

Summary
A female-sterile zebrafish maternal-effect mutation in cellular atoll (cea) results in defects in the initiation of cell division starting at the second cell division cycle. This phenomenon is caused by defects in centrosome duplication, which in turn affect the formation of a bipolar spindle. We show that cea encodes the centriolar coiled coil protein Sas-6, and that zebrafish Cea/Sas-6 protein localizes to centrosomes. cea also has a genetic paternal contribution, which when mutated results in an arrested first cell division followed by normal cleavage. Our data supports the idea that, in zebrafish, paternally-inherited centrosomes are required for the first cell division while maternally-derived factors are required for centrosomal duplication and cell divisions in subsequent cell cycles. DNA synthesis ensues in the absence of centrosome duplication, and the one-cycle delay in the first cell division caused by cea mutant sperm leads to whole genome duplication. We discuss the potential implications of these findings with regards to the origin of polyploidization in animal species. In addition, the uncoupling of developmental time and cell division count caused by the cea mutation suggests the presence of a time window, normally corresponding to the first two cell cycles, which is permissive for germ plasm recruitment.
Keywords: cellular atoll, sas-6, centrioles, centrosomes, cell division, tetraploid, gynogenesis, germ plasm, zebrafish
Introduction
Centrosomes are microtubule-organizing centers in animal cells that consist of a pair of centrioles surrounded by pericentriolar material (PCM). Previous studies show that centrioles are essential for the formation of the centrosomes (reviewed in Delattre and Gönczy, 2004). Due to their position at the center of the spindle aster, centrosomes have been traditionally attributed a role in spindle formation and cell division. In some organisms, such as in Caenorhabditis elegans, mutations in centrosomal components indeed result in the inability to organize a bipolar spindle (reviewed in Leidel and Gönczy, 2005).
Ultrastructural analysis has defined a stereotypic pathway for centrosome replication (reviewed in Delattre and Gönczy, 2004). In the first step in this duplication process, the two centrioles in a centrosome lose their characteristic orthogonal arrangement and split while remaining linked by a flexible connection. Secondly, a daughter centriole forms perpendicular to each parental centriole and elongates until it reaches the same size as the mother centriole. Finally, the PCM splits in two to form two different pairs of centrioles surrounded by PCM. Elegant genetic studies in C. elegans have defined a pathway for centriole formation in this organism, involving the kinase ZYG-1 and the coiled-coil proteins SPD-2, SAS-4, SAS-5 and SAS-6 (reviewed in Delattre and Gönczy, 2004; Leidel and Gönczy, 2005; see also Delattre et al., 2006; Pelletier et al., 2006). Human SAS-4 and SAS-6 homologues have been found to localize to the centrosomes (Andersen et al., 2003; Dammermann et al., 2004; Leidel et al., 2005), and siRNA-mediated inactivation of human SAS-6 results in centrosome duplication defects (Leidel et al., 2005). However, the components and mechanisms of assembly of centrioles and centrosomes remain incompletely understood, particularly in vertebrate species.
Maintaining two and only two centrosomes in the cell is important, as variations in this number can affect the reliability of chromosome segregation and cytokinesis (reviewed in Delattre and Gönczy, 2004; see also Kim and Ray, 2006). Thus, the cycle of centriolar duplication is coordinated with the cell cycle, such that centrioles duplicate once per cell division. Typically centriole duplication starts at the G1/S transition and is completed towards the end of mitosis. This strategy insures that each daughter cell will inherit one and only one centriolar pair, within one and only one centrosome. During fertilization, modifications of this scheme are required in order to prevent an increase in the number of centrosomes in the zygote. The most common strategy to solve this problem involves the differential contribution of centrosomes from the different gametes. In many animals, the sperm is thought to provide a pair of centrioles to a centriole-less oocyte. During the first cell cycle, these sperm-derived centrioles duplicate and recruit PCM materials from the egg immediately after fertilization to reconstitute a centrosome, and the reconstituted centrosomes continue to replicate in subsequent cell divisions.
During the oocyte-to-embryo transition, the faithful transmission of a constant number of centrioles, is coupled to the sequential processes of meiosis and pronuclear fusion, which insure the maintenance of a constant chromosomal number from one generation to the next. Because centrioles are important for cell division, but have been shown to be dispensable for DNA synthesis in a variety of systems, defects in parental centriolar duplication and/or function may be coupled to ploidy changes. Whole genome duplication, or polyploidization, has occurred multiple times during both plant and animal evolution (reviewed in Otto and Whitton, 2000). However, the mechanisms that generate and allow propagating such ploidy increases remain unclear.
In order to identify factors essential for early vertebrate development, we and others have carried out screens to isolate recessive maternal-effect mutations in the zebrafish (Dosch et al., 2004; Kishimoto et al., 2004; Pelegri et al., 2004; Pelegri and Schulte-Merker, 1999; Wagner et al., 2004). These efforts have lead to the identification of maternal genes involved in a variety of early developmental processes, including in the gene cellular atoll (cea), which affects the early cleavage divisions (Dosch et al., 2004). Here, we show that cea encodes the centriolar component Sas-6. Like centriolar genes in C. elegans (O’Connell et al., 2001), zebrafish Cea/Sas-6 is required both paternally for the first cell division, and maternally for the formation of centrosomes in subsequent cell cycles. We further demonstrate that sperm derived from cea mutant males can result in whole genome duplication, thus suggesting a specific novel molecular scenario, not involving unreduced gametes, where defects in paternal centriole duplication may facilitate the generation and propagation of polyploids in animal species. Our studies also constitute the first genetic analysis of centrosomal inheritance and duplication during the vertebrate oocyte-to-embryo transition, and highlight the high degree of conservation in this process in species as divergent as nematodes and the zebrafish.
Results
A mutation in cellular atoll leads to defects in the initiation of cell division beginning at the second cell cycle
Embryos genotypically homozygous for the cea mutation develop normally and appear indistinguishable from wild-type, but genotypically homozygous adult females produce clutches that are 100% inviable due to defects in cell division (Dosch et al., 2004). We analyzed these defects in more detail in embryos derived from females that are homozygous mutant for cea, which we refer to as “maternally mutant cea embryos”. Processes associated with egg activation, which occurs upon contact with water, such as the expansion of the chorionic membranes and ooplasmic streaming to form a blastodisc at the animal pole of the egg, occur normally in maternally mutant cea embryos (Dosch et al., 2004; Fig. 1E, compare to 1A). In wild-type embryos, fertilization normally results in the induction of a stereotypical cleavage pattern (Kimmel et al., 1995; Fig. 1A–D). Beginning at the second cleavage cycle, a fraction of cells in maternally mutant cea embryos fails to initiate furrow ingression (shown at the third cleavage cycle in Fig. 1F). However, in blastomeres of maternally mutant cea embryos, those furrows that do initiate cytokinesis, undergo maximal contraction and formation of a membrane septum as in wild-type embryos. Upon further development, this irregular cleavage pattern results in blastulae that have cells of uneven sizes (Fig. 1G), and which at the 1,000 cells stage show groups of cells sitting atop an abnormal uncellularized, syncytial region (Fig. 1H), a phenotype characteristic of other maternal-effect mutations that affect cell division (Dosch et al., 2004; Pelegri et al., 2004; Pelegri et al., 1999).
Figure 1.

Phenotype of maternally mutant cea embryos. (A–H) Side views of live wild-type (A–D) and mutant (E–H) embryos at the following time points: (A,E) 45 min post fertilization (p.f.), (B,F) 1.25 hours p.f., (C,G) 2.25 hours p.f., (D,H) 3 hours p.f. (corresponding, in wild-type, to the 2-, 8-, 128- and 1000-cell stages, respectively). Maternally mutant cea embryos show a normal first cell division (E) but fail in a fraction of subsequent divisions (F, arrowhead), leading to blastula with irregularly-sized blastomeres (G) and largely syncytial embryos (H). (I–L) Animal views of fixed wild-type (I,K) and mutant (J,L) embryos labeled to detect β-catenin (green) and DNA (red). At 1.25 hours p.f. (8-cell stage in wild-type; I,J), the mutant embryo reveals a failure of DNA and cell division in a fraction of the blastomeres (arrowheads indicate nuclei of cells that did not undergo division). At 3 hours p.f. (1,000 cell stage in wild-type; K,L), mutant embryos exhibit enlarged nuclei (inset in (L), compare to (K)).
Although our analysis clearly showed defects in cell division at the second and subsequent cell cycles, it did not allow us to determine whether maternally mutant cea embryos also result in a defective first cell division. This is because, in the zebrafish, clutches obtained through pair matings typically contain a variable fraction of unfertilized eggs, which fail to form a furrow at a time corresponding to the first cell cleavage and are therefore undistinguishable from embryos with potential defects in furrow initiation during the first cell cycle. In order to overcome this uncertainty, we analyzed clutches from cea mutant females after in vitro fertilization (IVF) with a concentrated sperm solution, which allowed us to obtain fertilization rates reaching nearly 100%. These clutches exhibit a normal first cell division in nearly 100% of the embryos, but in subsequent cell cycles show low fractions of successful cell divisions, ranging from 9% to 41% (Table 1). Moreover, in clutches exhibiting different strengths of the mutant phenotype, the percentages of successful cell divisions in the first and subsequent cell cycles do not correlate (compare crosses 1 and 2 in Table 1), suggesting that these two fractions depend on different variables (i.e. the effectiveness of the IVF method for the first cell division and maternal cea function for subsequent divisions). Together, our observations suggest that there is a differential requirement on maternal cea function during early development, such that it is not required during the first cell cycle but has a role in subsequent cell divisions.
Table 1.
Incidence of cell division in 1st and 2nd cell cycles in embryos from mutant cea females1.
| 1st cell cycle (45 min p.f.) | 2nd cell cycle (60 min p.f.) | |||||||
|---|---|---|---|---|---|---|---|---|
| no cleavage (%) 2 | normal 1st cleavage (2-cell) (%) | Two-cell (%) 3 | 3-cells (%) 4 | 4-cells (%) 5 | overall normal cleavage (%) 6 | cleavage fertilized eggs (%) 7 | n | |
| cea female #1 | 6 | 94 | 61 | 20 | 13 | 23 | 24 | 155 |
| cea female #2 | 3 | 97 | 5 | 31 | 61 | 77 | 79 | 150 |
| AB female 8 | 9 | 91 | 0 | 0 | 91 | 91 | 100 | 169 |
Embryos were fertilized through in vitro fertilization using wild-type (AB) sperm. Maternal-effect mutations often result in different degrees in the penetrance and expressivity of a phenotype (see, for example, Mohler and Wieschaus; 1986; Lehmann and Nüsslein-Volhard; 1991; Pelegri et al., 1999; Pelegri et al., 2004), as reflected in the clutches from the two cea mutant females shown in this table.
No furrow formation, characteristic of either unfertilized embryos or embryos defective in the 1st cell division. These embryos never undergo cleavage and are presumed unfertilized (see text).
2nd cell division cycle fails in both blastomeres
2nd cell division cycle fails in only one of the two blastomeres
Normal 2nd cell division in both blastomeres
Overall rate of successful cleavage for the 2nd cell cycle in the egg clutch, calculated using the percentages in (3–5) and considering there are two independent cell divisions during this cycle
Overall rate of successful cleavage for the 2nd cell cycle amongst fertilized embryos, assuming that all eggs without normal cleavage during the 1st cell cycle (in (2)) are unfertilized
Results pooled from two different AB females
Maternal cea function is required for mitosis
In order to determine whether the defect in maternally mutant cea embryos occurs in mitosis or the initiation of cytokinesis, we fixed such embryos at the 8-cell stage and labeled the fixed embryos to detect intercellular membranes, using an antibody against the cell adhesion component β-catenin (Jesuthasan, 1998; Urven et al., 2006), and nuclei using a DNA stain. Imaging of the labeled embryos shows that a fraction of the cells fail to accumulate β-catenin at the forming furrow (Fig. 1J, compare to I). Similar analysis to detect f-actin, which also accumulates at furrow cell adhesion junctions (Kishimoto et al., 2004; Urven et al., 2006), shows a similar defect (not shown). These observations further support a defect in cell division in maternally mutant cea embryos.
The DNA label allowed us to observe that in maternally mutant cea embryos the fraction of cells that fails to undergo cell division also exhibits defects in karyokinesis. In these cells, nuclei do not divide into two daughter nuclei, but remain instead as a single nuclear mass in the center of the cell (arrowheads in Fig. 1J). As expected from its nuclear and cell division defects, at later stages of development, maternally mutant cea embryos show a reduced number of cells containing a small number of nuclei (Fig. 1L, compare to K), which are typically larger than nuclei in wild-type at the same developmental stage (inset in Figs 1K,L; see below). Thus, the cell division phenotype observed in maternal cea mutants is associated with a defect in nuclear division.
Maternal cea function is essential for spindle formation and centrosome duplication
The mitotic spindle has been shown to be involved in both nuclear DNA segregation and, together with astral microtubules, the induction of the cleavage furrow (reviewed in Glotzer, 2005; Rappaport, 1996). We therefore tested for defects in spindle formation in maternally mutant cea embryos by labeling fixed samples using an anti α-tubulin antibody. During the first cell cleavage cycle, similar to the case of the live mutant phenotype, the spindle apparatus is indistinguishable between wild-type embryos (Fig. 2A) and maternally mutant cea embryos (Fig. 2C). However, at the second cell cleavage cycle, a fraction of cells in maternally mutant cea embryos shows defects in the formation of the mitotic spindle. Instead of exhibiting the bipolar spindle structure, which normally occurs synchronously in all cells of the embryo during the cleavage stages (Fig. 2B,B′ and not shown, see also Dekens et al., 2003), a fraction of cells in maternally mutant cea embryos contains a monopolar microtubule apparatus (shown in metaphase in figure Fig. 2D,D′). During telophase, astral microtubules increase in length as in wild-type embryos, with the exception that they form a monoaster and not the normal diastral arrangement (data not shown). Thus, maternally mutant cea embryos show defects in the organization of the mitotic spindle apparatus, and this defect likely underlies subsequent effects on karyokinesis and furrow initiation.
Figure 2.
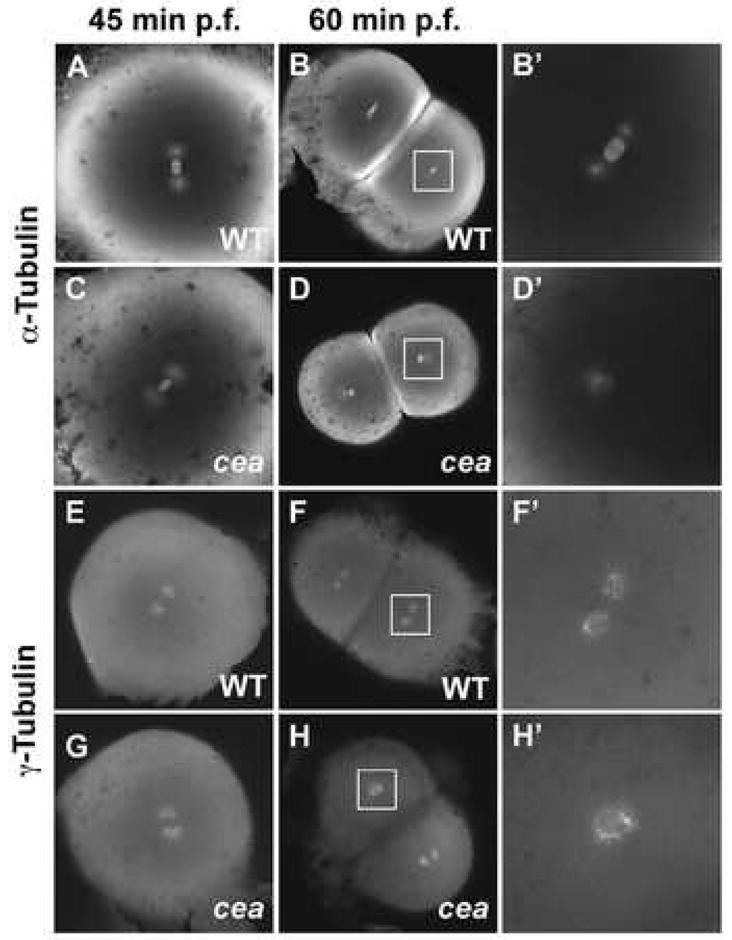
Spindle organization and centrosome duplication defects in maternally mutant cea embryos. (A–D′) Fixed wild-type (A–B′) and mutant (C–D′) embryos labeled with anti-α-tubulin antibody. Spindle organization is normal in the mutant immediately prior to the first cell division (35 min p.f.; (C); compare to (A)), but is defective in a fraction of blastomeres in the next cleavage cycle (65 min p.f.; (D,D′); compare to (B,B′)). (E–H′) Fixed wild-type (E–F′) and mutant (G–H′) embryos labeled with anti-γ-tubulin antibody. Centrosome duplication appears normal immediately prior to the first cleavage division (35 min. p.f.; (G), compare to (E)) but is defective in a fraction of the blastomeres in the following cleavage cycle (65 min. p.f.; (H,H′), compare to (F,F′)). Animal views. (B′,D′,F′,H′) are enlargements of the area indicated by the squares in (B,D,F,H), respectively.
Because in some cell types the organization of the mitotic spindle depends on the presence of functional pairs of centrosomes, we tested whether centrosomes are affected in maternally mutant cea embryos. Wild-type and maternally mutant cea embryos were fixed during the early cell division cycles and were labeled to detect centrosomes using antibodies against γ-tubulin (Bornens, 2002; Dekens et al., 2003). In wild-type embryos, a centrosomal pair can be observed in each blastomere (Fig. 2E,F,F′). At the one-cell stage, maternally mutant cea embryos show an apparently normal pair of centrosomes (Fig. 2G), which is consistent with the normal spindle morphology and cell division associated with the first cell cycle in these embryos. However, beginning at the second cell cycle, a fraction of the blastomeres in these embryos lack a centrosomal pair and instead exhibit a single centrosomal structure (Fig. 2H,H′). Thus, a mutation in cea results in defects in centrosome duplication, which likely results in defects in spindle organization and cell division. These phenotypes are similar to those produced by a reduction of the function of centriolar components in the C. elegans germ line (Dammermann et al., 2004; Delattre et al., 2004; Hammill et al., 2002; Kemp et al., 2004; Kirkham et al., 2003; Leidel et al., 2005; Leidel and Gönczy, 2003; O’Connell et al., 2001).
cellular atoll allele encodes Sas-6
In order to better understand the function of cea at the molecular level, we determined the identity of this gene through a candidate gene approach. Previous studies had established that the cea locus was linked to the SSLP marker Z10673, located in the zebrafish linkage group 22 (Dosch et al., 2004). Further mapping refined the critical region containing cea to an approximately 14 cM interval between markers z10673 and z51215 (Fig. 3A). Analysis of the available sequence databases showed the presence of the candidate gene sas-6 in this region, whose homologues in C. elegans and humans had been shown to encode a coiled-coil centriolar component required for centrosome duplication (Dammermann et al., 2004; Leidel et al., 2005). Sequencing of PCR-amplified sas-6 transcripts from maternal zebrafish cDNA shows the presence of two splice variants: a form corresponding to the Sas-6 protein reported in other organisms and a longer version with a stretch of 12 unique amino acids beginning at position 518 (Fig. 3B). We refer to these splice forms as Sas-6 Short (Sas-6S) and Sas-6 Long (Sas-6L).
Figure 3.
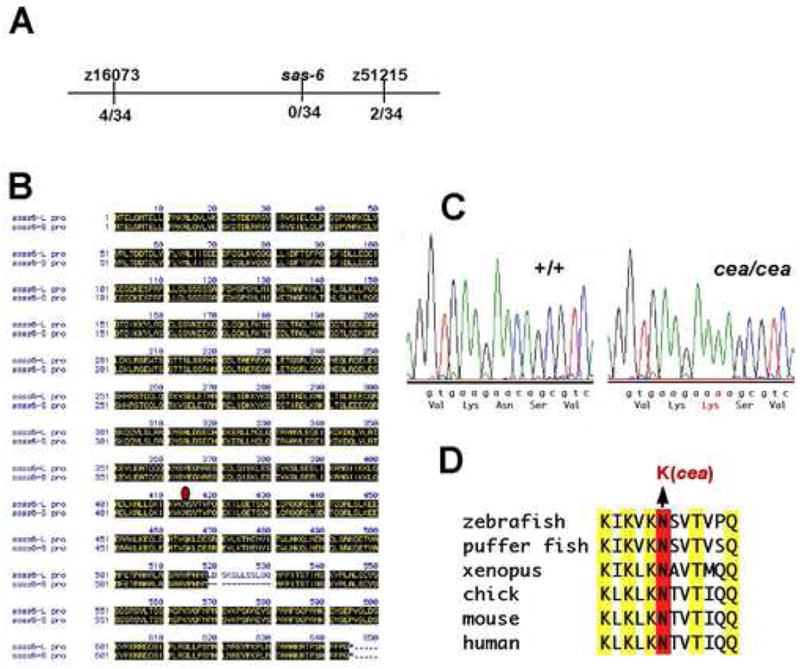
Molecular identification of cea. (A) Linkage map of cea. The number of recombinants in the indicated number of analyzed meiosis is indicated. (B) Protein sequence for the Sas-6L and Sas-6S alternative splice forms. The additional 12 amino acids in the Sas-6L form, starting in position 518, are shown. The position of the identified mutation at position 414 is indicated by a red diamond. (C) DNA sequence traces of the wild-type and mutated cea allele. The DNA base and amino acid substitutions are indicated in red. (D). Protein sequence comparison in the region encompassing the mutation, showing that the mutation results in the substitution of a highly conserved amino acid.
Sequencing of PCR-amplified mRNA from embryos derived from females homozygous for either the cea mutant allele or the wild-type allele (identified through their live phenotype and genotyping) showed the presence of one DNA base substitution that results in an Asn to Lys amino acid change at position 414 of the protein (Fig. 3B,C). Segregation analysis showed that this base substitution is fully linked to the cea mutation (Fig. 3A and data not shown). Sequence comparison of Sas-6 in other species showed that Asn at this position is absolutely conserved in all analyzed vertebrate species (Fig. 3D), suggesting that the replacement of this conserved amino acid may interfere with the function of the protein.
In order to verify that cea codes for Sas-6, we attempted to rescue the cea mutant phenotype through the expression of wild-type Sas-6 product. Clutches from cea mutant mothers were injected at the one-cell stage with either control β-galactosidase or a mixture of wild-type mRNA corresponding to Sas-6S and Sas-6L (Fig. 4; Table 2). As expected, uninjected embryos and control-injected embryos exhibited cell division defects during the cleavage stage, resulting in irregularly cellularized blastulae (Fig. 4A,E). On the other hand, embryos injected with the wild-type sas-6 mRNA mixture divided to produce apparently normal blastulae, which could proceed through gastrulation (Fig. 4B,F), showing that wild-type Sas-6 product can rescue the genetic defect corresponding to the cea mutation. mRNA corresponding to either splice form, when injected separately, was capable of rescuing the cea mutant phenotype to similar extents (Fig. 4C,D,G,H; Table 2), suggesting that, at least during the cleavage stage, both short and long forms of the zebrafish Sas-6 protein have similar functional activity.
Figure 4.

Genetic rescue of the maternal cea mutant phenotype by sas-6 expression. Side views of live embryos at the 1,000 cell stage (A–D,I–K) and animal views of fixed embryos labeled to detect β-catenin and DNA (E–H,L–N). Injection of mRNA coding for Sas-6L and Sas-6S results in a similar extent of rescue (B–D,F–H; compare to (A,E)). Injection of mRNA coding for the mutated cea protein results in significant rescue ((K,N); compare to (I,L)), which is nevertheless not as effective as the rescue conferred by wild-type mRNA injected in sibling embryos (J,M).
Table 2.
Genetic rescue of the maternal cea mutant phenotype by expression of sas-6 mRNA
| RNA injected 1 | Lysed (%) 2 | Defective epiboly (%) 2 | normal epiboly (%) 2 | n |
|---|---|---|---|---|
| β-gal (200pg) | 100 | 0 | 0 | 44 |
| sas-6S+L (100pg each) | 3 | 0 | 97 | 58 |
| sas-6S (200pg) | 3 | 3 | 94 | 39 |
| sas-6L (200pg) | 0 | 0 | 100 | 41 |
| sas-6S+L(mutant) (100pg each) | 31 | 65 | 4 | 93 |
| mCherry-sas-6S (400pg) | 4 | 7 | 89 | 35 |
| mCherry-sas-6L (400pg) | 7 | 0 | 93 | 28 |
RNA was injected in early one cell stage embryos obtained from same cross between a homozygous cea mutant female and AB male.
Phenotypes were determined at 11hpf. Lysed embryos represent a mixture of cellularization defects and failure to initiate epiboly.
We compared the ability of wild-type and mutant mRNA to rescue the cea mutant phenotype. At similar amounts of injected mRNA, the efficiency of rescue by mutant mRNA was lower than that caused by wild-type mRNA, although mutant mRNA did result in significant rescue when compared to control mRNA injections (Fig. 4K,N, compare to I,J,L,M; Table 2). These results indicate that the molecular lesion in the cea mutant allele produces a hypomorphic product that retains some level of partial function. Together, our results indicate that cea encodes Sas-6.
Expression of cellular atoll/sas-6
In order to better understand the function of cea/sas-6, we determined the expression pattern of this gene at various developmental stages during embryogenesis (Fig. 5). RT-PCR analysis shows that both sas-6 mRNA splice forms are present in freshly laid eggs and during embryogenesis (Fig. 5A). Whole mount in situ hybridization analysis indicated the presence of ubiquitously distributed cea/sas-6 transcripts during the early cleavage stages (Fig. 5B, compare to sense probe control in C). The presence of these transcripts immediately after fertilization and several hours prior to the activation of the zygotic genome at the midblastula transcription (2.5–3.3 hours p.f.; Kane and Kimmel, 1993; Mathavan et al., 2005; Zamir et al., 1997) indicate their maternal origin. High levels of uniformly distributed transcripts are present until the end of the blastula period (Fig. 5D). During the gastrulation stages, embryos exhibit significantly lower, yet detectable, levels of ubiquitously distributed transcript (Fig. 5E,F). At the tail bud stage, transcripts begin to become enriched in the prospective brain region (Fig. 5G). Levels of expression appear to increase in the central nervous system of the 24-hour embryo (Fig. 5H). Thus, consistent with its maternal effect, cea/sas-6 exhibits a strongly enriched maternal expression pattern, in addition to expression in a subset of zygotic structures (see Discussion).
Figure 5.
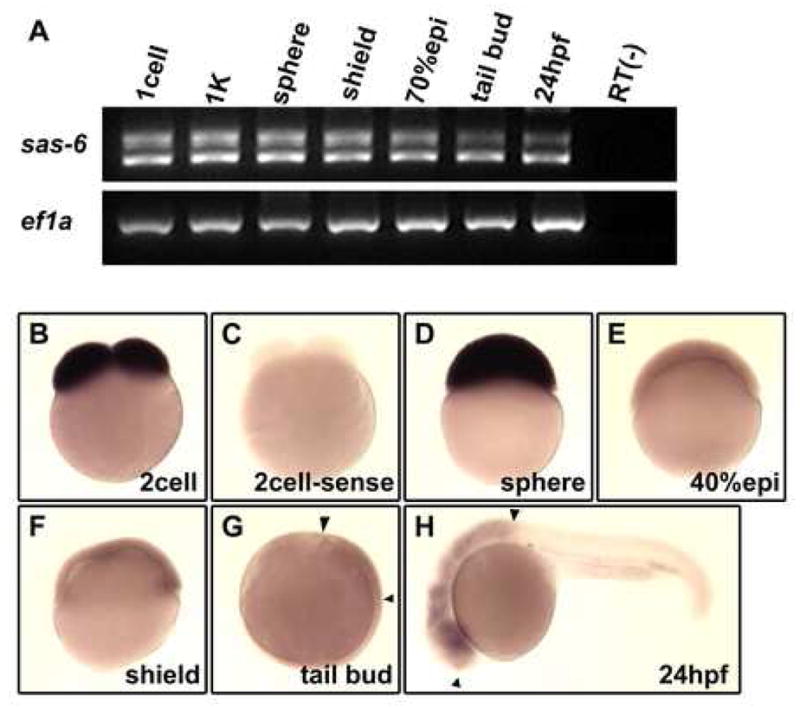
Expression of zebrafish cea/sas-6 during early embryogenesis. (A) RT-PCR analysis shows that both splice forms are expressed maternally and present during early embryogenesis. (B–H) In situ hybridization analysis shows the presence of maternal transcripts ((B); compared to control sense probe in (C)), which remain until the sphere stage (D), and subsequently a lower level of ubiquitous expression during gastrulation (E–F) and enrichments in the brain region (G,H; flanked by arrowheads).
Cea/Sas-6 protein localizes to centrosomes
We next tested whether zebrafish Sas-6 protein localizes to the centrosome. We constructed in-frame fusions between both zebrafish Sas-6S and Sas-6L splice forms and the Red Fluorescent Protein (RFP) monomeric derivative mCherry, and mRNA for these constructs were separately injected into one-cell stage embryos. Injected embryos were fixed at 50% epiboly (5 hours p.f.), when the translation of the chimeric product resulted in sufficient RFP fluorescence for analysis, and were additionally processed to label γ-tubulin (Dekens et al., 2003), and DNA. Imaging of the colabelled embryos showed that, for both Sas-6 splice forms, the RFP tag colocalizes with γ-tubulin in specific subcellular structures (Fig. 6 and data not shown). Separate injections showed that the mCherry-Sas-6 fusion proteins are functional (Table 2). These results indicate that zebrafish Sas-6 splice forms encode centrosomal components.
Figure 6.
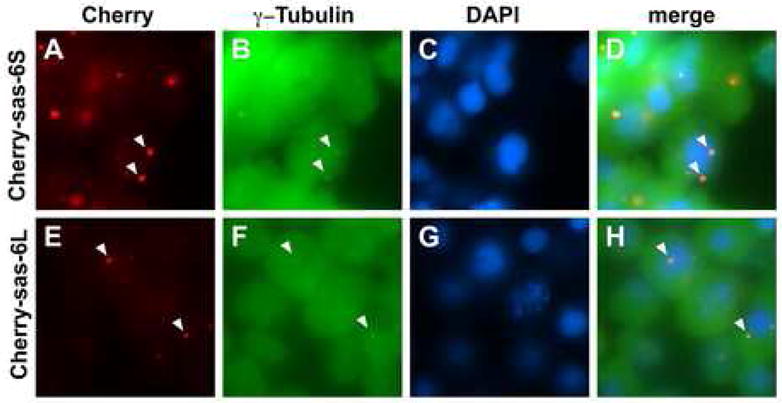
Cea/Sas-6 localizes to centrosomes. Expressed fusions of mCherry and Sas-6 splice forms colocalize cytoplasmic structures containing γ-tubulin (arrowheads), a marker for centrosomes. Fields of cells in embryos fixed at 50% epiboly.
cellular atoll is required paternally for the first cell cleavage division
Previous studies have shown that mutations in centriolar components in C. elegans, in addition to resulting in maternal-effect defects, also result in paternal effects because of defects in centriole production during spermatogenesis (O’Connell et al., 2001). Moreover, zebrafish sperm, as is the case in C. elegans and many other animal species, contain a pair of centrioles (Kessel et al., 1983), indicating that centriole duplication is likely essential for functional sperm formation in this vertebrate system. We therefore tested the possibility that cea might also have a function during zebrafish spermatogenesis, and consequently that zygotes derived from sperm from cea fathers may exhibit defects in the first cell division. As in C. elegans, if such zygotes are derived from wild-type eggs, such paternal-effect defects may be temporary, since centriole duplication would ensue in subsequent cycles due to the presence of maternally provided centrosomal components. Thus, embryos derived from crosses between cea mutant fathers and wild-type females, which we refer to as “paternally mutant cea embryos”, would be expected to undergo a delay in the first cell division, and initiate, at a time coincident with the second cell cycle, an otherwise normal cleavage pattern.
In order to test this hypothesis, we analyzed paternally mutant cea embryos that were synchronized by the collection of freshly laid eggs at 3-minute intervals. Under normal conditions, wild-type embryos exhibit synchronous cleavage cycles, initiating at 40 minutes post fertilization (p.f.) and undergoing subsequent cell cycles every 20 minutes (Kimmel et al., 1995). Embryos from wild-type females and cea mutant males were sorted at 45–50 minutes p.f. into two classes, those that had undergone a normal first cell division, and those that did not show sign of furrow ingression. The latter class of embryos was expected to consist of unfertilized eggs, which do not show a symmetrically located cell furrow, as well as, potentially, embryos that were experiencing a delay in their first cell division cycle. As predicted by this hypothesis, a fraction of eggs derived from mutant cea sperm that did not undergo the first cell cleavage (Fig. 7C, compare to A) initiated a normal cell division pattern at a time corresponding to the second cell division in unaffected siblings (Fig. 7D,D′, compare to B,B′; see also Table 3). This fraction was variable in crosses from different homozygous mutant males, ranging from 0 to 5%. This incompletely penetrant phenotype is likely a consequence of partial function in the cea hypomorphic allele, possibly coupled to the requirement for at least one centriole for the formation of sperm flagella (reviewed in Snell et al., 2004). Screening of a large number of similarly scored embryos from a wild-type AB stock or sibling heterozygous cea/+ females failed to show any instances of a similar cell division delay in these embryos. Similar to the case of C. elegans embryos, after the one-cycle delay in the first cell division, all paternally mutant cea embryos show a normal cleavage pattern in subsequent cell cycles (Table 3 and data not shown), suggesting that the second and subsequent cell divisions do not depend on paternal cea function. Together with the apparently exclusive effect of maternal cea/sas-6 function starting at the two-cell cycle, our data are consistent with the idea that cea/sas-6 function during spermatogenesis is involved in the formation of centriole pairs in sperm cells, which drive the first cell division cycle, and that maternally provided Cea/Sas-6 product is required for centriole duplication in subsequent cell cycles.
Figure 7.
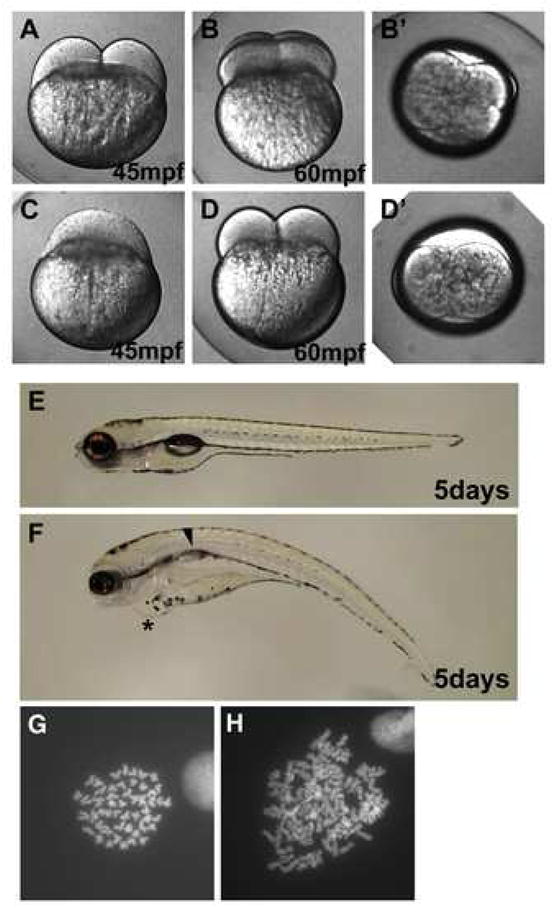
One-cycle cell division delay in embryos from mutant cea fathers. Live embryos at the indicated time points. (A,B,C,D,E,F) are side views, (B′,D′) are animal views of the same embryos shown in (B,D). A fraction of paternally mutant cea embryos show a one-cycle delay in cell division ((C); compare to (A)), but subsequently resume normal cleavage (D,D′). At 5 days p.f., embryos that undergo a one-cycle delay in cell division (F) exhibit heart edema (asterisk) and lack swim-bladder inflation (arrow). (G–H) Metaphase spreads from 24-hour embryos. Wild-type embryos contain a normal complement of 50 chromosomes (G), while paternally mutant cea embryos that experience a one-cell cycle delay in the first cell division contain a duplicated number of chromosomes (H).
Table 3.
Paternally mutant cea embryos can exhibit a one-cycle delay in their first cellular division
| Cross 1 | Delayed 1st cleavage (%) | n 2 |
|---|---|---|
| cea/cea male 1 | 5.0 | 259 |
| cea/cea male 2 | 1.4 | 358 |
| cea/cea male 3 | 0.9 | 336 |
| cea/cea male 4 | 0.3 | 318 |
| cea/cea male 5 | 0 | 211 |
| +/+ male 1 | 0 | 307 |
| +/+ male 2 | 0 | 331 |
| +/+ male 3 | 0 | 220 |
| +/+ male 4 | 0 | 169 |
| AB males (pool) | 0 | 745 |
All crosses are between individual genotypically identified males that are either homozygous for the mutant (cea/cea) or wild-type (+/+) cea alleles, against phenotypically normal sibling females (either cea/+ heterozygotes or +/+ homozygotes), with the exception of the AB males, which were collected as a mixed pool from multiple crosses between wild-type males and females of the AB stock.
Total number of individuals that showed a regular cleavage pattern and are thus presumed to be derived from fertilized eggs, either with or without a delayed 1st cleavage.
Mutant cea/sas-6 sperm promote changes in ploidy number
Paternally mutant cea embryos that resume cleavage after stalling in the first cell cycle develop an apparently normally patterned body plan. However, such embryos are not viable, showing an invariable phenotype at 5 days p.f. consisting of heart edema and a lack of swim bladder inflation (Fig. 7F, compare to E; see below). This lethal phenotype suggested the possibility that, in such embryos, DNA synthesis in the absence of the first cytokinesis results in a genome-wide duplication leading to tetraploidy, a condition which in animal embryos is often associated with specific syndromes and inviability (Kaufman, 1991; Pellestor, 1995). We tested this possibility by chromosome counts of metaphase spreads from 24-hour embryos. Cells from wild-type embryos contained the normal diploid chromosome complement, i.e. a total of 50 chromosomes (Daga et al., 1996; Fig. 7G; 36 metaphase spreads from 8 embryos). On the other hand, cells from all tested embryos derived from cea mutant males and which experienced a one-cycle delay in the first cell division contained a doubled number of chromosomes, consistent with these embryos being tetraploid (Fig. 7H; 24 metaphase spreads from 3 embryos). In metaphase spreads from these embryos, chromosomal counts higher than the tetraploid component were not observed, supporting the notion that the genome duplication event occurred only once, during the first cell cycle. As expected, the ploidy change in paternally mutant cea embryos is dependent on the delay in the first cell division, as sibling embryos that did not experience this delay contained a normal (diploid) chromosomal content (not shown, 17 metaphase spreads from 5 embryos).
We further addressed the hypothesis that the stalling of the first cell division observed in paternally mutant cea/sas-6 embryos results in whole genome diploidization by testing whether cea mutant sperm can induce gynogenetic development. Mature zebrafish eggs can be fertilized in vitro with UV-treated sperm, where the DNA has been inactivated, leading to the production of haploid embryos (Streisinger et al., 1981). Although haploid embryos do not survive past the third day of development, haploid zygotes can become fully viable diploids after genome duplication through the inhibition of the first cell division by heat shock or hydrostatic pressure (Streisinger et al., 1981). We reasoned that, if sperm derived from cea fathers can result in the inhibition of the first cell division without interfering with DNA synthesis, such sperm may promote gynogenetic development in the absence of additional treatment to prevent cell division. We tested this possibility using mature eggs from wild-type females homozygous for the albino mutation, a genetic marker used to corroborate that the resulting diploid embryos do not contain paternal DNA (Fig. 8A). As expected, albino-derived eggs fertilized with UV-treated wild-type sperm did not exhibit a delay in the first cell division, and all of them developed into albino haploid embryos (Fig. 8D; Table 4). On the other hand, and similar to the case using untreated sperm, fertilization of albino-derived eggs using UV-treated sperm from cea mutant fathers resulted in clutches where a fraction of the embryos exhibited a one-cycle delay in the first cell division (Table 4). As predicted by our hypothesis, such zygotes developed into albino diploid embryos, as judged from their morphology at days 3–5 p.f. (Fig. 8E, compare to B,C; Table 4). Together, our data show that the one-cycle delay of the first cell division observed in embryos from cea mutant fathers results in the duplication of the full genomic complement.
Figure 8.

Induction of gynogenesis by sperm from cea mutant males. (A) Diagram of gynogenesis procedure. (B–E) Side views of 3 day p.f. embryos. For comparison, (B) and (C) show, respectively, control diploid wild-type and albino pigmented derived from standard natural crosses. (D) albino haploid embryo derived from fertilization of albino females with UV-inactivated wild-type sperm. (E) A fraction of embryos fertilized with UV-inactivated sperm from cea mutant males develop into albino diploid gynogenotes.
Table 4.
One-cycle delay in the 1st cell division induced by UV-irradiated sperm from cea mutant fathers is associated with the promotion of diploid gynogenotes.
| Source of sperm (1) | 1st cleavage | Phenotype at 3 days p.f. | ||
|---|---|---|---|---|
| Lysed (2) or haploid | Diploid | |||
| cea/cea male | Normal cleavage | 1558 | 1558 | 0 |
| delayed 1st cleavage | 11 | 1 (3) | 10 | |
| AB male | Normal cleavage | 1249 | 1249 | 0 |
| delayed 1st cleavage | 0 | 0 | 0 | |
Sperm was UV-irradiated and used for in vitro fertilization of oocytes from females homozygous for the albino mutation. A small fraction of non-albino embryos, likely derived from sperm whose DNA was incompletely affected by the UV treatment, has been excluded from the analysis.
Typically a large fraction of haploid embryos lyse prior to 24 hours p.f. and do not survive to 3 days p.f. (Walker, 1999).
This specific embryo died early in development (< 24 hours p.f.) and it is presumed to be a diploid gynogenote that is inviable due to ineffective diploidization or homozygosis for a preexisting lethal mutation.
Germ plasm recruitment in cells affected by the cea mutation
In maternally mutant cea embryos, cells that fail to undergo mitosis during a given cell cycle can also resume mitosis in subsequent cycles. Cycles that occur after the resumption of mitosis occur in a manner synchronized with other cells in the embryo, suggesting that cells are responding to a global cell cycle oscillator. The stalling and resumption of cell division that occurs in maternally mutant cea embryos allows uncoupling developmental time from cell division count during early embryogenesis. This ability led us to investigate the role of cell division versus developmental time in the recruitment of germ plasm to the forming furrows. In wild-type embryos, germ plasm mRNA components become recruited as stable structures to the furrows of the first and second cleavage cycles, but not the furrows of subsequent cell cycles (Yoon et al., 1997; Fig. 9A–C). The differential germ plasm recruitment during different cell cycles may depend on the number of cell divisions. Such a model predicts that germ plasm should retain the ability to become recruited to furrows of cells that resume cleavage after a one-cycle delay in the second cell division. Alternatively, the ability of germ plasm aggregates to become recruited to the furrow may depend on developmental time rather than the number of cell divisions experienced by the cell, a model that predicts a failure of germ plasm recruitment in cells that stall in the second cleavage, even if they resume cell division in subsequent cycles. As expected, in maternally mutant cea embryos, germ plasm recruitment occurs normally during the first cell division (Fig. 9D), and does not occur when the furrow fails to form in the second cell cycle (Fig. 9E, arrowhead). Remarkably, in such mutants, germ plasm recruitment is absent in cells that resume cell division after a one-cycle delay in the second cleavage (Fig. 9F, arrowhead). This finding suggests that germ plasm recruitment to the furrow depends on developmental time rather than the absolute number of cell divisions (see Discussion).
Figure 9.

Germ plasm recruitment fails after a one-cycle delay and cleavage resumption. In situ hybridization to label vasa mRNA in wild-type (A–C) and maternally mutant cea embryos (D–F) at the 45 min p.f. (2-cell in wild-type; A,D), 1 hour p.f. (4-cell in wild-type; B,E) and 1.25 hours p.f. (8-cell in wild-type; C,F). In wild-type embryos, germ plasm accumulates at the furrows of the first and second cell divisions (A,B), but not for the third cell division (C). In mutant embryos, germ plasm accumulates normally in the furrow of the first cell division (D), but fails to accumulate in both furrows that do not form in the second cell cycle (E, arrowhead) and furrows that form after a one-cycle delay of the second cleavage division (F, arrowhead). 1st, 2nd and 3rd indicate the cleavage furrows of the first, second and third cleavage cycle.
Discussion
Our analysis indicates that cellular atoll encodes the centriolar component Sas-6, which when mutated results in centrosome duplication defects in the embryo that likely underlie defects associated with this mutation, such as in spindle formation, and nuclear and cell division. To our knowledge, these studies also constitute the first report of a gene identified as maternal-effect mutation for which a specific molecular lesion can be attributed to the mutant phenotype, demonstrating the applicability of forward genetics to the analysis of early vertebrate development.
cellular atoll encodes the centriolar component Sas-6
Molecular identification of the maternal-effect gene cea indicates that it encodes the centriolar component Sas-6. sas-6 is one of a handful of genes that have been shown in several systems to encode centrosomal components (reviewed in Bornens, 2002). One of the best studied systems of centrosome duplication is in the nematode C. elegans, where the kinase ZYG-1 and the coiled-coil proteins SAS-4, SAS-5, SAS-6 and SPD-2 have been found to be components of the centriole and to be required for its duplication (reviewed in Delattre and Gönczy, 2004; Leidel and Gönczy, 2005). Studies in this organism suggest a pathway of sequential incorporation of centriolar components in the following order: SPD-2, ZYG-1, SAS5/SAS-6 complexes, and SAS-4 (Delattre et al., 2006; Pelletier et al., 2006). Potential human homologues of SAS-4 (CPAP; Andersen et al., 2003; Hung et al., 2000) and SAS-6 (HsSAS-6; Andersen et al., 2003; Dammermann et al., 2004; Leidel et al., 2005) have also been found localized to centrosomes. Both in C. elegans and human cells, a reduction in SAS-6 function results in defects in centrosome duplication and spindle formation (Dammermann et al., 2004; Leidel et al., 2005), and ultrastructural studies have shown that this defect is based on a requirement for SAS-6 in daughter centriole tube formation and elongation (Dammermann et al., 2004; Pelletier et al., 2006).
We find that cea/sas-6 is also required for centrosomal duplication in the zebrafish embryo, and that expressed Cea/Sas-6 protein becomes part of the centrosome. By analogy to the centriolar pathway established in C. elegans, we hypothesize that the defect in centrosomal duplication observed in cea mutants derives from a defect in the formation of a daughter centriole from a mother centriole. However, ultrastructural studies will be required to conclusively test this hypothesis. The failure in centrosome duplication in embryos from cea mutant mothers is associated with defects in spindle formation, and this, in turn, explains the lack of segregation of DNA during karyokinesis as well as the lack of cell furrow formation.
The molecular identification of a maternal-effect mutation in cellular atoll gene has led us to find a precise lesion in a conserved amino acid of the gene sas-6. However, our studies indicate that the maternal-effect mutated cea allele retains partial functional activity, as shown by the weak rescue of the mutant phenotype by expression of the mutated protein. This interpretation is also consistent with the incomplete penetrance of the maternal-effect cell division defects in the early embryo.
Centrosomal inheritance in the zebrafish oocyte-to-embryo transition
The maternal-effect cea phenotype is observed only in the second and subsequent cell cycles, and not in the first cell cycle. Conversely, paternal cea function is required for the first cell division of the embryo, but not for subsequent cell divisions. The maternal and paternal genetic requirements for the zebrafish cea gene are similar to those for the function of centriolar components in C. elegans (O’Connell et al., 2001), and corroborate classical work in sea urchin that established that the ability of centrosomes to lead to a productive cell division correlates with the presence of a pair of centrioles prior to the mitotic cycle (Mazia et al., 1960; Sluder and Rieder, 1985). Furthermore, these functional requirements are in agreement with the known behavior of centrioles during fertilization in many animal species, including the zebrafish (Kessel et al., 1983), where a pair of centrioles is provided by the sperm to a centriole-less oocyte (Fig. 10A, based on previous studies in C. elegans by O’Connell et al., 2001; reviewed in Leidel and Gönczy, 2005). In this strategy, which is essential to avoid an increase in the number of centrioles every generation, the first cell division depends on sperm-derived centrioles, while subsequent cell divisions depend on centrioles produced in the developing embryo. As with all early developmental processes that occur prior to the initiation of zygotic transcription at the midblastula transition (MBT), which in zebrafish occurs at about the 1,000 cell stage (Kane and Kimmel, 1993; Mathavan et al., 2005; Zamir et al., 1997), the formation of new centrioles and centrosomes in the early embryo is expected to rely solely on maternal components produced during oogenesis.
Figure 10.

Model for the inheritance of centrosomal components during the oocyte-to-embryo transition, based on previous studies in C. elegans (O’Connell et al., 2001; reviewed in Leidel and Gönczy, 2005), and its consequences for cell division and ploidy number. (A–C) Fertilization with untreated sperm: wild-type embryos (A), maternally mutant cea embryos (B), paternally mutant cea embryos (C). (D,E) Fertilization with UV-inactivated sperm from wild-type (D) and cea mutant fathers. Fertilization of sperm from cea mutant fathers associated with ongoing DNA synthesis and a one-cell cycle delay in cell division is proposed to result in ploidy changes: tetraploidy (C) with untreated sperm (after the initial production of a diploid zygote), and gynogenetic diploidization (F) with UV-inactivated sperm (after the initial formation of a haploid zygote). Sperm-derived centrioles are shown in green, newly synthesized centrioles in red, DNA in blue.
As in the case of C. elegans (Dammermann et al., 2004; Leidel et al., 2005), the formation of a pair of centrosome-like structures and the bipolar spindle for the first cell cleavage does not appear to be affected by the lack of maternal zebrafish cea/sas-6 function, a condition expected to result in the presence of a single centriole in each centrosome (Fig. 10B). Thus, in both organisms centrosome-like structures containing a single centriole appear capable of organizing each pole of the spindle apparatus. On the other hand, the first cell cycle is defective in paternally mutant cea embryos, which is consistent with the idea that these embryos are derived from sperm that contain a single centriole, and which can neither undergo centrosomal splitting nor form a bipolar spindle (Fig. 10C).
The function of cea/sas-6 during later stages of embryonic development remains unclear. Individuals zygotically homozygous for the cea mutation appear to be fully viable and morphologically normal, suggesting the possibility that only maternal cea/sas-6 genetic contribution is essential for embryonic development. However, given that the cea allele retains some wild-type function, and that cea/sas-6 mRNA is also present after zygotic gene activation at MBT, it is possible that Cea/Sas-6 product derived from new zygotic transcription is also required for embryonic development, and that the apparent strict maternal effect is a consequence of a higher functional threshold requirement for cea/sas-6 activity during the rapid early cell cleavage divisions. As a non-exclusive alternative, a small fraction of cells in zygotically homozygous cea mutant embryos may exhibit defective cell divisions, which nevertheless do not significantly affect embryonic viability. Morpholino-conjugated oligonucleotide (MO)-mediated knock-down (Nasevisius and Ekker, 2000) targeted against cea/sas-6 has failed to yield apparent specific defects on early zebrafish embryos, and searches of the zebrafish genome databases have failed to reveal a second cea gene copy, which may redundantly contribute maternal and/or zygotic cea function (T.Y., X.G. and F.P., unpublished). A better understanding of the role of cea/sas-6 function awaits the identification and analysis of null alleles of this gene.
Paternal cellular atoll/sas-6 dysfunction promotes genome duplication
At the blastula stage, maternally mutant cea/sas-6 embryos exhibit nuclei whose size, which is normally correlated to DNA content (Cavalier-Smith, 1978), is significantly larger than that observed in wild-type embryos. This suggests that, in the zebrafish embryo, the inhibition of cell division caused by the lack of centrosome duplication does not affect entry into S-phase and DNA replication. Wild-type eggs fertilized with untreated sperm from cea mutant males (i.e. which initiate development as diploid zygotes), can experience a one-cycle delay of the first cell division which, coupled to ongoing DNA replication, would be expected to lead to tetraploidy (Fig. 10C). Although O’Connell et al. (2001) reported that mutations in centriolar components result in a paternally-induced cell cycle delay similar to that we observe in the zebrafish, to our knowledge no study has tested whether the resulting embryos are tetraploid. We show that embryos with such a one-cycle delay in their first cell division indeed have a tetraploid chromosome complement. Moreover, we used the zebrafish system to determine whether cea mutant sperm can promote full genome diploidization by testing whether it can promote gynogenetic development. While fertilization by UV-treated wild-type sperm invariably leads to haploid development (Streisinger et al., 1981; Fig. 10D), fertilization with UV-treated sperm derived from cea mutant males can induce gynogenetic development (Fig. 10E). These findings, together with previous studies that show that zebrafish embryogenesis does not tolerate chromosomal aneuploidy (Poss et al., 2004), indicate that, in the absence of centrosome duplication, the DNA replication cycle proceeds undisturbed and results in the faithful duplication of the genome.
Earlier studies have begun to address the effects of chromosome number abnormalities in the zebrafish embryo. Mutations in the mitotic checkpoint gene mps1 and the mismatch repair gene mlh1 result in chromosomal aneuploidy in the embryonic progeny (Poss et al., 2004; Feitsma et al., 2007). Such aneuploidy results in a wide range of severe defects that occur within the first few days of development, causing gross alterations in body plan morphology and early embryonic lethality. Our studies show that tetraploid zebrafish develop an apparently normal body plan but exhibit late embryonic lethality. To our knowledge, this is the first description of the effects of tetraploidy in zebrafish embryos, as previous studies of genome diploidization in this organism involved physical methods to inhibit the first cell division (Streissinger et al., 1981; Walker, 1999), which themselves produce a wide range of developmental abnormalities that likely obscured the identification of this syndrome.
Our results also indicate that zebrafish embryos are better able to tolerate full ploidy changes than genetic imbalances caused by aneuploidy for individual or groups of chromosomes. A similar conclusion has been recently proposed by Feitsma et al, (2007), who find that a fraction of embryos from mlh1 mutant mothers contain a full triploid chromosome content and are able to survive to become viable adults. The increased severity of defects observed in zebrafish tetraploids compared to those reported in triploids suggests that increments in ploidy number may cause escalating gene network imbalances in the developing embryo, which beyond a certain threshold are incompatible with viability.
Single-centriole-mediated polyploidization may facilitate the propagation of new polyploid variants
In species that undergo sexual reproduction, whole genome duplication results in a saltatory change in chromosome number that is incompatible with the parental cytotype. This is because fusion of gametes of the prevalent, parental cytotype with gametes of the new, diploidized cytotype result in individuals with odd ploidy number, which are typically sterile due to defects in chromosome segregation during meiosis (Stebbins, 1971; Husband, 2000). Three scenarios have been proposed, which may result in both the production of polyploid individuals and the propagation of the new cytotype (reviewed in Mallet, 2007). A first scenario occurs in hermaphroditic plants, whose indeterminate growth allows for a somatic diploidization event in a single individual to be represented in both male and female gametes. Such a scenario, however, is not easily applicable to animal species due to the early separation of germ line and somatic cell lineages during animal development (reviewed in Wylie, 1999), as well as with the fact that hermaphroditism is the exception and not the norm in the animal kingdom. A second scenario for the origin of polyploidization involves the fusion of unreduced gametes, each derived from defective cytokinesis during the meiotic divisions (and, in non-hermaphroditic species, from multiple individuals). A third proposed mechanism for polyploidization relies on a “triploid bridge”, where an odd-numbered ploidy individual, derived from the fusion of an unreduced and a normal gamete, produces in the next generation a small fraction of tetraploid offspring due to stochastic segregation of unpaired chromatids.
All the above mechanisms depend on the formation of unreduced gametes, derived from an individual of normal ploidy. Another type of scenario for polyploidization could be envisioned by the duplication of the zygotic genome, namely through the inhibition of cytokinesis corresponding to the first embryonic cell cycle. Given that most early embryonic processes depend on maternally derived processes, however, it is difficult to imagine how genetic mutations in maternal genes would be able to specifically affect the first embryonic cell division while leaving intact subsequent divisions, which are necessary for embryonic viability. The nature of the centriolar cycle during the oocyte to embryo transition, namely centriolar inheritance through the sperm during fertilization and centriolar duplication relying on maternally-derived products after fertilization, provides a unique developmental situation. In this case, when a single centriole (instead of a normal centriolar pair) is provided by the sperm, cytokinesis corresponding to the first and only the first cell cycle is inhibited, thus resulting in embryonic polyploidization. This situation also allows the production of multiple polyploid individuals in the same generation, since male individuals carrying centriolar mutations may produce multiple affected sperm. Moreover, because the first cell division delay caused by paternal centriolar dysfunction is independent of the embryonic genetic constitution and therefore sex determination, such polyploid offspring may grow to be adults of either sex, which may be able to intercross and thus propagate the ploidy change. Thus, whole genome duplication caused by defects in paternal centriolar duplication provides a simple scenario in which polyploidy can arise and propagate in a sexual animal species.
In our experiments, sperm from cea mutant males promotes diploidization in a small fraction of embryos (up to 5% in a our sample of cea males), which is likely a consequence of the incomplete penetrance of the cea mutation. It is possible that mutations that specifically interfere with centriole duplication during spermatogenesis (e.g. specific alleles of general centriolar genes, or mutations in spermatogenesis-specific centriolar genes) may result in higher fractions of polyploidized progeny. However, even relatively low yields of such single-centriole-induced genome duplications may be sufficient for the propagation of the polyploidized genome through genetic crosses in the next generation, especially in species that produce large numbers of offspring. For reasons that are poorly understood, polyploidy is typically poorly tolerated in higher organisms (Storchová and Pellman, 2004; Comai, 2005), a phenomenon that we also observe in the zebrafish. Nevertheless, our results suggest a mechanism by which polyploidy could arise in a population. Mutations in centriolar components in the zebrafish may allow using this model organism as a genetic system to test this hypothesis.
Resumption of cell division after cell cycle delay: uncoupling of developmental time and cell cycle count and implications for germ plasm recruitment
Cells in cea mutant embryos that fail to divide are capable of undergoing mitosis in subsequent cell cycles at the same time as unaffected cells in the same embryo. In Xenopus egg extracts, centriole duplication has been shown to require Cdk2/cyclinE activity (Tsou and Stearns, 2006), which is also thought to drive zebrafish early divisions (Yarden and Geiger, 1996). It is possible that the reduced function of the hypomorphic cea allele leads to a reduced growth rate of daughter centrioles during interphase, which results in the inability to complete centriole duplication and prevents bipolar spindle formation and consequently cytokinesis. The timing of cell cycle resumption in subsequent cell cycles, which is precisely coordinated with cell divisions in unaffected sibling cells, is likely dependent on the presence of global signals leading to synchronized cell division in the early zebrafish embryo (Kane and Kimmel, 1993).
The stalling and resumption of cell cycles in cea mutant embryos allows for the experimental uncoupling of developmental time and the cell division cycles. This ability has allowed us to test the cellular requirements for the recruitment of the zebrafish germ plasm to the furrows in the first and second cell cycles. We find that germ plasm aggregates fail to recruit to resuming furrows during the third cycle, even if cell cycle count and furrow morphology for the dividing cell corresponds to that of a second cleavage cycle. Our previous studies have shown that the recruitment of cortical germ plasm aggregates to the furrow is regionalized, occurring only in the immediate vicinity of the forming furrow (Theusch et al., 2006). Thus, it seems unlikely that the failure of germ plasm recruitment in the delayed second furrow is caused by depletion due to long-range recruitment at furrows forming in other regions of the blastodisc. Rather, our observations suggests the presence of an optimal time window, normally corresponding to the first two cell cycles but independent of cell cycle count, which is permissive for germ plasm recruitment to the forming furrow. This conclusion is consistent with our recent proposal that furrow recruitment of germ plasm aggregates is coupled to the progressive peripheral displacement of an f-actin cytoskeletal network bound to these aggregates, which is normally largely completed by the end of the second cell cycle (Theusch et al., 2006).
In summary, cea has distinct paternal and maternal roles for centrosome duplication during the early zebrafish cell divisions, and encodes a component of the centriole, Sas-6. Our findings suggest a potential mechanism of genome duplication, involving whole genome duplication at the level of the zygote, which may have facilitated the generation and propagation of polyploidy during animal evolution. Our studies also highlight a remarkable degree of conservation of the genetic pathway that drives centrosomal inheritance and reconstitution during the oocyte-to-embryo transition, providing an essential bridge for our understanding of this process from invertebrate model systems to vertebrate species.
Materials and Methods
Genetic stocks and methods
Wild-type stocks were the standard AB and an AB stock carrying the albino mutation. The cellular atollp35mfa allele (Dosch et al., 2004) was in an AB/Tübingen hybrid background. Fish and embryos were raised and maintained under standard conditions at 28.5 °C (Brand et al., 2002). Fish from the cea stock were genotyped by using by using the flanking linked markers z10673 and z51215. In vitro fertilization with untreated and UV-inactivated sperm was carried out as previously described (Pelegri and Schulte-Merker, 1999). Metaphase chromosome spreads were carried out as in Amores and Postlethwait (1999) and mounted in VectaShield mounting medium with DAPI (Vector Laboratories).
Positional cloning of cea
Males heterozygous or homozygous for the cea mutation were mated to wild-type WIK females to generate F1 families, which were incrossed to obtain homozygous F2 fish (Pelegri and Mullins, 2004). The early cell cleavage phenotype of F3 embryos allowed determining the genotype of F2 females with respect to the cea mutation, which was compared to the segregation of flanking SSLP markers. To find the mutation in sas-6, we amplified two fragments of sas-6 cDNA from cea mutant and wild-type embryo cDNA by RT-PCR with two primer sets: for the N-terminal region: 5′-TCGTTGCTTTGTGAATGAATTAGG-3′ and 5′-TTTCCTTCCACAGAGTTCTTCTG-3′, for the C-terminal region: 5′-TCAAAGATAAAGACCAGCTTGTGC-3′ and 5′-CAGTGATGAATCTGGATTAACCTG-3′. All PCR products were cloned into pGEM-T easy and sequenced. To confirm linkage between the cea mutation and the mutation in sas-6 gene, we amplified a partial sequence of sas-6 containing the mutation region by PCR using the following primers: 5′-GTCAGAGGAGCTCATCAAGG-3′ and 5′-CTCCTCCTTCAGTGAGAGTC-3′, and the PCR fragments were directly sequenced.
Immunofluorescence and imaging
For the detection of tubulins and β--catenin, dechorionated embryos were fixed and labeled as in Theusch et al. (2006) using microtubule fix buffer and the following primary antibodies: anti-α-tubulin (Sigma, monoclonal B5–1–2, 1:2500), anti-γ-tubulin (Sigma, monoclonal GTU-88, 1:2000), and anti-β-catenin (Sigma, rabbit polyclonal, 1:1000). The primary antibodies were recognized using an Alexa 488-conjugated rabbit anti-mouse antibody or Alexa 488-conjugated goat anti-Rabbit antibody (Molecular probes). Staining of f-actin was determined using Alexa-488 phalloidin (Molecular Probes) as in Urven et al. (2006). Subsequently, embryos were labeled for DNA using DAPI (0.5 μg/ml in PBS) for 10 minutes at room temperature or propidium iodide (described in Pelegri et al., 1999). Expressed mCherry-Cea fusions were detected by their fluorescence in fixed embryos. In situ hybridizations were carried out as described previously (Pelegri and Maischein, 1998) using vasa (Yoon et al., 1997) and sas-6 cloned into a pGEM-T easy vector.
Live and fluorescently labeled embryos were imaged using a Zeiss Axioplan2 fluorescent microscope and Open Lab imaging software. Images of embryos labeled using in situ hybridization were acquired using a Leica-FLIII and a color camera (Diagnostic Instruments Spot Insight).
Molecular methods and RNA injections
Wild-type and mutant zebrafish sas-6 cDNAs were cloned into the BamH1 and Xho1 site of pCS2+ vector for in vitro transcription. To create the in-frame fusions between mCherry and Sas-6, we amplified mCherry cDNA by PCR and cloned it into the BamH1 and Xho1 site of pCS2+ (pCS2+cherry), and PCR-amplified sas-6-S and sas-6-L fragments were cloned into Xho1 and Xba1 site of pCS2+cherry. RNA for injection was prepared and injected at the one-cell stage as previously described (Pelegri and Maischein, 1998) using a Phemtojet microinjector (Eppendorf).
Acknowledgments
We are grateful to Dr. Mary Mullins and coworkers (U. Pennsylvania) for generously providing us the cellular atoll mutation. We also thank Drs. John White, Chris Wiese and Bret Payseur (U. Wisconsin – Madison), as well as members of our laboratory, for helpful discussions and comments on the manuscript. This work was funded by grant 1RO1GM/HD65303-01 from NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amores A, Postlethwait JH. Banded chromosomes and the zebrafish karyotype. In: Detrich W, Zon LI, Westerfield M, editors. The Zebrafish: Genetics and Genomics. Vol. 60. Academic Press; San Diego: 1999. pp. 323–338. [DOI] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol. 2002;14:25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- Brand M, Granato M, Nüsslein-Volhard C. Keeping and raising zebrafish. In: Nüsslein-Volhard C, Dahm R, editors. Zebrafish. Vol. 261. Oxford University Press; Oxford: 2002. pp. 7–37. [Google Scholar]
- Cavalier-Smith T. Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C-value paradox. J Cell Sci. 1978;34:247–278. doi: 10.1242/jcs.34.1.247. [DOI] [PubMed] [Google Scholar]
- Comai L. The advantages and disadvantages of being polyploid. Nature Rev Genet. 2005;6:836–846. doi: 10.1038/nrg1711. [DOI] [PubMed] [Google Scholar]
- Daga RR, Thode G, Amores A. Chromosome complement, C-banding, Ag-NOR and replication banding in the zebrafish Danio rerio. Chromosome Res. 1996;4:29–32. doi: 10.1007/BF02254941. [DOI] [PubMed] [Google Scholar]
- Dammermann A, Müller-Reichert T, Pelletier L, Habermann B, Desai A, Oegema K. Centriole assembly requires both centriolar and pericentriolar material proteins. Dev Cell. 2004;7:815–829. doi: 10.1016/j.devcel.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Dekens MPS, Pelegri FJ, Maischein HM, Nüsslein-Volhard C. The maternal-effect gene futile cycle is essential for pronuclear congression and mitotic spindle assembly in the zebrafish zygote. Development. 2003;130:3907–3916. doi: 10.1242/dev.00606. [DOI] [PubMed] [Google Scholar]
- Delattre M, Canard C, Gönczy P. Sequential protein recruitment in C. elegans centriole formation. Curr Biol. 2006;16:1844–1849. doi: 10.1016/j.cub.2006.07.059. [DOI] [PubMed] [Google Scholar]
- Delattre M, Gönczy P. The arithmetic of centrosome biogenesis. J Cell Sci. 2004;117:1619–1629. doi: 10.1242/jcs.01128. [DOI] [PubMed] [Google Scholar]
- Delattre M, Leidel S, Wani K, Baumer K, Bamat J, Schnabel H, Feichtinger R, Schnabel R, Gönczy P. Centriolar SAS-5 is required for centrosome duplication in C. elegans. Nature Cell Biol. 2004;6:656–664. doi: 10.1038/ncb1146. [DOI] [PubMed] [Google Scholar]
- Dosch R, Wagner DS, Mintzer KA, Runke G, Wiemelt AP, Mullins MC. Maternal control of vertebrate development before the midblastula transition: mutants from the zebrafish. Dev Cell. 2004;6:771–780. doi: 10.1016/j.devcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Feitsma H, Leal MC, Moens PB, Cuppen E, Schulz RW. Mlh1 deficiency in zebrafish results in male sterility and aneuploid as well as triploid progeny in females. Genetics. 2007;175:1561–1569. doi: 10.1534/genetics.106.068171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M. The molecular requirements for cytokinesis. Science. 2005;307:1735–1739. doi: 10.1126/science.1096896. [DOI] [PubMed] [Google Scholar]
- Hammill DR, Severson AF, Carter JC, Bowerman B. Centrosome maturation and mitotic spindle assembly in C. elegans require SPD-5, a protein with multiple coiled-coil domains. Dev Cell. 2002;3:673–684. doi: 10.1016/s1534-5807(02)00327-1. [DOI] [PubMed] [Google Scholar]
- Husband BC. Constraints on polyploid evolution: a test of the minority cytotype exclusion principle. Proc R Soc Lond B. 2000;267:217–223. doi: 10.1098/rspb.2000.0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung LY, Tang CJ, Tang TK. Protein 4.1 R-135 interacts with a novel centrosomal protein (CPAP) which is associated with the γ-tubulin complex. Mol Cell Biol. 2000;20:7813–7825. doi: 10.1128/mcb.20.20.7813-7825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesuthasan S. Furrow-associated microtubule arrays are required for the cohesion of zebrafish blastomeres following cytokinesis. J Cell Sci. 1998;111:3695–3703. doi: 10.1242/jcs.111.24.3695. [DOI] [PubMed] [Google Scholar]
- Kane DA, Kimmel CB. The zebrafish midblastula transition. Development. 1993;119:447–456. doi: 10.1242/dev.119.2.447. [DOI] [PubMed] [Google Scholar]
- Kaufman MH. New insights into triploidy and tetraploidy, from an analysis of model systems for these conditions. Hum Reprod. 1991;6:8–16. doi: 10.1093/oxfordjournals.humrep.a137263. [DOI] [PubMed] [Google Scholar]
- Kemp CA, Kopish KR, Zipperlen P, Ahringer J, O’Connell KF. Centrosome maturation and duplication in C. elegans require the coiled-coil protein SPD-2. Dev Cell. 2004;6:511–523. doi: 10.1016/s1534-5807(04)00066-8. [DOI] [PubMed] [Google Scholar]
- Kessel RG, Beams HW, Tung HN, Roberts R. Unusual particle arrays in the plasma membrane of zebrafish spermatozoa. J Ultrastruct Res. 1983;84:268–274. [Google Scholar]
- Kim DY, Ray R. Cell cycle regulators control centrosome elimination during oogenesis in Caenorhabditis elegans. J Cell Biol. 2006;174:751–757. doi: 10.1083/jcb.200512160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development in the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kirkham M, Müller-Reichert T, Oegema K, Grill S, Hyman AA. SAS-4 is a C. elegans centriolar protein that controls centrosome size. Cell. 2003;112:575–587. doi: 10.1016/s0092-8674(03)00117-x. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Koshida S, Furutani-Seiki M, Kondoh H. Zebrafish maternal-effect mutations causing cytokinesis defects without affecting mitosis or equatorial vasa deposition. Mech Dev. 2004;121:79–89. doi: 10.1016/j.mod.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Lehmann R, Nüsslein-Volhard C. The maternal gene nanos has a central role in posterior pattern formation of the Drosophila embryo. Development. 1991;112:679–691. doi: 10.1242/dev.112.3.679. [DOI] [PubMed] [Google Scholar]
- Leidel S, Delattre M, Cerutti L, Baumer K, Gönczy P. SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nature Cell Biol. 2005;7:115–124. doi: 10.1038/ncb1220. [DOI] [PubMed] [Google Scholar]
- Leidel S, Gönczy P. SAS-4 is essential for centrosome duplication in C. elegans and is recruited to daughter centrioles once per cell cycle. Dev Cell. 2003;4:431–439. doi: 10.1016/s1534-5807(03)00062-5. [DOI] [PubMed] [Google Scholar]
- Leidel S, Gönczy P. Centrosome duplication and nematodes: recent insights from an old relationship. Dev Cell. 2005;9:317–325. doi: 10.1016/j.devcel.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Mallet J. Hybrid speciation. Nature. 2007;446:279–283. doi: 10.1038/nature05706. [DOI] [PubMed] [Google Scholar]
- Mathavan S, Lee SGP, Mak A, Miller LD, Murthy KRK, Govindarajan KR, Tong Y, Wu YL, Lam SH, Yang H, Ruan Y, Korzh V, Gong Z, Liu DT, Lufkin T. Transcriptome analysis of zebrafish embryogenesis using microarrays. PLOS Genetics. 2005;1:260–276. doi: 10.1371/journal.pgen.0010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazia D, Harris P, Bilbring T. The multiplicity of the mitotic centers and the time-course of their duplication and separation. J Bioph Bioch Cyt. 1960;7:1–20. doi: 10.1083/jcb.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler J, Wieschaus EF. Dominant maternal effect mutations of Drosophila melanogaster causing the production of double-abdomen embryos. Genetics. 1986;112:808–822. doi: 10.1093/genetics/112.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevisius A, Ekker SC. Effective targeted gene “knockdown” in zebrafish. Nature Genetics. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- O’Connell KF, Caron C, Kopish KR, Hurd DD, Kemphues KJ, Li Y, White JG. The C. elegans zyg-1 gene encodes a regulator of centrosome duplication with distinct maternal and paternal roles in the embryo. Cell. 2001;105:547–558. doi: 10.1016/s0092-8674(01)00338-5. [DOI] [PubMed] [Google Scholar]
- Otto SP, Whitton J. Polyploid incidence and evolution. Annu Rev Genet. 2000;34:401–437. doi: 10.1146/annurev.genet.34.1.401. [DOI] [PubMed] [Google Scholar]
- Pelegri F, Dekens MPS, Schulte-Merker S, Maischein HM, Weiler C, Nüsslein-Volhard C. Identification of recessive maternal-effect mutations in the zebrafish using a gynogenesis-based method. Dev Dyn. 2004;231:325–336. doi: 10.1002/dvdy.20145. [DOI] [PubMed] [Google Scholar]
- Pelegri F, Knaut H, Maischein HM, Schulte-Merker S, Nüsslein-Volhard C. A mutation in the zebrafish maternal-effect gene nebel affects furrow formation and vasa RNA localization. Curr Biol. 1999;9:1431–1440. doi: 10.1016/s0960-9822(00)80112-8. [DOI] [PubMed] [Google Scholar]
- Pelegri F, Maischein HM. Function of zebrafish β-catenin and TCF-3 in dorsoventral patterning. Mech Dev. 1998;77:63–74. doi: 10.1016/s0925-4773(98)00132-4. [DOI] [PubMed] [Google Scholar]
- Pelegri F, Mullins M. Genetic screens for maternal-effect mutations. In: Detrich HW, Westerfield M, Zon LI, editors. The Zebrafish: 2nd Edition. Genetics, Genomics and informatics. Vol. 77. Elsevier Academic Press; San Diego: 2004. pp. 21–51. [DOI] [PubMed] [Google Scholar]
- Pelegri F, Schulte-Merker S. A gynogenesis-based screen for maternal-effect genes in the zebrafish, Danio rerio. In: Detrich W, Zon LI, Westerfield M, editors. The Zebrafish: Genetics and Genomics. Vol. 60. Academic Press; San Diego: 1999. pp. 1–20. [DOI] [PubMed] [Google Scholar]
- Pellestor. The cytogenetic analysis of human zygotes and preimplantation embryos. Hum Reprod Update. 1995;1:581–5. doi: 10.1093/humupd/1.6.581. [DOI] [PubMed] [Google Scholar]
- Pelletier L, O’Toole E, Schwager A, Hyman AA, Müller-Reichert T. Centriole assembly in Caenorhabditis elegans. Nature. 2006;444:619–623. doi: 10.1038/nature05318. [DOI] [PubMed] [Google Scholar]
- Poss KD, Nechiporuk A, Stringer KF, Lee C, Keating MT. Germ cell aneuploidy in zebrafish with mutations in the mitotic checkpoint gene mps1. Genes Dev. 2004;18:1527–1532. doi: 10.1101/gad.1182604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport R. Cytokinesis in animal cells. Cambridge University Press; Cambridge: 1996. [Google Scholar]
- Sluder G, Rieder CL. Experimental separation of pronuclei in fertilized sea urchin eggs: chromosomes do not organize a spindle in the absence of centrosomes. J Cell Biol. 1985;76:35–51. doi: 10.1083/jcb.100.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell WJ, Pan J, Wang Q. Cilia and flagella revealed: from flagellar assembly in Chlamydomonas to human obesity disorders. Cell. 2004;117:693–697. doi: 10.1016/j.cell.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Stebbins GL. Processes of organic evolution. Prentice-Hall; Englewood Cliffs, New Jersey: 1971. [Google Scholar]
- Storchová Z, Pellman D. From plyploidy to aneuploidy, genome instability and cancer. Nature Rev Mol Cell Biol. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- Streisinger G, Walker C, Dower N, Knauber D, Singer F. Production of clones of homozygous diploid zebra fish (Brachydanio rerioI) Nature. 1981;291:293–296. doi: 10.1038/291293a0. [DOI] [PubMed] [Google Scholar]
- Theusch EV, Brown KJ, Pelegri F. Separate pathways of RNA recruitment lead to the compartmentalization of the zebrafish germ plasm. Dev Biol. 2006;292:129–141. doi: 10.1016/j.ydbio.2005.12.045. [DOI] [PubMed] [Google Scholar]
- Tsou MFB, Stearns T. Mechanism limiting centrosome duplication to once per cell cycle. Nature. 2006;442:947–951. doi: 10.1038/nature04985. [DOI] [PubMed] [Google Scholar]
- Urven LE, Yabe T, Pelegri F. A role for non-muscle myosin II function in furrow maturation in the early zebrafish embryo. J Cell Sci. 2006;119:4342–4352. doi: 10.1242/jcs.03197. [DOI] [PubMed] [Google Scholar]
- Wagner DS, Dosch R, Mintzer KA, Wiemelt AP, Mullins MC. Maternal control of Development at the midblastula transition and beyond: mutants from the zebrafish II. Dev Cell. 2004;6:781–790. doi: 10.1016/j.devcel.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Walker C. Haploid screens and gamma-ray mutagenesis. In: Detrich W, Zon LI, Westerfield M, editors. The Zebrafish: Genetics and Genomics. Vol. 60. Academic Press; San Diego: 1999. pp. 43–70. [DOI] [PubMed] [Google Scholar]
- Wylie C. Germ cells. Cell. 1999;96:165–174. doi: 10.1016/s0092-8674(00)80557-7. [DOI] [PubMed] [Google Scholar]
- Yarden A, Geiger B. Zebrafish cyclin E regulation during early embryogenesis. Dev Dyn. 1996;206:1–11. doi: 10.1002/(SICI)1097-0177(199605)206:1<1::AID-AJA1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Yoon C, Kawakami K, Hopkins N. Zebrafish vasa homologue RNA is localized to the cleavage planes of 2- and 4-cell-stage embryos and is expressed in the primordial germ cells. Development. 1997;124:3157–3165. doi: 10.1242/dev.124.16.3157. [DOI] [PubMed] [Google Scholar]
- Zamir E, Kam Z, Yarden A. Transcription-dependent induction of G1 phase during the zebra fish midblastula transition. Mol Cell Biol. 1997;17:529–536. doi: 10.1128/mcb.17.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]