cellular pool of TrkA during the same time-course of NGF treatment shows a decrease in total cellular gp140TrkA preceded by a decrease of mature TrkA at the cell surface. By contrast, gp110TrkA levels remain constant.
Taken together, these data suggest that NGF does not affect the rate of receptor synthesis during this time because precursor levels remain constant. Rather, the presence of growth factor modifies receptor trafficking from the cell surface, probably by shortening the time of receptor residence at the plasma membrane as shown by pulse-chase analysis. The decrease in the amount of total gp140TrkA observed during NGF treatment is therefore most probably the result of an increase in the amount of receptor internalization and subsequent degradation in the lysosome. Experiments presented herein show that the inhibition of lysosomal enzymes provokes a decrease of NGF-induced TrkA degradation to the same extent as that observed for EGF-induced EGF receptor degradation. It is well documented that the EGF receptor is degraded in lysosomes (51). This study indicates that TrkA degradation most probably occurs in the same compartment.
Phorbol 12-myristate 13-acetate (PMA) treatment has been shown to induce cleavage of the TrkA extracellular domain at the plasma membrane in Chinese hamster ovary cells transfected with the receptor (52, 53). In this cell line, NGF also provokes a cleavage but to a lesser extent than that observed with PMA. Therefore, this phenomenon is probably not involved in the massive degradation of gp140TrkA induced by NGF observed in the present study. In cerebellar neurons it has been shown that BDNF induces degradation of TrkB, whereas no NGF-induced TrkA degradation was detected (54). This suggests that, within the highly conserved Trk receptor family, differential regulation of receptor trafficking may occur.
Kinetics of Activated TrkA Cellular Localization
Signaling by growth factor receptors is believed to be affected by the duration of ligand-induced activation and also by the location of activated receptors (55-57). The present study suggests that signaling by TrkA from the cell surface is brief, because the receptor is cleared rapidly from the cell surface after the addition of NGF. It also indicates that trafficking from the cell surface to the site of degradation occurs with a half-life of ∼40-50 min. During this transition, the receptor travels through endosomes in its activated state. These data are in agreement with the observation of activated TrkA in the endosomal fraction of PC12 cells and within the axonal structure (16, 20, 58, 59).
Expression and Role of p75NTR in NGF Internalization
Contrasting results of the p75NTR-mediated internalization of NGF have been reported. Depending on the cell type, p75NTR may or may not promote internalization of NGF. In Madin-Darby canine kidney (MDCK) cells, the expression of wild type p75NTR does not permit internalization, whereas a truncated form of p75NTR lacking its cytoplasmic domain is able to produce NGF-internalization (39). In PC12 cells, numerous studies indicate that p75NTR cannot internalize NGF (5, 60, 61). The observation that exposure to NGF does not modify cell surface levels of p75NTR agrees with the conclusion that this receptor does not take part in the actual physical internalization of NGF in PC12 cells. Other possible explanations are that the amount internalized is very low compared with the total surface p75NTR or that recycling is very efficient and, therefore, the amounts are below the levels of detection in the assays used. The results presented herein do suggest, however, a role for p75NTR in the internalization of TrkA either via the putative NGF-concentrating function of p75NTR (62) or via a direct interaction of p75NTR with TrkA (63).
Two antibodies directed toward the extracellular domain of p75NTR, REX (32) and MC192 (36), were used in these studies. The association of MC192 to p75NTR leads to a 2.5-fold increase of NGF binding to this receptor, whereas REX inhibits NGF binding. REX only inhibits NGF-induced TrkA internalization at low growth factor concentrations. These results offer support to the NGF-concentrating role of p75NTR in the internalization of TrkA. The availability of a mutant NGF with decreased affinity for p75NTR made it possible to further test the proposed NGF-concentrating function of p75NTR in this process (33). Data presented herein show that the reduced ability of the mutated NGF to promote TrkA internalization is also only detectable at low neurotrophin concentrations. Together with the observations obtained with REX, these data indicate that p75NTR is required for NGF-induced TrkA internalization in the low range of neurotrophin concentrations.
The results obtained with MC192 suggest that p75NTR has a role in addition to that of concentrating NGF at low growth factor concentrations. MC192 inhibits TrkA internalization even at high NGF concentrations, i.e. under conditions where NGF is not limiting and where both the low and high affinity binding sites are fully saturated. Binding of MC192 may modify the interaction between p75NTR and TrkA, increasing the affinity of individual members of the complex and thereby retaining TrkA at the cell surface. In this regard, the analyses of cell surface receptor levels using flow cytometry or biotinylation consistently reveal a slight but reproducible increase (10%) in the amount of TrkA at the cell surface after exposure to MC192 in the absence of NGF. Ross and co-workers (25, 26) have shown that the co-expression of p75NTR with TrkA leads to a decreased mobility of the latter receptor at the cell surface, forming patches and suggesting that p75NTR may be involved in stabilization of TrkA receptor in the plasma membrane.
The growing number of studies describing a physical association between these two receptors supports the possibility that the formation of high affinity NGF binding sites may be mediated by the direct interaction of p75NTR and TrkA (37, 63). It may be hypothesized that p75NTR maintains TrkA in an internalization-competent state at the plasma membrane. MC192 switches this to an internalization-incompetent state perhaps by increasing the interaction between these two receptors. Interestingly, it has been shown that the inhibition of TrkA internalization with a mutant dynamin disrupts NGF-induced differentiation without affecting the survival effect of this growth factor (17). It has been reported that MC192 impaired TrkA-mediated cell differentiation, whereas it enhanced TrkA-mediated cell survival (36, 64). The differential effect of MC192 on these two NGF-mediated responses (50, 65) may also be the result of the impairment in TrkA internalization reported herein.
The impact of NGF on TrkA trafficking parameters is summarized in Fig. 9A. The major effect of NGF is to shorten the time of residence of TrkA at the plasma membrane. An observation of receptor pools suggests that signaling by the TrkA-NGF complex takes place essentially intracellularly during receptor transfer to the lysosome. Upon primary NGF treatment, two phases of receptor signaling can be distinguished (Fig. 9B). An initial phase of signaling is supported by recruitment of the bulk of TrkA receptors located at the cell surface before growth factor treatment. Modification of receptor trafficking properties by NGF then leads to a new equilibrium with fewer receptors located at the cell surface. After this first transition step, the number of TrkA receptors available for NGF binding at the cell surface is reduced. Thus, the second phase of signaling is comprised of a continuous flux of receptor to and from the cell surface, thereby maintaining a low level of receptors at this location. The time required to switch between these two equilibrium phases is believed to be the inverse of the sum of the equilibrium constants for the translocation to and from the cell surface (66). Thus, the relative shortness of receptor half-life may permit rapid accession to conditions of a new equilibrium.
Fig. 9. NGF-induced modification of kinetic parameters of TrkA trafficking.
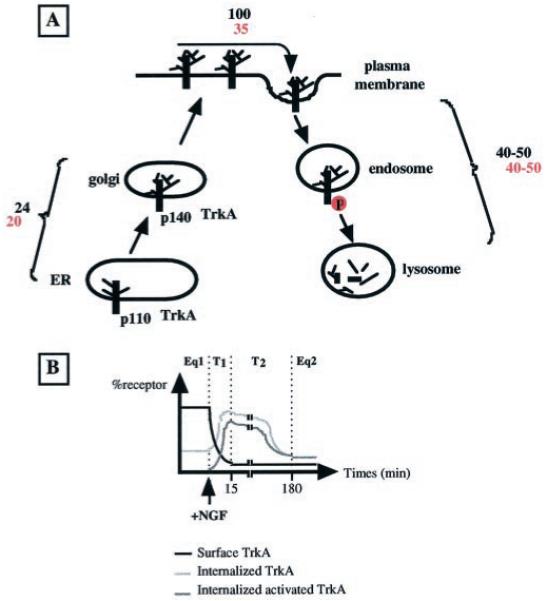
A, schematic representation of the different TrkA locations in PC12 cells. Half-lives (min) of receptor movements between compartments are indicated (values in red and black are with and without NGF, respectively). B, the cell surface level of TrkA receptor is the result of an equilibrium between movements of the receptor to and from the cell surface. Equilibrium constant values are different in the absence or presence of NGF. Upon the modification of kinetic constants by NGF, there is a two-phase transition (T1 and T2) to reach a new equilibrium. The T1 transition step corresponds to the establishment of a new equilibrium at the cell surface. During this phase, the bulk of the TrkA receptor located at the cell surface before growth factor treatment is internalized. The T2 phase corresponds to lysosomal targeting of this initial wave of internalized receptor. The differences between the E1 (Eq1) and E2 (Eq2) states depend on the decrease in the overall quantity of cellular receptor and the inversion of the ratio of cell surface to the internalized receptor (2 :0.7).
In conclusion, these studies open the way for identification of molecules that play a role in the specific processes regulating cell surface expression of neurotrophic factor receptors, an essential aspect of their signaling process.
Acknowledgments
We are extremely grateful to Dr. F. X. Real for advice concerning metabolic labeling experiments. We thank Dr. M. Robinson-Rechalvi for his help in the statistical analysis and Dr. P. Colas for helpful comments on the manuscript. We thank Dr. Carlos Ibanez for the generous gift of mutant NGF and Dr. Ian Towbridge for the anti-transferrin receptor antibody.
Footnotes
This work was supported by grants from the Ligue Nationale Contre le Cancer, the committee of the Ligue from the Rhône, the Rhône-Alpes Region, the Association for Research against Cancer (ARC), and the Fondation de France.
- NGF
- nerve growth factor
- BDNF
- brain-derived neurotrophic factor
- NT-3
- neurotrophin-3
- NTR
- neotrophin receptor
- RTA
- anti-TrkA extracellular domain
- REX
- anti-p75NTR extracellular domain
- WGA
- wheat germ agglutinin
- PBS
- phosphate-buffered saline
- EGFP
- enhanced green fluorescent protein
- EGF
- epidermal growth factor
- PDGF
- platelet-derived growth factor
REFERENCES
- 1.Patapoutian A, Reichardt LF. Curr. Opin. Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- 2.Chao MV. J. Neurosci. Res. 2000;59:353–355. [PubMed] [Google Scholar]
- 3.Patel TD, Jackman A, Rice FL, Kucera J, Snider WD, Frade JM, Barde YA. Neuron. 2000;25:345–357. doi: 10.1016/s0896-6273(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 4.Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 5.Kahle P, Barker PA, Shooter EM, Hertel C. J. Neurosci. Res. 1994;38:599–606. doi: 10.1002/jnr.490380512. [DOI] [PubMed] [Google Scholar]
- 6.Greene LA, Tischler AS. Proc. Natl. Acad. Sci. U. S. A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman WJ, Greene LA. Exp. Cell Res. 1999;253:131–142. doi: 10.1006/excr.1999.4705. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan DR, Martin-Zanca D, Parada LF. Nature. 1991;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 9.Soltoff SP, Rabin SL, Cantley LC, Kaplan DR. J. Biol. Chem. 1992;267:17472–17477. [PubMed] [Google Scholar]
- 10.Vetter ML, Martin-Zanca D, Parada LF, Bishop JM, Kaplan DR. Proc. Natl. Acad. Sci. U. S. A. 1991;88:5650–5654. doi: 10.1073/pnas.88.13.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wood KW, Sarnecki C, Roberts TM, Blenis J. Cell. 1992;68:1041–1050. doi: 10.1016/0092-8674(92)90076-o. [DOI] [PubMed] [Google Scholar]
- 12.Rudkin BB, Lazarovici P, Levi B-Z, Abe Y, Fujita K, Guroff G. EMBO J. 1989;8:3319–3325. doi: 10.1002/j.1460-2075.1989.tb08493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Grunsven LA, Thomas A, Urdiales JL, Machenaud S, Choler P, Durand I, Rudkin BB. Oncogene. 1996;12:855–862. [PubMed] [Google Scholar]
- 14.Urdiales JL, Becker E, Andrieu M, Thomas A, Jullien J, van Grunsven LA, Menut S, Evan GI, Martin-Zanca D, Rudkin BB. J. Neurosci. 1998;18:6767–6775. doi: 10.1523/JNEUROSCI.18-17-06767.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang CS, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, Mobley WC. J. Biol. Chem. 1999;274:36707–36714. doi: 10.1074/jbc.274.51.36707. [DOI] [PubMed] [Google Scholar]
- 16.Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. J. Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Moheban DB, Conway BR, Bhattacharyya A, Segal RA, Grimes ML, Beattie E, Mobley WC. J. Neurosci. 2000;20:5671–5678. doi: 10.1523/JNEUROSCI.20-15-05671.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ, Ure DR, Campenot RB, Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Mol. Cell. Biol. 2000;20:8069–8083. doi: 10.1128/mcb.20.21.8069-8083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reynolds AJ, Bartlett SE, Hendry IA, Grimes ML, Beattie E, Mobley WC. Brain Res. Brain Res. Rev. 2000;33:169–178. doi: 10.1016/s0165-0173(00)00028-x. [DOI] [PubMed] [Google Scholar]
- 20.Grimes ML, Beattie E, Mobley WC. Proc. Natl. Acad. Sci. U. S. A. 1997;94:9909–9914. doi: 10.1073/pnas.94.18.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ure DR, Campenot RB, Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. J. Neurosci. 1997;17:1282–1290. [Google Scholar]
- 22.Senger DL, Campenot RB. J. Cell Biol. 1997;138:411–421. doi: 10.1083/jcb.138.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Zanca D, Oskam R, Mitra G, Copeland T, Barbacid M. Mol. Cell. Biol. 1989;9:24–33. doi: 10.1128/mcb.9.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Valletta JS, Grimes ML, Mobley WC. J. Neurochem. 1995;65:1146–1156. doi: 10.1046/j.1471-4159.1995.65031146.x. [DOI] [PubMed] [Google Scholar]
- 25.Ross AH, Daou MC, McKinnon CA, Condon PJ, Lachyankar MB, Stephens RM, Kaplan DR, Wolf DE. J. Cell Biol. 1996;132:945–953. doi: 10.1083/jcb.132.5.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf DE, McKinnon-Thompson C, Daou MC, Stephens RM, Kaplan DR, Ross AH. Biochemistry. 1998;37:3178–3186. doi: 10.1021/bi9719253. [DOI] [PubMed] [Google Scholar]
- 27.Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 28.Hanisch FG. Biol. Chem. 2001;382:143–149. doi: 10.1515/BC.2001.022. [DOI] [PubMed] [Google Scholar]
- 29.Keller P, Simons K. J. Cell Sci. 1997;110:3001–3009. doi: 10.1242/jcs.110.24.3001. [DOI] [PubMed] [Google Scholar]
- 30.Rohn WM, Rouille Y, Waguri S, Hoflack B. J. Cell Sci. 2000;113:2093–2101. doi: 10.1242/jcs.113.12.2093. [DOI] [PubMed] [Google Scholar]
- 31.Reilly JF, Maher PA. J. Cell Biol. 2001;152:1307–1312. doi: 10.1083/jcb.152.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clary DO, Weskamp G, Austin LR, Reichardt LF. Mol. Biol. Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ibanez CF, Ebendal T, Barbany G, Murray-Rust J, Blundell TL, Persson H. Cell. 1992;69:329–341. doi: 10.1016/0092-8674(92)90413-7. [DOI] [PubMed] [Google Scholar]
- 34.van Grunsven LA, Billon N, Savatier P, Thomas A, Urdiales JL, Rudkin BB. Oncogene. 1996;12:1347–1356. [PubMed] [Google Scholar]
- 35.Arenas MI, Royuela M, Fraile B, Paniagua R, Wilhelm B, Aumuller G. J. Androl. 2001;22:79–87. [PubMed] [Google Scholar]
- 36.Barker PA, Shooter EM. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- 37.Mischel PS, Smith SG, Vining ER, Valletta JS, Mobley WC, Reichardt LF. J. Biol. Chem. 2001;276:11294–11301. doi: 10.1074/jbc.M005132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keating MT, Williams LT. J. Biol. Chem. 1987;262:7932–7937. [PubMed] [Google Scholar]
- 39.Le Bivic A, Sambuy Y, Patzak A, Patil N, Chao M, Rodriguez-Boulan E. J. Cell Biol. 1991;115:607–618. doi: 10.1083/jcb.115.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ekstrand AJ, Liu L, He J, Hamid ML, Longo N, Collins VP, James CD. Oncogene. 1995;10:1455–1460. [PubMed] [Google Scholar]
- 41.Daniel TO, Milfay DF, Escobedo J, Williams LT. J. Biol. Chem. 1987;262:9778–9784. [PubMed] [Google Scholar]
- 42.Huang SS, Koh HA, Konish Y, Bullock LD, Huang JS. J. Biol. Chem. 1990;265:3340–3346. [PubMed] [Google Scholar]
- 43.Supino-Rosin L, Yoshimura A, Altaratz H, Neumann D. Eur. J. Biochem. 1999;263:410–419. doi: 10.1046/j.1432-1327.1999.00516.x. [DOI] [PubMed] [Google Scholar]
- 44.van Weering DH, Moen TC, Braakman I, Baas PD, Bos JL. J. Biol. Chem. 1998;273:12077–12081. doi: 10.1074/jbc.273.20.12077. [DOI] [PubMed] [Google Scholar]
- 45.Bejcek BE, Voravud N, Deuel TF. Biochem. Biophys. Res. Commun. 1993;196:69–78. doi: 10.1006/bbrc.1993.2217. [DOI] [PubMed] [Google Scholar]
- 46.Watson FL, Porcionatto MA, Bhattacharyya A, Stiles CD, Segal RA. J. Neurobiol. 1999;39:323–336. doi: 10.1002/(sici)1097-4695(199905)39:2<323::aid-neu15>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 47.Ellgaard L, Molinari M, Helenius A. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- 48.Parodi AJ. Biochem. J. 2000;348:1–13. [PMC free article] [PubMed] [Google Scholar]
- 49.Yeaman C, Le Gall AH, Baldwin AN, Monlauzeur L, Le Bivic A, Rodriguez-Boulan E. J. Cell Biol. 1997;139:929–940. doi: 10.1083/jcb.139.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saragovi HU, Zheng W, Maliartchouk S, DiGugliemo GM, Mawal YR, Kamen A, Woo SB, Cuello AC, Debeir T, Neet KE. J. Biol. Chem. 1998;273:34933–34940. doi: 10.1074/jbc.273.52.34933. [DOI] [PubMed] [Google Scholar]
- 51.Kil SJ, Hobert M, Carlin C. J. Biol. Chem. 1999;274:3141–3150. doi: 10.1074/jbc.274.5.3141. [DOI] [PubMed] [Google Scholar]
- 52.Cabrera N, Diaz-Rodriguez E, Becker E, Martin-Zanca D, Pandiella A. J. Cell Biol. 1996;132:427–436. doi: 10.1083/jcb.132.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diaz-Rodriguez E, Cabrera N, Esparis-Ogando A, Montero JC, Pandiella A. Eur. J. Neurosci. 1999;11:1421–1430. doi: 10.1046/j.1460-9568.1999.00552.x. [DOI] [PubMed] [Google Scholar]
- 54.Sommerfeld MT, Schweigreiter R, Barde YA, Hoppe E. J. Biol. Chem. 2000;275:8982–8990. doi: 10.1074/jbc.275.12.8982. [DOI] [PubMed] [Google Scholar]
- 55.Carpenter G. Bioessays. 2000;22:697–707. doi: 10.1002/1521-1878(200008)22:8<697::AID-BIES3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 56.Ceresa BP, Schmid SL. Curr. Opin. Cell Biol. 2000;12:204–210. doi: 10.1016/s0955-0674(99)00077-0. [DOI] [PubMed] [Google Scholar]
- 57.Wiley HS, Burke PM. Traffic. 2001;2:12–18. doi: 10.1034/j.1600-0854.2001.020103.x. [DOI] [PubMed] [Google Scholar]
- 58.Bhattacharyya A, Watson FL, Bradlee TA, Pomeroy SL, Stiles CD, Segal RA. J. Neurosci. 1997;17:7007–7016. doi: 10.1523/JNEUROSCI.17-18-07007.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. J. Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eveleth DD, Bradshaw RA. J. Cell Biol. 1992;117:291–299. doi: 10.1083/jcb.117.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gargano N, Levi A, Alema S. J. Neurosci. Res. 1997;50:1–12. doi: 10.1002/(SICI)1097-4547(19971001)50:1<1::AID-JNR1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 62.Mahadeo D, Kaplan L, Chao MV, Hempstead BL. J. Biol. Chem. 1994;269:6884–6891. [PubMed] [Google Scholar]
- 63.Esposito D, Patel P, Stephens RM, Perez P, Chao MV, Kaplan DR, Hempstead BL. J. Biol. Chem. 2001;276:32687–32695. doi: 10.1074/jbc.M011674200. [DOI] [PubMed] [Google Scholar]
- 64.Kimpinski K, Jelinski S, Mearow K. Neuroscience. 1999;93:253–263. doi: 10.1016/s0306-4522(99)00156-6. [DOI] [PubMed] [Google Scholar]
- 65.Maliartchouk S, Saragovi HU. J. Neurosci. 1997;17:6031–6037. doi: 10.1523/JNEUROSCI.17-16-06031.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burrone J, Murthy VN. Curr. Biol. 2001;11:R274–R277. doi: 10.1016/s0960-9822(01)00137-3. [DOI] [PubMed] [Google Scholar]