

Abstract
The TrkA receptor is activated primarily by nerve growth factor (NGF), but it can also be activated by high concentrations of neurotrophin 3 (NT-3). The pan-neurotrophin receptor p75NTR strongly inhibits activation of TrkA by NT-3 but not by NGF. To examine the role of p75NTR in regulating the specificity of TrkA signaling, we expressed both receptors in Xenopus oocytes. Application of NGF or NT-3 to oocytes expressing TrkA alone resulted in efflux of 45Ca2+ by a phospholipase C-γ-dependent pathway. Coexpression of p75NTR with TrkA inhibited 45Ca2+ efflux in response to NT-3 but not NGF. The inhibitory effect on NT-3 activation of TrkA increased with increasing expression of p75NTR. Coexpression of a truncated p75NTR receptor lacking all but the first 9 amino acids of the cytoplasmic domain inhibited NT-3 stimulation of 45Ca2+ efflux, whereas coexpression of an epidermal growth factor receptor/p75NTR chimera (extracellular domain of epidermal growth factor receptor with transmembrane and cytoplasmic domains of p75NTR) did not inhibit NT-3 signaling through TrkA. These studies demonstrated that the extracellular domain of p75NTR was necessary to inhibit NT-3 signaling through TrkA. Remarkably, p75NTR binding to NT-3 was not required to prevent signaling through TrkA, since occupying p75NTR with brain-derived neurotrophic factor or anti-p75 antibody (REX) did not rescue the ability of NT-3 to activate 45Ca2+ efflux. These data suggested a physical association between TrkA and p75NTR. Documenting this physical interaction, we showed that p75NTR and TrkA could be coimmunoprecipitated from Xenopus oocytes. Our results suggest that the interaction of these two receptors on the cell surface mediated the inhibition of NT-3-activated signaling through TrkA.
Nerve growth factor (NGF),1 brain-derived neurotrophic factor (BDNF), neurotrophin-4 (NT4), and neurotrophin-3 (NT-3) initiate their actions by binding to Trk receptors and p75NTR (1). NGF binds to TrkA; BDNF and NT-4 bind to TrkB, and NT-3 binds to TrkC. NT-3 also binds TrkA (and TrkB), although with significantly lower affinity (1), and can activate TrkA signaling and cellular responses (2-7). In vivo, NT-3 signaling through other Trk receptors is required for nervous system development, since NT3-/- mice exhibit significantly greater neuronal loss than TrkC-/- mice (8). Recently, NT-3 has been shown to be required for the survival of TrkA-expressing postmitotic sympathetic neurons (9) and sensory neurons (10) in vivo.
p75NTR also binds each of the neurotrophins (11) and can signal independently of TrkA in response to neurotrophin stimulation (12-14). In addition, p75NTR regulates Trk receptor signaling. p75NTR potentiates NGF activation of TrkA at low ligand concentrations (15, 16) and collaborates with TrkA to form high affinity binding sites (17-19). Furthermore, the signaling pathways initiated by p75NTR block Trk receptor signaling in certain contexts (20, 21) and synergize with Trk receptor signaling in other contexts (22). These opposing actions suggest a complex interaction between p75NTR and Trk receptors in regulating neurotrophin signaling.
NT-3 does not activate TrkA signaling in PC12 cells (4, 23), unless p75NTR levels are reduced (23), suggesting that p75NTR directly suppresses the ability of TrkA to respond to NT-3 (4). In support of this hypothesis, isolated sympathetic neurons from p75NTR-/- mice are more responsive to NT-3 in culture than are wild type sympathetic neurons (24). These data suggest that p75NTR can prevent NT-3 signaling through TrkA, but the molecular nature of this interaction remains to be characterized.
We used a Xenopus oocyte microinjection assay to study NGF and NT-3 signaling through TrkA and to assess the role of p75NTR in regulating this signaling. We showed that p75NTR prevented the signaling of NT-3 through TrkA but not that of NGF. We demonstrated that the extracellular domain of p75NTR, but not the cytoplasmic domain, was necessary to inhibit NT-3 signaling through TrkA. Furthermore, p75NTR binding to NT-3 was not required to inhibit NT-3 activation of TrkA. Finally, we showed that p75NTR and TrkA could be coimmunoprecipitated from Xenopus oocytes, suggesting that interaction of these receptors mediates the inhibition of NT-3 signaling through TrkA.
EXPERIMENTAL PROCEDURES
Growth Factors and Antibodies
Purified recombinant human neurotrophins were obtained from Amgen Inc. (Thousand Oaks, CA) and Genentech Corp. (South San Francisco, CA). Rabbit polyclonal anti-p75NTR antibody was raised against the extracellular domain of rat p75NTR (25). Rabbit polyclonal antibody raised against the cytoplasmic domain of p75NTR was purchased from Promega (Madison WI). Rabbit polyclonal antibody (26) was raised to the C terminus of human TrkA. M2 monoclonal anti-FLAG antibody was purchased from Stratagene (La Jolla, CA), and monoclonal anti-EGFR antibody (LA22) was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY).
cDNA Constructs and Plasmid Generation
cDNAs encoding the full length rat TrkA and rat p75NTR receptors were subcloned into PGEMHE vector, which contains 5′ and 3′ Xenopus beta-globin untranslated sequences for RNA stability (gift from Dr. E. R. Liman) (27). Truncated p75NTR receptor was generated from the rat p75NTR cDNA using PCR with forward primer 5′GAA TTC ATG AGG AGG GCA GGT GCT GCC and reverse primer 5′CAC GGG TCT AGA CTA GGC GCC TTG TTT ATT TTG TTT GCA GCT G to amplify the extracellular and transmembrane domains, and the first nine amino acids of the cytoplasmic domain, flanked by an 5′ EcoRI site and a 3′ XbaI site. The amplified product was subcloned into PGEMHE. All constructs used in these experiments were verified by sequencing.
A chimeric EGF receptor/p75NTR cDNA encoding the extracellular domain of the human EGF receptor with the transmembrane and cytoplasmic domains of human p75NTR (gift of Dr. Moses Chao) (28) was subcloned from pCMVEN10 into the PGEMHE vector. This was done using PCR with forward primer 5′ GCG CGC GTC GAC GCG ATG CGA CCC TCC GGG ACG GCC and reverse primer 5′ GCG CGC TCT AGA GGC TCA CAC CGG GGA TGA GGC AGT GG to amplify the chimeric EGFR/p75NTR construct flanked by a 5′ SalI site and a 3′ XbaI site. The amplified product was digested with SalI and XbaI and subcloned into PGEMHE.
The C-terminal FLAG-tagged TrkA receptor was generated by PCR using forward primer 5′CA GGG ACT AGT GGT CAA GAT GAT TGG A and reverse primer 5′ CTT ATC ATC ATC ATC CTT GTA ATC GCC CAG AAC GTC CAG G to amplify a fragment encoding the last 405 base pairs of rat TrkA receptor coding sequence (minus the stop codon) including the 5′ SpeI site (part of the coding sequence) and the first FLAG epitope on the 3′ end. This product was then used as a template for a second PCR using the same forward primer and a reverse primer encoding the second FLAG tag followed by a stop codon and an XhoI restriction site 5′CTG CTC GAG CTA CTT ATC ATC ATC ATC CTT GTA ATC CTT ATC ATC ATC. The amplified product was digested with SpeI and XhoI and subcloned into similarly digested full-length TrkA in PGEMHE.
In Vitro Transcription of cRNA and Expression in Xenopus Oocytes
DNA templates were linearized and transcribed in vitro using the T7 RNA polymerase promoter with mMessage in vitro transcription kit from Ambion (Austin, TX). Transcripts were purified using an RNAeasy kit purchased from Qiagen Inc. (Valencia, CA) and then analyzed on agarose gels and quantified by spectrophotometry (as well as by densitometric comparison of bands to known RNA standards).
Xenopus laevis and Preparation of Oocytes
Mature Xenopus oocytes (Dumont stage V-VI) were harvested and defolliculated enzymatically with 1-2 mg/ml collagenase (type I or II) for 2-3 h (Worthington). Oocytes were maintained at 17-18 °C in ND96 (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH 7.5) plus 1 μg/ml ciprofloxacin, 6 μg/ml ceftazidime, and 1 μg/ml gentamicin.
45Ca2+ Efflux Assays
For calcium efflux assays, oocytes were injected with a total of 0.2 ng of cRNA per oocyte. When cRNA encoding a single receptor was injected, 0.1 ng of GFP cRNA was coinjected to equalize the total amount of injected cRNA. Efflux experiments were conducted according to methods published previously (29, 30). In brief, 2 days after injection, oocytes were incubated in 45Ca2+ (from Amersham Pharmacia Biotech), 100 mCi/ml for 2.5 h in calcium-free ND96. Groups of 8 (or 4) oocytes were washed and transferred to 24-well dishes with 0.5 ml of ND96 per well. Medium was collected and replaced every 10 min, and radioactivity was measured by liquid scintillation counting. After stabilization of background 45Ca2+ efflux, 100 ng/ml either NT-3 or NGF was added to each group of oocytes. Data were collected from each of the experimental conditions by using 4-8 oocytes per condition (same number of oocytes in each experiment), with each condition run in triplicate for each experiment. The initial NT-3 dose-response curve (Fig. 2) was run in duplicate. Each experiment was repeated two or three times, using oocytes from two or three different frogs. Therefore, every data point for each condition in one experiment consisted of the average of three measurements obtained from two or three groups of four to eight oocytes. Signaling curves were graphed using Microsoft Excel, and mean and standard errors of measure were determined using Microsoft Excel. For analysis of dose-response curve, mean peak counts per min were compared using analysis of variance methods and allowing for unequal variances. The Tukey-Fisher criterion was used to compute post hoc t statistics and their corresponding p values. Similar results were obtained using a log scale transformation of the data where equal variances could be assumed.
Fig. 2. Dose-response curve of 45Ca efflux after NT-3 application to TrkA-expressing oocytes.

Oocytes expressing TrkA (0.1 ng of cRNA/oocyte) were loaded with 45Ca2+ and washed (see “Experimental Procedures”). After 45Ca2+ efflux levels stabilized, NT-3 was added (at time 0) to the medium at final concentrations ranging from 10 to 300 ng/ml. Peak mean counts/min were plotted against the dose of NT-3. Peak mean response to NT-3 was seen after 30 min in response to 10 ng/ml NT-3, after 20 min for 25 and 50 ng/ml of NT-3, and 10 min after addition of 100, 200, or 300 ng/ml of NT-3 (data not shown).
Immunoprecipitations and Immunoblotting
For analysis of TrkA, p75NTR, and truncated p75NTR- receptors, Xenopus oocytes were injected with each cRNA (1-5 ng per oocyte of each cRNA). Thirty six hours post-injection, 25 oocytes from each condition were manually homogenized in 250 μl of ice-cold solubilization buffer (65 mm Tris-HCl, pH 7.5, 50 mm NaCl, 5 mm EDTA, 2% Triton X-100, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mm sodium orthovana-date, and 10 mm sodium fluoride). Lysates were incubated at 4 °C for 1 h, and the soluble phase was partitioned from the insoluble and lipid phases by three rounds of centrifugation at 18,370 × g for 5 min at 4 °C. 250 μl of 2× IP buffer (solubilization buffer with 10 mg/ml of bovine serum albumin) was added to each sample and pre-cleared with protein A- or protein G-Sepharose beads for 1 h. TrkA receptors were immunoprecipitated with the anti-FLAG monoclonal antibody (M2), and P75NTR and p75-receptors were precipitated using wheat germ lectin-agarose beads (Amersham Pharmacia Biotech). Precipitated samples were washed four times with 1× solubilization buffer, after which precipitated proteins were removed from the beads by boiling for 5 min in 3× Laemmli sample buffer, and SDS-PAGE was performed on 7.5% polyacrylamide gels. After transfer to nitrocellulose membranes (Hybond, Amersham Pharmacia Biotech), samples were blocked in TBST with nonfat dried milk and incubated with primary antibodies against the FLAG epitope of TrkA (Stratagene, La Jolla CA), the extracellular domain of p75NTR (REX), or the cytoplasmic domain of p75NTR (Promega, Madison, WI) overnight at 4 °C. After washing, blots were incubated with horseradish peroxidase-conjugated goat anti-mouse (Promega, Madison, WI or New England Biolabs, Beverly, MA) or horseradish peroxidase-conjugated goat anti-rabbit (Promega, Madison, WI or New England Biolabs, Beverly, MA) at a dilution of 1:5000 for 1-2 h at room temperature. After washing, bands were visualized by incubating with Supersignal (Pierce) or ECL (Amersham Pharmacia Biotech), followed by exposure to Kodak BioMax film (Eastman Kodak Co.).
For analysis of the chimeric EGFR/p75NTR receptor, Xenopus oocytes were injected with 5 ng per oocyte of EGFR/p75NTR cRNA. Thirty six hours post-injection, 25 oocytes were manually homogenized in ice-cold homogenization buffer (65 mm Tris-HCl, pH 7.5, 10 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mm sodium orthovanadate, and 10 mm sodium fluoride), and lysates were centrifuged at 735 × g for 5 min at 4 °C to separate a heavy fraction (containing the plasma membrane and cortical granules) from the cytoplasmic fraction. The heavy fraction was solubilized in 250 μl of RIPA buffer (with 2.5% Triton X-100) for 1 h at 4 °C and centrifuged at 18,370 × g for 20 min at 4 °C. Samples were pre-cleared with protein G-Sepharose beads. EGFR/p75NTR receptors were immunoprecipitated with anti-EGFR monoclonal antibody (clone LA22, Upstate Biotechnology, Inc., Lake Placid, NY) overnight at 4 °C. Precipitated samples were washed four times with 1× solubilization buffer, after which precipitated proteins were removed from the beads by boiling for 5 min in 3× Laemmli sample buffer, and SDS-PAGE was performed on 7.5% polyacrylamide gels. After transfer to nitrocellulose membranes (Hybond, Amersham Pharmacia Biotech), samples were blocked in TBST with nonfat dried milk and incubated with anti-EGFR antibody.
For coimmunoprecipitation experiments, equal amounts of cRNA for FLAG-tagged TrkA and p75NTR (5 ng of each cRNA) were microinjected into each oocyte. After 48 h, oocytes were incubated with NT-3 (100 ng/ml), NGF (100 ng/ml), or no neurotrophin for 20 min at room temperature, after which they were placed on ice. Oocytes were manually homogenized in ice-cold homogenization buffer, and samples were centrifuged at 735 × g for 5 min at 4 °C. The pellet (containing the membrane) was solubilized in 500 μl of a buffer containing 130 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2% Triton X-100, and 10 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm sodium orthovanadate. The cytoplasmic fraction was solubilized by adding a 2× concentration of this solubilization buffer to the 250 μl of supernatant. Fractions were solubilized for 15 min at 37 °C and centrifuged at 18,370 × g for 20 min at 4 °C. Samples were pre-cleared with protein A/G-Sepharose beads and immunoprecipitated with anti-p75NTR (REX) (25). After SDS-PAGE and transfer, nitrocellulose membranes (Hybond, Amersham Pharmacia Biotech) were blocked in TBST with nonfat dried milk and incubated with primary antibodies against p75NTR extracellular domain (REX) (25) or anti-FLAG M2 monoclonal antibody (Stratagene, La Jolla, CA).
RESULTS
Expression of TrkA and p75NTR in Xenopus Oocytes
The Xenopus oocyte 45Ca2+ efflux assay has been used to quantify receptor activation and signal transduction (29, 30). Mature oocytes are capable of translating injected cRNA, targeting proteins to correct subcellular locations, and inserting receptors into the plasma membrane (31). Because the phospholipase C-γ pathway is conserved in oocytes, activation of receptor tyrosine kinases, such as Trk receptors, stimulates phospholipase C-γ and phosphatidylinositol turnover and stimulates intracellular calcium release (32, 33). The increased intracellular calcium activates a chloride current in Xenopus oocytes and causes cytoplasmic calcium to be released into the extracellular medium (32, 34, 35). Receptor activation and signal transduction through the phospholipase C-γ pathway can therefore be quantified by using a 45Ca2+ efflux assay (29, 30). For this reason, we chose to express TrkA (with or without p75NTR) in Xenopus oocytes to quantitatively study NT3 activation of TrkA and the role of p75NTR in regulating it.
To determine whether TrkA and p75NTR protein expression correlate with the amount of each cRNA injected, we microinjected Xenopus oocytes with 0, 1, 2.5, or 5 ng of TrkA per oocyte and determined the level of TrkA protein expressed. There was a proportional increase in the amount of TrkA receptor protein expressed by oocytes in response to increasing amounts of injected TrkA cRNA (Fig. 1A). Similarly, oocytes injected with 0, 1, or 2.5 ng of p75NTR cRNA demonstrated that p75NTR protein expression correlated with the amount of p75NTR cRNA injected (Fig. 1B). To determine whether coexpression of p75NTR interferes with TrkA receptor expression, oocytes were coinjected with TrkA cRNA (1 ng per oocyte) and increasing amounts of p75NTR cRNA (0, 1, or 2.5 ng per oocyte). Injection of increasing amounts of p75NTR cRNA resulted in increasing p75NTR expression; however, the amount of TrkA expressed was unaffected by p75NTR coexpression (Fig. 1C). Thus, the amount of TrkA and p75NTR protein expressed correlated with the amount of each cRNA injected, similar to other proteins exogenously expressed in Xenopus oocytes (29, 30, 36).
Fig. 1. TrkA and p75NTR receptor expression are highly correlated with the amount of cRNA injected into Xenopus oocytes, and coexpression of p75NTR does not interfere with TrkA receptor expression.

A, the amount of TrkA receptor protein expressed correlates with the amount of TrkA receptor cRNA injected. Xenopus oocytes were injected with 0, 1, 2.5, or 5 ng of TrkA cRNA, and the level of TrkA protein expressed was determined by immunoprecipitation followed by immunoblotting. There is a proportional increase in the amount of TrkA receptor, 140- and 110-kDa doublet, expressed in response to increasing amounts of TrkA cRNA injected. B, the amount of p75NTR protein expressed correlates with the amount of p75NTR cRNA injected. Oocytes were injected with 0, 1, or 2.5 ng of p75NTR cRNA, and p75NTR protein expression was determined by precipitation with wheat germ lectin-agarose followed by immunoblotting. A proportional increase in p75NTR protein, 75-80 kDa, is seen with increasing p75NTR cRNA injection. C, increasing p75NTR expression does not alter the level of TrkA receptor expressed. Oocytes were injected with 1 ng of TrkA and increasing amounts of p75NTR (0, 1, and 2.5 ng per oocyte). p75NTR expression is noted to increase proportionally; however, TrkA expression is not altered by p75NTR coexpression.
NT-3 Activates Signaling through TrkA Receptors
Having determined that TrkA and p75NTR receptor expression correlate with the amount of each cRNA injected, we next studied the signaling of TrkA-expressing Xenopus oocytes in response to a range of NT-3 concentrations. NT-3 activated TrkA signaling through the phospholipase C-γ pathway in a dose-dependent fashion (Fig. 2). Peak response was seen 30 min after addition of 10 ng/ml NT-3, 20 min after addition of 25 or 50 ng/ml of NT-3 and 10 min after addition of 100, 200, or 300 ng/ml NT-3 (data not shown). The peak response to NT-3 plateaued at about 100 ng/ml, suggesting that 100 ng/ml was functionally saturating. Similar results were obtained using a log scale transformation of the data where equal variances could be assumed. In addition, differences in mean peak signaling were compared using one-way analysis of variance. There were no statistically significant mean differences in NT-3 signaling at 100, 200, and 300 ng/ml (p > 0.52 or larger for the three comparisons). However, the mean response at 100 ng/ml was significantly different from the response at 50 ng/ml (p = 0.002). All subsequent experiments were done using 100 or 200 ng/ml of NT-3.
p75NTR Specifically Inhibited NT-3 Signaling through TrkA
To determine whether p75NTR inhibited NT-3 signaling through TrkA and to assess whether this inhibition was specific to NT-3, we coexpressed TrkA and p75NTR in oocytes. Equal amounts of cRNA encoding p75NTR and TrkA were injected. Oocytes that expressed TrkA alone were also injected with an equal amount of green fluorescent protein (GFP) cRNA to control for total RNA injected. (GFP had no effect on the signaling response to neurotrophins, data not shown.) After loading the oocytes with 45Ca2+ and allowing for stabilization of 45Ca2+ efflux, 100 ng/ml of either NGF or NT-3 was added, and 45Ca2+ efflux was measured every 10 min. Application of NGF or NT-3 to TrkA-expressing oocytes elicited 45Ca2+ efflux. NGF signaling was unaffected by coexpression of p75NTR, and p75NTR had no effect on the timing or peak level of NGF signaling (Fig. 3A). In contrast, p75NTR completely inhibited NT-3 signaling through TrkA (Fig. 3B). The inhibitory effect of p75NTR on NT-3 signaling through TrkA increased with increasing expression of p75NTR (Fig. 3C). Remarkably, almost full potency of TrkA signaling was recovered when the p75NTR: TrkA ratio was decreased from 1:1 to 1:2. These data are evidence that rather small changes in p75NTR expression have marked effects on TrkA signaling. Increased expression of p75NTR relative to TrkA did not impair the response of oocytes to subsequent NGF stimulation (Fig. 3C). These data demonstrate that p75NTR specifically inhibits NT-3 signaling through TrkA and that the level of inhibition depends on the ratio of p75NTR relative to TrkA.
Fig. 3. p75NTR inhibits NT-3 signaling through TrkA but not NGF signaling.

A, oocytes expressing TrkA alone or in combination with p75NTR (see “Experimental Procedures”) were loaded with 45Ca2+. After stabilization of 45Ca2+ efflux, 100 ng/ml NGF (A) or 100 ng/ml NT-3 (B) was added at time 0 (indicated by arrow), and calcium efflux was measured every 10 min. C, inhibitory effect of p75NTR on NT-3 signaling through TrkA increases with increasing expression of p75NTR. Oocytes injected with TrkA receptor cRNA (0.2 ng/oocyte) and p75NTR (ranging from 0.04 to 0.2 ng/oocyte) were loaded with 45Ca2+. After stabilization of 45Ca2+ efflux, 100 ng/ml of NT-3 was added at time 0 (indicated by arrow), and calcium efflux was measured every 10 min. Subsequent response of oocytes to application of 100 ng/ml of NGF (arrow) demonstrates that the oocytes are capable of responding to NGF stimulation.
The Extracellular Domain of p75NTR Inhibited NT-3 Signaling through TrkA
To determine which structural domain of p75NTR mediated the inhibitory effect on NT-3 signaling through TrkA, we constructed a mutant p75NTR receptor with a truncated cytoplasmic domain (containing only the first nine amino acids of the cytoplasmic domain). We coexpressed this truncated p75NTR with TrkA to assess its effect on NT-3 signaling. Expression of the truncated p75NTR receptor was confirmed by immunoblotting (Fig. 4B), and surface expression was further confirmed by 125I-NT-3 binding and cross-linking studies (data not shown). After stabilization of 45Ca2+ efflux, 200 ng/ml of NT-3 was added, and 45Ca2+ efflux was measured every 10 min. The truncated p75NTR completely inhibited NT-3 signaling through TrkA (Fig. 4A). This suggested that the cytoplasmic domain of p75NTR (excluding the juxtamembrane region) is not required to inhibit NT-3 signaling through TrkA. To determine whether the extracellular domain of p75NTR is necessary, we coexpressed TrkA along with an EGFR/p75NTR chimera, containing the extracellular domain of the EGFR fused to the transmembrane and cytoplasmic domains of p75NTR. This chimeric receptor has been used to study p75NTR signaling in PC12 cells and has been shown to be expressed on the cell surface (28). Expression in oocytes was confirmed by the presence of a 130-kDa band, consistent with the predicted size (28) (Fig. 4D). NT-3 signaling in Xenopus oocytes expressing the EGFR/p75NTR receptor along with TrkA was as robust as in oocytes expressing TrkA alone (Fig. 4B). From these experiments, we conclude that the extracellular domain of p75NTR is necessary for inhibition of NT-3 signaling through TrkA. We also conclude that the transmembrane and cytoplasmic domains of p75NTR are not sufficient to inhibit NT-3 signaling through TrkA.
Fig. 4. The extracellular domain of p75NTR is necessary to prevent NT-3 signaling through TrkA, and the transmembrane and cytoplasmic domains are not sufficient to inhibit signaling.

A, a truncated mutant p75NTR receptor with a minimal cytoplasmic domain (9 amino acids) inhibits NT-3 signaling through TrkA. Oocytes expressing TrkA alone in combination with truncated p75NTR-were loaded with 45Ca2+. After stabilization of 45Ca2+ efflux levels, 200 ng/ml of NT-3 was added to the medium (indicated by arrow), and 45Ca2+ efflux was measured every 10 min. B, oocytes injected with p75NTR-cRNA express the truncated p75NTR receptor protein. Lysates from oocytes injected with p75NTR- cRNA (+) and from uninjected oocytes (-) were precipitated with wheat germ lectin-agarose followed by immunoblotting with an antibody against the extracellular domain of p75NTR (REX). C, a chimeric EGFR/p75NTR receptor containing the extracellular domain of the EGF receptor fused to the transmembrane and cytoplasmic domains of p75NTR does not inhibit NT-3 signaling through TrkA. Oocytes expressing TrkA alone or in combination with EGFR/p75NTR were loaded with 45Ca2+. After stabilization of 45Ca2+ efflux levels, 200 ng/ml NT-3 was added to the medium (indicated by arrow), and calcium efflux was measured every 10 min. D, oocytes injected with the EGFR/p75NTR cRNA express the chimeric receptor protein. Lysates from oocytes injected with EGFR/p75NTR cRNA (+) and from uninjected oocytes (-) were immunoprecipitated with monoclonal antibody that recognizes the extracellular domain of the EGF receptor followed by immunoblotting using the anti-EGF receptor antibody.
Antagonistic interactions between the signaling pathways of Trk receptors and p75NTR have been described (21, 37, 38). However, our finding that the extracellular domain of p75NTR inhibited NT-3 signaling, whereas most of the cytoplasmic domain was dispensable, suggests that events occurring between the receptors on the plasma membrane and between their extracellular domains are critical for inhibiting NT-3 signaling through TrkA.
p75NTR Inhibited NT-3 Activation of TrkA Even When It Did Not Bind NT-3
This raised two possibilities as follows: either p75NTR interacts with NT-3 in a manner that inhibits its ability to activate TrkA or p75NTR interacts with TrkA to inhibit activation by NT-3. The first possibility requires p75NTR binding to NT-3 to suppress signaling. Under the second possibility, NT-3 binding to p75NTR is dispensable. To distinguish between these possibilities, we performed a 45Ca2+ efflux assay using oocytes coexpressing both receptors, in the presence or absence of anti-p75NTR antibody or excess BDNF (1 μg/ml), each of which prevents NT-3 binding to p75NTR. Oocytes expressing TrkA and p75NTR or TrkA alone (along with GFP) were loaded with 45Ca2+ as described above. Oocytes expressing both receptors were incubated for 2 h prior to the addition of NT-3 with 1 μg/ml of BDNF, or with 50 μg/ml of anti-p75NTR antibody (REX), or in medium alone. BDNF and anti-p75NTR antibody at these concentrations nearly abolished NT-3 binding to p75NTR in PC12 cells overexpressing TrkA (data not shown), and the ability of the anti-p75NTR antibody to block NT-3 binding to p75NTR in oocytes was verified by binding and cross-linking studies (data not shown). 200 ng/ml of NT-3 was added to the oocytes, and 45Ca2+ efflux was measured every 10 min. The data show that blocking NT-3 binding to p75NTR had no effect on the ability of this receptor to block TrkA activation (Fig. 5). No response to NT-3 was seen in the presence of the p75NTR antibody or BDNF. This experiment shows that p75NTR inhibited NT-3 signaling through TrkA, even in the absence of direct binding to NT-3. Because BDNF and the anti-p75NTR antibody may act as p75NTR ligands, this experiment does not exclude the possibility that p75NTR liganding may alter TrkA responsiveness to NT-3. However, it does indicate that direct interaction of p75NTR with NT-3 is not required to inhibit TrkA activation.
Fig. 5. p75NTR does not need to directly bind to NT-3 to inhibit activation of signaling through TrkA.
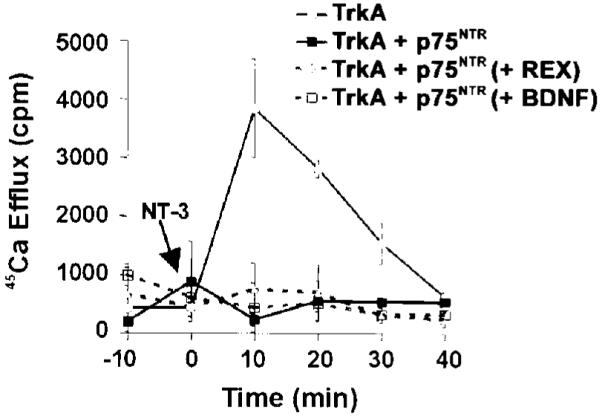
Oocytes expressing TrkA alone or in combination with p75NTR were loaded with 45Ca2+. Oocytes expressing TrkA and p75NTR were treated for 2 h with 50 μg/ml of anti-p75NTR antibody (REX), 1 μg/ml BDNF, or were left untreated as indicated. The anti-p75NTR antibody and BDNF at this concentration have both been shown to prevent NT-3 binding to p75NTR (see “Results”). After stabilization of 45Ca2+ efflux, 200 ng/ml of NT-3 was added at time 0 (indicated by arrow), and calcium efflux was measured every 10 min.
p75NTR and TrkA Physically Interacted in Xenopus Oocytes Coexpressing Both Receptors
Our results suggested that TrkA and p75NTR may physically interact in surface membranes. To address this possibility, we coexpressed a C-terminal FLAG-tagged TrkA receptor along with p75NTR in Xenopus oocytes. The TrkA construct consisted of full-length TrkA with two FLAG epitopes in tandem at the C terminus. Expression of the FLAG-tagged receptor was verified by homogenizing 15 oocytes microinjected with cRNA encoding the receptor, immunoprecipitating with a monoclonal antibody against the FLAG epitope (M2), and immunoblotting with the same anti-FLAG antibody. A specific doublet corresponding to TrkA was identified, which was not present in uninjected oocytes (Fig. 6).
Fig. 6. Expression of FLAG-tagged TrkA receptor in microinjected Xenopus oocytes.

Fifteen oocytes were injected with 1 ng of TrkA-FLAG cRNA (+) or were left uninjected (-). Thirty six hours after injection, oocytes were lysed, immunoprecipitated with anti-FLAG antibody, separated by 7.5% SDS-PAGE, and immunoblotted with the anti-FLAG antibody (see “Experimental Procedures”). Bands of 140 and 110 kDa are seen in the TrkA-injected oocytes.
We next asked if the FLAG-tagged TrkA receptor functionally responded to neurotrophin stimulation. NGF activation of TrkA results in activation of the phospholipase C-γ pathway with resultant intracellular calcium release (39). Because a calcium-activated chloride current occurs in Xenopus oocytes in response to release of intracellular calcium (32, 33, 35), we performed electrophysiologic recordings from single oocytes in the presence of NGF to confirm that FLAG-tagged TrkA was functional. Oocytes expressing this receptor responded specifically to 100 ng/ml of NGF,2 demonstrating that this receptor is indeed functional.
To determine whether p75NTR physically interacted with TrkA, we performed coprecipitation studies in microinjected oocytes. Equal amounts of cRNA for FLAG-tagged TrkA and p75NTR (5 ng of each cRNA) were injected, and after 48 h, oocytes were exposed to NT-3 (100 ng/ml), NGF (100 ng/ml), or no neurotrophin for 20 min at room temperature, after which they were placed on ice (to prevent further membrane trafficking events).
Oocytes were manually homogenized and fractionated into a heavy fraction, containing the membrane and cortical granules, and a cytoplasmic fraction (see “Experimental Procedures”). Fractions were solubilized in a buffer containing 2% Triton X-100 (and protease inhibitors) for 15 min at 37 °C and were then centrifuged. Supernatants were pre-cleared with protein A/G-Sepharose beads and immunoprecipitated with a polyclonal antibody to p75NTR (25). SDS-PAGE was performed on samples (see “Experimental Procedures”). After transfer to nitrocellulose membranes, samples were immunoblotted with the monoclonal antibody to the FLAG epitope (Stratagene, La Jolla, CA). As seen in Fig. 7, TrkA was coprecipitated with p75NTR from the membrane fractions of oocytes injected with TrkA and p75NTR (lanes 2-4) but not from uninjected oocytes (lane 5) or from the antibody plus protein A/G bead control sample (lane 1). p75NTR and TrkA were coprecipitated in oocytes expressing these receptors regardless of whether they were treated with 100 ng/ml of NT-3 (lane 4), NGF (lane 3), or with no neurotrophin (lane 2). The anti-p75NTR serum has previously been shown to bind p75NTR and to lack detectable binding to TrkA (25). We further verified that the anti-p75NTR serum did not immunoprecipitate TrkA from oocytes expressing TrkA alone and that the anti-FLAG monoclonal antibody did not recognize a specific band in oocytes expressing only p75NTR (data not shown). Because high levels of expression of both p75NTR and TrkA were required to observe this interaction, and only a relatively small percentage of each receptor appears to be in this complex, we cannot fully determine the role of this physical interaction at physiological concentrations. However, these results do suggest that p75NTR and TrkA can physically interact in the plasma membrane and that this interaction appears to be independent of the presence of neurotrophins.
Fig. 7. p75NTR and TrkA can be coimmunoprecipitated from the membrane fractions of microinjected Xenopus oocytes expressing both receptors.

Oocytes were injected with TrkA-FLAG and p75NTR (5 ng each). Thirty six hours later, 12 oocytes per condition were treated with 100 ng/ml NGF (lane 3), 100 ng/ml NT-3 (lane 4), or no neurotrophin (lane 2) for 20 min at room temperature (see “Experimental Procedures”). Oocytes were lysed, immunoprecipitated with anti-p75NTR antibody, and immunoblotted with anti-FLAG antibody. TrkA is coprecipitated with p75NTR in oocytes injected with TrkA and p75NTR cRNA (lanes 2-4), but not in uninjected oocytes (lane 5), or in antibody and protein A/G-Sepharose beads only control (lane 1). Anti-p75NTR serum has previously been shown to bind p75NTR and to lack detectable binding to TrkA (25). We have also observed that anti-p75NTR anti-serum does not precipitate TrkA from oocytes expressing TrkA only, nor does the anti-FLAG monoclonal antibody recognize a specific band from oocytes expressing p75NTR alone (data not shown).
DISCUSSION
In this paper, we examined the role of p75NTR in inhibiting NT-3 signaling through TrkA. By using a calcium efflux assay to assess signaling in Xenopus oocytes microinjected to express both receptors, we demonstrated quantitatively that p75NTR completely suppressed NT-3 signaling through TrkA. The inhibition was specific to NT-3, because NGF signaled equally well through TrkA, regardless of whether or not p75NTR was present. Our data are consistent with studies in PC12 cells which demonstrate that p75NTR suppresses NT-3 signaling through TrkA (4, 23) and with the observation that TrkA-expressing sympathetic neurons from p75NTR-/- mice are more responsive to NT-3 than wild type sympathetic neurons (24). Our findings are also consistent with the recent evidence that p75NTR can inhibit the ability of NT-3 to activate TrkA in vivo (40). Compared with control littermates, NGF+/- mice have a 50% deficit in sympathetic neurons. This deficit is rescued by NT-3 in the absence of p75NTR (NGF+/- and p75NTR-/-) but not in mice that express normal levels of p75NTR (40). The rescue of these TrkA-expressing sympathetic neurons depends on NT-3, since many of these neurons are lost in NGF+/-, p75NTR-/-, NT-3+/- mice (40).
In addition to inhibiting TrkA signaling, p75NTR may also inhibit signaling through TrkB. In transfected A293 cells expressing TrkB, coexpression of p75NTR decreased TrkB phosphorylation in response to NT-3 and NT-4 but not in response to BDNF (41). In contrast to this finding, another group (42) has recently demonstrated that coexpression of p75NTR with TrkB inhibited phosphorylation in response to BDNF and NT-4 but not NT-3. The full effect of p75NTR on TrkB signaling in response to BDNF, NT-4, and NT-3 signaling through TrkB will need to be further elucidated. However, the mechanism by which p75NTR suppresses TrkB activation may be similar to the way p75NTR suppresses NT-3 signaling through TrkA.
Structural Domains of p75NTR Involved in the Inhibition of NT-3 Signaling through TrkA
In these experiments, we showed that the extracellular domain of p75NTR was necessary to inhibit NT-3 signaling through TrkA. NT-3 could not signal through TrkA in Xenopus oocytes coexpressing a mutant p75NTR receptor that lacks all but the first nine amino acids of its cytoplasmic domain. In contrast, oocytes coexpressing an EGFR/p75NTR chimeric receptor, which contained full p75NTR transmembrane and cytoplasmic domains, did not inhibit NT-3 signaling through TrkA. Based on these findings, we conclude that the extracellular domain of p75NTR is necessary to inhibit NT-3 signaling through TrkA. These experiments also clearly showed that the transmembrane and cytoplasmic domains of p75NTR are not sufficient to inhibit NT-3 signaling through TrkA.
The truncated p75NTR receptor contains the juxtamembrane region of p75NTR, including cysteine 279, which is critical for palmitoylation of p75NTR (43). Because palmitoylation of p75NTR appears to be important for targeting p75NTR (as well as TrkA) to caveolae-like membranes where much of TrkA receptor signaling is initiated (44), it is possible that the juxtamembrane region of p75NTR may be important for inhibiting NT-3 signaling through TrkA. However, our experiments clearly show that this region, in the absence of the extracellular domain, is not sufficient to inhibit NT-3 signaling through TrkA.
p75NTR and Trk receptors may engage in cross-talk through the interaction of their cytoplasmic signaling pathways. To date, much of this interaction appears to be inhibitory. NGF activation of TrkA can block the p75NTR-mediated cell death pathway (21), and activation of p75NTR by BDNF suppresses cellular responses to NGF activation of TrkA (37, 38). Our data do not entirely exclude the possibility that p75NTR initiates a cytoplasmic signal through its juxtamembrane region, either directly or by interacting with another cytoplasmic protein. However, our observation that the p75NTR receptor, lacking almost the entire cytoplasmic domain, inhibited NT-3 signaling through TrkA suggests that p75NTR did not act downstream from the receptor to suppress TrkA signaling.
Interaction of p75NTR and TrkA to Mediate the Inhibition of NT-3 Signaling through TrkA
By coexpressing TrkA and p75NTR in oocytes and occupying p75NTR with either BDNF of anti-p75NTR antibody, we determined that p75NTR inhibited activation of TrkA even in the absence of direct binding to NT-3. Because BDNF and REX may act as p75NTR ligands, these results do not exclude the possibility that liganding of p75NTR alters the ability of TrkA to respond to NT-3. More importantly, these results suggest strongly that events occurring between p75NTR and TrkA, either directly or indirectly, are critical for inhibiting TrkA response to NT-3.
In oocytes coexpressing both receptors at significantly higher levels than used in the 45Ca2+ efflux assays, we coimmunoprecipitated p75NTR and TrkA from the membrane fraction of oocytes. Although we could only detect this interaction when both receptors were expressed at these relatively higher levels, these findings suggest that p75NTR and TrkA can physically interact in the surface membrane and that this interaction does not require the presence of neurotrophins.
The structural basis for p75NTR and TrkA interactions has not been defined. Evidence to support their interaction has been presented (41, 45-49), and most reports have focused on the role of p75NTR in increasing the affinity of TrkA for NGF (19, 48, 49). Our results suggest a potential role for the physical interaction of p75NTR and TrkA in regulating the specificity of TrkA for NGF relative to NT-3. The coimmunoprecipitation data suggest the existence of either a direct physical interaction between p75NTR and TrkA or the formation of a complex bridged by one or more mutually interacting proteins. p75NTR and TrkA are both significantly enriched in caveolae-like domains at the plasma membrane, and the two receptors may interact (at least functionally) within this micro-environment (44). Furthermore, indirect interaction between p75NTR and TrkA via the mutually interacting protein caveolin-1 has been suggested (50).
It is also possible that p75NTR inhibits NT-3 signaling by inhibiting NT-3 binding to TrkA. However, preliminary data from our laboratory using 125I-NT-3 binding and cross-linking in PC12 cells that overexpress TrkA indicate that NT-3 can still bind TrkA in the presence of p75NTR.3 In these cells, occupying p75NTR with BDNF or REX did not alter the amount of NT-3 bound to TrkA.3 However, since BDNF and REX may act as p75NTR ligands, it is possible that liganded p75NTR alters the conformation of the TrkA-binding site. An allosteric model for p75-TrkA interactions has been proposed that may account for this property (51). In the future, it will be important to examine NT-3 binding in a cell line that expresses TrkA alone, or in combination with p75NTR, to fully determine the effect of p75NTR on NT-3 binding to TrkA.
In summary, our experiments have shown that p75NTR interacts with TrkA to inhibit NT-3 signaling. Our work demonstrates that the extracellular domain of p75NTR is necessary for this inhibition and suggests that this interaction occurs at the level of p75NTR-TrkA interaction on the surface membrane. Our study has important implications for understanding the regulation of NT-3 signaling through TrkA in vivo. During late embryonic and early postnatal development, nearly 50% of sympathetic neurons that are NGF-dependent develop an additional requirement for NT-3, mediated by TrkA (8, 9, 52, 53). These neurons express low levels of p75NTR relative to TrkA (53), which our data suggest would enable NT-3 to promote survival by activating TrkA (2, 5, 53). As targets are reached, p75NTR expression increases and responsiveness to NT-3 declines (2). In vivo studies of the neonatal sympathetic ganglia show that p75NTR regulates the survival of TrkA-expressing sympathetic neurons by regulating the responsiveness of these neurons to NT-3 (40). In fact, it appears that one of the most important roles for p75NTR in the developing sympathetic ganglia is to regulate TrkA responsiveness to NT-3 (40). In addition to sympathetic neurons, a subset of late embryonic TrkA-expressing sensory neurons that are NGF-dependent also develop a requirement for NT-3 that is mediated by TrkA (5, 10). Unlike postnatal sympathetic neurons, postnatal sensory neurons lose their ability to respond to NT-3 via TrkA. It will be interesting to determine whether p75NTR plays a role in decreasing the responsiveness of postnatal sensory neurons to NT-3. Finally, although our work has focused on the role of p75NTR in regulating NT-3 signaling through TrkA, p75NTR may interact with TrkB to regulate its responsiveness. It will be important in the future to determine whether p75NTR regulates TrkB signaling by a similar mechanism.
Acknowledgments
We thank Dr. Moses Chao for the EGFR/p75NTR construct. We also thank Drs. Eric Huang, Song Hu, and Ardem Patapoutian for helpful discussions and Kan Lu for help with preparation of the figures.
Footnotes
This work was supported in part by United States Public Health Service Research Grants MH48200 (to L. F. R.), NRSA NS10301 (to P. S. M.), and NS24054 (to W. C. M.), the Howard Hughes Medical Institute, a Stein Oppenheimer award (to P. S. M.), the McGowen Charitable Fund (to W. C. M.), the Adler Foundation (to W. C. M.), and the John Douglas French Alzheimer’s Foundation (to W. C. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- NGF
- nerve growth factor
- NT-3
- neurotrophin-3
- BDNF
- brain-derived neurotrophic factor
- NT-4
- neurotrophin-4
- p75NTR
- p75 neurotrophin receptor
- EGFR
- epidermal growth factor receptor
- PAGE
- polyacrylamide gel electrophoresis
- PCR
- polymerase chain reaction
P. S. Mischel and J. Umbach, S. Eskandari, S. G. Smith, C. B. Gundersen, and G. A. Zampighi, submitted for publication.
P. S. Mischel, J. S. Valletta, W. C. Mobley, and L. F. Reichardt, unpublished observations.
REFERENCES
- 1.Reichardt LF, Fariñas I. In: Molecular Approaches to Neural Development. Cowan MW, Jessell TM, Zipursky FL, editors. Oxford University Press; New York: 1997. pp. 220–263. [Google Scholar]
- 2.Belliveau DJ, Krivko I, Kohn J, Lachance C, Pozniak C, Rusakov D, Kaplan D, Miller FD. J. Cell Biol. 1997;136:375–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birren SJ, Lo L, Anderson DJ. Development. 1993;119:597–610. doi: 10.1242/dev.119.3.597. [DOI] [PubMed] [Google Scholar]
- 4.Clary DO, Reichardt LF. Proc. Natl. Acad. Sci. U. S. A. 1994;91:11133–11137. doi: 10.1073/pnas.91.23.11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies AM, Minichiello L, Klein R. EMBO J. 1995;14:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verdi JM, Anderson DJ. Neuron. 1994;13:1359–1372. doi: 10.1016/0896-6273(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 7.Wyatt S, Pinon LG, Ernfors P, Davies AM. EMBO J. 1997;16:3115–3123. doi: 10.1093/emboj/16.11.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farinas I, Jones KR, Backus C, Wang XY, Reichardt LF. Nature. 1994;369:658–661. doi: 10.1038/369658a0. [DOI] [PubMed] [Google Scholar]
- 9.Francis N, Farinas I, Brennan C, Rivas-Plata K, Backus C, Reichardt L, Landis S. Dev. Biol. 1999;210:411–427. doi: 10.1006/dbio.1999.9269. [DOI] [PubMed] [Google Scholar]
- 10.Huang EJ, Wilkinson GA, Farinas I, Backus C, Zang K, Wong SL, Reichardt LF. Development. 1999;126:2191–2203. doi: 10.1242/dev.126.10.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chao MV, Hempstead BL. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- 12.Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. J. Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 14.Davey F, Davies AM. Curr. Biol. 1998;8:915–918. doi: 10.1016/s0960-9822(07)00371-5. [DOI] [PubMed] [Google Scholar]
- 15.Barker PA, Shooter EM. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- 16.Hantzopoulos PA, Suri C, Glass DF, Goldfarb MP, Yancopoulos GD. Neuron. 1994;13:187–201. doi: 10.1016/0896-6273(94)90469-3. [DOI] [PubMed] [Google Scholar]
- 17.Battleman DS, Geller AI, Chao MV. J. Neurosci. 1993;13:941–951. doi: 10.1523/JNEUROSCI.13-03-00941.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- 19.Mahadeo D, Kaplan L, Chao MV, Hempstead BL. J. Biol. Chem. 1994;269:6884–6891. [PubMed] [Google Scholar]
- 20.MacPhee IJ, Barker PA. J. Biol. Chem. 1997;272:23547–23551. doi: 10.1074/jbc.272.38.23547. [DOI] [PubMed] [Google Scholar]
- 21.Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV. J. Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM. Mol. Cell. Neurosci. 1999;14:28–40. doi: 10.1006/mcne.1999.0770. [DOI] [PubMed] [Google Scholar]
- 23.Benedetti M, Levi A, Chao MV. Proc. Natl. Acad. Sci. U. S. A. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KF, Davies AM, Jaenisch R. Development. 1994;120:1027–1033. doi: 10.1242/dev.120.4.1027. [DOI] [PubMed] [Google Scholar]
- 25.Weskamp G, Reichardt LF. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J, Valletta JS, Grimes ML, Mobley WC. J. Neurochem. 1995;65:1146–1156. doi: 10.1046/j.1471-4159.1995.65031146.x. [DOI] [PubMed] [Google Scholar]
- 27.Liman ER, Tytgat J, Hess P. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 28.Yan H, Schlessinger J, Chao MV. Science. 1991;252:561–563. doi: 10.1126/science.1850551. [DOI] [PubMed] [Google Scholar]
- 29.Ueno H, Escobedo JA, Williams LT. J. Biol. Chem. 1991;268:22814–22819. [PubMed] [Google Scholar]
- 30.Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. J. Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soreq H, Seidman S. Methods Enzymol. 1992;207:225–265. doi: 10.1016/0076-6879(92)07016-h. [DOI] [PubMed] [Google Scholar]
- 32.Filtz TM, Paterson A, Harden TK. J. Biol. Chem. 1996;271:31121–31126. doi: 10.1074/jbc.271.49.31121. [DOI] [PubMed] [Google Scholar]
- 33.Schultz P, Stannek P, Bischoff SC, Dahinden CA, Gierschik P. Cell. Signal. 1992;4:153–161. doi: 10.1016/0898-6568(92)90079-n. [DOI] [PubMed] [Google Scholar]
- 34.Gillo B, Lass Y, Nadler E, Oron Y. J. Physiol. (Lond.) 1987;392:349–361. doi: 10.1113/jphysiol.1987.sp016784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartzell HC. J. Gen. Physiol. 1996;108:157–175. doi: 10.1085/jgp.108.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richter JD, Smith LD. Cell. 1981;27:183–191. doi: 10.1016/0092-8674(81)90372-x. [DOI] [PubMed] [Google Scholar]
- 37.Kimpinski K, Jelinski S, Mearow K. Neuroscience. 1999;93:253–263. doi: 10.1016/s0306-4522(99)00156-6. [DOI] [PubMed] [Google Scholar]
- 38.Kohn J, Aloyz RS, Toma JG, Haak-Frendscho M, Miller FD. J. Neurosci. 1999;19:5393–5408. doi: 10.1523/JNEUROSCI.19-13-05393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Segal RA, Greenberg ME. Annu. Rev. Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 40.Brennan C, Rivas-Plata K, Landis SC. Nat. Neurosci. 1999;2:699–705. doi: 10.1038/11158. [DOI] [PubMed] [Google Scholar]
- 41.Bibel M, Hoppe E, Barde YA. EMBO J. 1999;18:616–622. doi: 10.1093/emboj/18.3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vesa J, Kruttegen, Shooter EM. J. Biol. Chem. 2000;275:24414–24420. doi: 10.1074/jbc.M001641200. [DOI] [PubMed] [Google Scholar]
- 43.Barker PA, Barbee G, Misko TP, Shooter EM. J. Biol. Chem. 1994;269:30645–30650. [PubMed] [Google Scholar]
- 44.Huang CS, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, Mobley WC. J. Biol. Chem. 1999;274:36707–36714. doi: 10.1074/jbc.274.51.36707. [DOI] [PubMed] [Google Scholar]
- 45.Gargano N, Levi A, Alema S. J. Neurosci. Res. 1997;50:1–12. doi: 10.1002/(SICI)1097-4547(19971001)50:1<1::AID-JNR1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 46.Huber LJ, Chao MV. J. Neurosci. Res. 1995;40:557–563. doi: 10.1002/jnr.490400415. [DOI] [PubMed] [Google Scholar]
- 47.Ross AH, Daou MC, McKinnon CA, Condon PJ, Lachyankar MB, Stephens RM, Kaplan DR, Wolf DE. J. Cell Biol. 1996;132:945–953. doi: 10.1083/jcb.132.5.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ross GM, Shamovsky IL, Lawrance G, Solc M, Dostaler SM, Weaver DF, Riopelle RJ. Eur. J. Neurosci. 1998;10:890–898. doi: 10.1046/j.1460-9568.1998.00094.x. [DOI] [PubMed] [Google Scholar]
- 49.Wolf DE, McKinnon CA, Daou MC, Stephens RM, Kaplan DR, Ross AH. J. Biol. Chem. 1995;270:2133–2138. doi: 10.1074/jbc.270.5.2133. [DOI] [PubMed] [Google Scholar]
- 50.Bilderback TR, Gazula VR, Lisanti MP, Dobrowsky RT. J. Biol. Chem. 1999;274:257–263. doi: 10.1074/jbc.274.1.257. [DOI] [PubMed] [Google Scholar]
- 51.Bothwell M. Annu. Rev. Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 52.Ernfors P, Lee KF, Kucera J, Jaenisch R. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 53.Wyatt S, Davies AM. J. Cell Biol. 1995;130:1435–1446. doi: 10.1083/jcb.130.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]