

During development, neurons migrate and undergo extensive morphological changes necessary to establish functional connections. Extracellular cues function through surface receptors to control signaling pathways whose interplay results in the remodeling of the cytoskeleton and thereby changes in cell shape and motility. Past research has identified numerous receptors as important for promoting or directing cell and growth cone movements. Compared with the explosion in information on these surface interactions, however, little is known about the intracellular signaling mechanisms that direct cell motility. Recent work, published in this journal and described in this minireview, has identified a receptor (tyrosine phosphatase, Dlar) and a signaling pathway (Abl kinase) important in regulating axon guidance. These studies provide a provocative link from the cell surface to the actin cytoskeleton through the Ena/VASP family of proteins and Profilin, both of which are known to regulate actin dynamics and are shown in these papers to play important roles in axon growth and guidance.
To understand the significance of these papers, it is important to review briefly our current knowledge about regulation of actin dynamics (see Figure 1). Actin filaments form by the polymerization of monomeric ATP-actin, which is followed by slow hydrolysis of the bound ATP to ADP within the filaments. Filament growth is polarized because of a higher affinity of ATP-actin for the “barbed” than for the “pointed” filament end.
Figure 1.
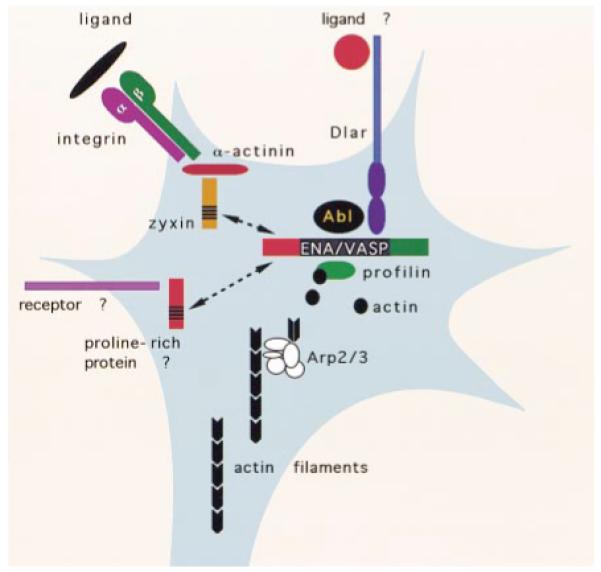
Selected Interactions between Membrane Receptors and the F-Actin Cytoskeleton Likely to be Important in Neural Development
Proteins described in the text are illustrated in the figure. It is important to note that not every one of these proteins is necessarily present in every growth cone.
More than 50 proteins have been identified that bind actin and control filament dynamics (reviewed by Mitchison and Cramer, 1996). Some affect monomers by controlling sequestration or nucleotide exchange; others control filament formation and stability by regulating capping, nucleating, cross-linking, bundling, and severing. For example, Profilin, a monomeric globular actin sequestering protein (see Figure 1), is believed to enhance polymerization by catalyzing exchange of ADP for ATP in globular actin, thereby increasing the concentration of polymerizable actin. In some circumstances, though, Profilin may instead inhibit actin polymerization, possibly by sequestering actin monomers. The Arp2/3 complex nucleates filament assembly by stabilizing actin dimers (Figure 1). Arp2/3 also has been shown to bind to the sides of existing filaments, thereby creating branch points in the actin network at the leading edges of migrating cells (Mullins et al., 1998). In contrast, Cofilin binds to ADP regions of filamentous polymerized actin, severing and thereby disassembling filaments.
Cellular signaling molecules control actin remodeling by regulating all of these activities. In particular, families of proline-rich proteins, the WASP/SCAR and Ena/VASP families, play crucial roles in actin dynamics, and are believed to act, at least in part, by binding and thereby regulating Profilin’s activity (Figure 1). In addition, the C termini of WASP/SCAR proteins recruit the Arp2/3 complex to nucleate actin filaments (Machesky and Insall, 1998).
Integrins and other adhesion molecules may promote actin filament assembly by concentrating Ena/VASP proteins (see Figure 1). The Integrin-associated proteins Zyxin and Vinculin interact with Ena/VASP proteins (Beckerle, 1998). There are other pathways for actin control. Small Rho family GTPases (Cdc42, Rac, and Rho) normally control the formation of cellular actin structures, such as filopodia, lamellipodia, and stress fibers, by regulating the activities of numerous direct and indirect effectors (reviewed by Mackay and Hall, 1998). Receptor tyrosine kinases and serpentine receptors control the activity of these small G proteins through regulation of GDP/GTP exchange factors. Thus, multiple receptors activate signaling pathways that impinge upon actin regulatory elements and thereby remodel the cytoskeleton.
Profilin in Axon Extension
As mentioned above, Profilin has been implicated as a key regulator in many of the model systems used to investigate actin motility. Insights into the complex functions of this actin-binding protein have recently been provided by studies on axon pathfinding in Drosophila published the February 1999 issue of Neuron (Wills et al., 1999b). Mutant alleles in Chickadee (chic), the Drosophila profilin gene, were isolated in a screen for genes necessary for motor neuron pathfinding. Those motor axons, which together constitute intersegmental nerve branch b (ISNb), exit the CNS in the ISN nerve. Near the muscle field, they defasiculate from ISN and turn toward the ventral muscles. Growth cones explore the muscle surfaces and form synaptic connections with muscles 6/7, 12, and 13. Morphological observations and mutant analyses have identified a series of choice points for these growth cones at the sites of defasiculation from ISN, entry into the muscle field, and extension to the most distal muscle 12.
In a chic mutant, all major motor neuron nerve branches display a “stranded” phenotype where axons stall at a choice point. Wills et al. (1999b) provide convincing evidence, however, that Profilin is required for general axon extension. The deficiency in Profilin was shown to reduce axonal outgrowth by nerve cord explants on a nonphysiological substrate in vitro, and the “stranded” phenotype in the chic mutant was shown to result from a depletion of maternally deposited Profilin in a zygotic null background. In neurons, Profilin appears to have a general function in promoting axon extension, which is most stringent at choice points where axon growth is typically slowed even in wild-type embryos. Intriguingly, the morphologies of “stranded” growth cones are quite complex and include multiple filipodia. Thus, it is not clear whether Profilin is acting within them to promote or inhibit actin assembly.
Not surprisingly, the murine profilin-1 gene has been shown to be essential for normal mouse development: profilin-/- embryos die before implantation. When appropriate conditional alleles are generated allowing analysis at a later developmental stage, it seems likely that it will prove to be as essential for normal axon growth in mouse as in Drosophila. As will be described below, a role of Profilin-1 in vertebrate neurulation has been uncovered because of a genetic interaction with Murine enabled, or Mena (Lanier et al., 1999).
Role of Ena/VASP Proteins in Axon Guidance
Originally identified as a genetic suppressor of the Abelson tyrosine kinase (abl) mutation, Drosophila enabled (ena) encodes a proline-rich protein in the Ena/VASP family whose members also include three vertebrate paralogs, Vasodilator-stimulated phosphoprotein (VASP), Mena, and Ena/VASP-like (Evl). Ena/VASP proteins regulate actin filament formation (Reinhard et al., 1995). The N termini of Ena/VASP proteins contain a unique proline-binding domain, which interacts with Vinculin and Zyxin, two cytoskeletal proteins localized with Integrins to focal adhesions (Gertler et al., 1996). The central proline-rich regions of Ena/VASP proteins directly interact in vitro with Profilin and with SH3 domain-containing adaptor proteins and kinases, such as Abl and Src. The Ena/VASP proteins are also phosphorylated by both tyrosine and serine/threonine kinases. Therefore, The Ena/VASP proteins modulate actin dynamics, and their activities are likely to be regulated by multiple intracellular signaling pathways.
Previous work has demonstrated that the Drosophila Ena protein is required for embryonic axonogenesis. Ena mutant embryos display pleiotropic axon patterning defects, by a reduction in longitudinal nerve bundle fasciculation, a thinning of these nerve bundles, and an increase in the number of nerves exiting CNS (Gertler et al., 1995). Wills et al. (1999a) have reported a specific guidance role of Ena within the growth cones of the motor neurons that form the ISNb nerve branch. In the Drosophila ena mutant, some ISNb growth cones fail to defasciculate from ISN, while others turn abnormally after defasciculating from ISN and “bypass” the ventral target muscles. Thus, Ena plays an important role in making growth cone decisions at two choice points of ISNb.
A comparison of the ena and chickadee phenotypes suggest that there must be other proteins that regulate Profilin function. While mutation in chic results in a disruption of all motor neuron branches, a mutation in ena appears to affect only a subset of the motor neuron growth cones. Despite the ubiquitously high expression of Ena in all neurons, alternative signaling molecules must be available to ensure appropriate growth cone guidance in those motor neurons that make correct pathfinding decisions in its absence. The involvement of other pathways is not surprising since it has been shown that Rac and Cdc42 play a role in axonogenesis (Kaufmann et al., 1998), and one of their effectors, WASP, also binds and thereby regulates Profilin function (Suetsugu et al., 1998).
Is Ena required for general actin dynamics or for interpreting specific guidance cues? Evidence supports a guidance role because Ena selectively interacts with cellular signaling molecules, and because ena embryos show a “bypass” phenotype only in a subset of nerves, although it has guidance defects in nerves not described above. However, the phenotype is also consistent with Ena playing a more general role, as ena mutants have other guidance defects in other nerves (Gertler et al., 1995), and disruption of axon extension by low doses of cytochalasin D has been shown to result in a “bypass” pathfinding phenotype (Kaufmann et al., 1998).
In a recent contribution in Neuron, Lanier et al. (1999) provide direct evidence that Mena, a murine homolog of Ena, is also crucial for axon guidance during vertebrate development. Consistent with a role in axon guidance, Mena can be detected in the growth cones, including tips of filopodia. Despite high expression of Mena in many tissues during embryogenesis, mena-/- embryos are viable but have several specific axon guidance defects at the midline, where fibers in the corpus callosum and fornix reach but fail to cross the midline. Not all axons require Mena to cross the midline, however, as several other commissures form normally. The deficits in mena-/- embryos are much more limited than those in Drosophila ena mutants. While not tested, it seems likely that compensation provided by VASP or Evl accounts for this difference. The Ena/VASP proteins share many common interactions and extensively overlap in expression. Similar to murine mena-/-, murine VASP-/- mutants are also viable and have no identified CNS deficits, very likely because of compensation (Aszódi et al., 1999).
Lanier et al. (1999) further demonstrate that murine Mena and Profilin-1 cooperate to promote closure of the neural tube. While heterozygous profilin+/- or homozygous mena-/- mutants are viable, profilin+/-; mena-/- embryos die before birth. In half of these embryos, the neural tube is not closed at E9.5 or later. Neurulation involves extensive reshaping and movement of cells, processes that have been shown to be mediated by actin-dependent mechanisms. The result demonstrated that, in the absence of Mena, a Profilin-limited pathway is needed for neurulation. Among the possibilities, this path may involve VASP or Evl. Interestingly, the neurulation deficit resembles that of the double mutant of murine abl and abl related gene (arg), corroborating a model in which murine Mena and Profilin activities are regulated by Abl family kinases (Koleske et al., 1998; see below).
Regulation of Ena/VASP Function: The Abl Tyrosine Kinase
The modifications and interactions described above provide many loci for regulation of Ena/VASP function by various signaling pathways. Genetics has provided compelling evidence that Abl interacts with Ena to regulate axon guidance. Abl is a cytoplasmic tyrosine kinase with C-terminal binding sites for globular and filamentous actin. Initial evidence implicating Drosophila Abl in axon guidance decisions was provided by Elkins et al. (1990), who demonstrated that in a fasciclin I-abl double mutant, major commissural axons fail to project across the midline. Evidence that Abl regulates activity of Ena was obtained by Gertler et al. (1995), who demonstrated that reductions in Ena suppress the larval lethality and CNS defects observed in abl mutants in a sensitized background. They also demonstrated that Abl directly associates with and phosphorylates Ena (see Figure 1). Together, these interactions suggest that Abl inhibits Ena function. More recently, Giniger (1998) demonstrated that Drosophila abl and Notch mutations act synergistically to result in defects in both CNS and PNS axon patterning. Thus, Abl is required for the axon extension controlled by Notch, and this regulation is postulated to be mediated by an interaction between Notch and Disabled, a previously identified genetic modifier of Abl function.
Wills et al. (1999b) have demonstrated that Abl and Ena have both antagonistic and distinct functions in motor neuron pathfinding. Consistent with an antagonistic relationship, overexpression of Abl or loss of Ena both result in abnormal “bypass” type turning of axons associated with the ISNb nerve. In contrast, loss of Ena also prevents normal defasciculation of the ISNb branch in some mutant embryos, which is not mimicked in embryos overexpressing Abl. Moreover, loss of Abl interferes with axonal extension to the most distal muscle 12, resulting in a “stranded” phenotype, but overexpression of Ena does not affect this decision. While supporting earlier data indicating that Abl regulates Ena, these data suggest also that Abl regulates and Ena is regulated by other molecules.
A paper in the December 1998 issue of Neuron has shown that Abl and Arg, two closely related tyrosine kinases, are required for vertebrate neurulation (Koleske et al., 1998). While single mutations of abl or arg survive to birth, abl-/-; arg-/- double mutants die at E10.25 and have dramatically delayed closure of the rostral neural tube. In the neuroepithelium of the wild-type embryos, both Abl and Arg proteins are localized to the apical actin latticework; in double mutant homozygotes, the actin network is very disorganized. Collectively, these results demonstrate that at least one member of the Abl family must be present to promote normal neurulation and suggest that these kinases direct neurulation through regulation of the cytoskeleton.
Regulation of Abl and Ena: A Receptor Protein Phosphatase
The surface receptors that control Ena activity were completely unknown prior to a recent publication in Neuron implicating the receptor tyrosine phosphatase Dlar (Wills et al., 1999a). Dlar belongs to the Lar subfamily of receptor protein tyrosine phosphatases (PTP), which have extracellular domains containing immunoglobulin and fibronectin type III domains. These phosphatases also contain a cytoplasmic D1 domain with catalytic activity and a structurally related but catalytically inactive D2 domain. Many Lar family phosphatases are expressed in the nervous system, but few have known ligands or cytoskeletal associations. In Drosophila, previous work has demonstrated that the phosphatases Dlar, DPTP69D, and DPTP99A are required for entry of the ISNb growth cones into the muscle field and for ISNb defasciculation (e.g., Krueger et al., 1996). In the mouse, a hypomorphic lar mutation has been shown to also have a modest innervation deficit in the hippocampus, possibly related to pathfinding.
Combining genetics and biochemistry, Wills et al. (1999a) present strong evidence that Drosophila Dlar directly regulates both Abl and Ena. When heterozygous, a mutant abl allele effectively increases the percentage of Dlar null growth cones exhibiting normal turning, reducing the percentage of growth cones displaying a “bypass” phenotype. Overexpression of Abl in neurons also is shown to cause a pathfinding phenotype similar to that observed in the loss-of-function Dlar mutant. These investigators further show that Dlar through its D2 domain directly interacts with both Abl and Ena in vitro. Moreover, association is accompanied by reciprocal phosphorylation of Dlar and Ena by Abl and dephosphorylation of phosphorylated Abl and Ena by Dlar. Thus, as depicted in Figure 1, these results suggest that Abl and Dlar exert opposing effects on Ena activity.
Together, these results suggest that the phosphatase activity of Dlar is required for ISNb growth cones to initiate a turning decision and that Abl inhibits turning by inhibiting this phosphatase or its target Ena. One might speculate that an unidentified “guidance ligand” activates Dlar phosphatase activity or promotes its ability to dephosphorylate Ena or Abl. A functional ligand for Dlar has not yet been identified, but in vertebrates, LAR isoforms have been shown to interact with matrix ligands and to localize to focal adhesions (e.g., Serra-Pages et al., 1995). By analogy with vertebrate homologs, ligand-induced Dlar dimerization seems likely to strongly reduce its phosphatase activity, so one possibility is that the “guidance” ligand relieves dimerization-promoted interactions of Dlar with a more ubiquitously expressed ligand, such as a cell adhesion molecule or an extracellular matrix protein.
Besides associating with Dlar, Ena probably interacts with additional cytoskeletal or membrane-associated proteins to promote normal axonal pathfinding. An ena mutation in the conserved N-terminal domain that strongly affects motor neuron guidance decisions (Wills et al., 1999a) has been shown to consist of a single conservative amino acid substitution that disrupts its interaction with Zyxin. Thus, Ena must almost certainly interact with Zyxin or a similar proline-rich protein to promote normal pathfinding. As illustrated in Figure 1, Zyxin has been shown to bind to α-actinin, which in turn associates with both Integrins at focal adhesions and with the actin cytoskeleton elsewhere in the cell (Beckerle, 1998). As speculation, it seems possible that Zyxin and Ena collaborate to cocluster Dlar with Integrins, so that stimulation of Dlar activity by a “guidance ligand” promotes localized actin polymerization at adhesive contacts.
The axon guidance roles of Drosophila Abl and Dlar clearly indicated the importance of tyrosine phosphorylation as a means of regulation in the Ena pathway. It is, however, not clear whether the phosphorylation is direct in vivo. Phosphorylated Tyr residues are present in both Ena and one isoform of Mena; however, these sites are not conserved in the Ena/VASP proteins. One model is that the tyrosine phosphorylation control is unique to Ena and Mena. An alternative model is that the pathway is general, but additional signaling step(s) are involved between the tyrosine kinases/phosphatases and the Ena/VASP proteins. Besides the tyrosine phosphorylation control, other regulatory mechanisms most likely exist to modulate the activity of the Ena/VASP proteins. One such pathway involves the serine and threonine phosphorylation by cGMP- and cAMP-dependent protein kinases. VASP and Mena are both serine phosphorylated, and one Ser residue is conserved among all Ena/VASP members. The effect of serine phosphorylation on the activity of the Ena/VASP protein is not understood, except that it has been shown that phosphorylation of purified VASP does not alter its association with Profilin. The possible involvement of cAMP and cGMP as regulators of Ena/VASP function is particularly intriguing, as these second messagers have recently been shown to switch the responses of growth cones to chemoattractants and repellents (Song et al., 1998). Whether the Ena/VASP proteins are involved in growth cone responses to chemoattractants and repelents remains an interesting possibility.
Concluding Remarks
Recent cell biology studies have resulted in many exciting discoveries about the actin cytoskeleton. These include discovery of the central role of the Arp2/3 complex in actin filament formation and clarification of the functions of many actin-binding proteins such as Profilin and the Ena/VASP family. The plethora of recent papers in Neuron has greatly expanded our knowledge of how these proteins function in a neural context where they regulate neurulation, cell migration, and axon guidance. They have documented important roles for cell surface receptors with links to proteins that control dynamics or localization of the actin cytoskeleton. Of particular interest are demonstrations of the conserved axon guidance functions of the Ena/VASP proteins and their regulation by protein kinases and receptor phosphatases. Many opportunities remain for utilizing neurons to increase our understanding of the many regulatory circuits that control the shape and dynamics of the actin cytoskeleton.
Selected Reading
- Aszódi A, Pfeifer A, Ahmad M, Glauner M, Zhue Z-H, Ny L, Andersson K-E, Kehrel B, Offermanns S, Fässler R. EMBO J. 1999;18:37–48. doi: 10.1093/emboj/18.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerle MC. Cell. 1998;95:741–748. doi: 10.1016/s0092-8674(00)81697-9. [DOI] [PubMed] [Google Scholar]
- Elkins T, Zinn K, McAllister L, Hoffmann FM, Goodman CS. Cell. 1990;60:565–575. doi: 10.1016/0092-8674(90)90660-7. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Comer AR, Juang J-L, Ahern SM, Clark MJ, Liebl EC, Hoffmann FM. Genes Dev. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- Giniger E. Neuron. 1998;20:667–681. doi: 10.1016/s0896-6273(00)81007-7. [DOI] [PubMed] [Google Scholar]
- Kaufmann N, Wills ZP, Van Vactor D. Development. 1998;125:453–461. doi: 10.1242/dev.125.3.453. [DOI] [PubMed] [Google Scholar]
- Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson BT, Miczak KA, Baltimore D. Neuron. 1998;21:1243–1257. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- Krueger NX, Van Vactor D, Wan HI, Gelbart WM, Goodman CS, Saito H. Cell. 1996;84:611–622. doi: 10.1016/s0092-8674(00)81036-3. [DOI] [PubMed] [Google Scholar]
- Lanier LM, Gates MA, Witke W, Menzies AS, Wehman AM, Macklis JD, Kwiatkowski D, Soriano P, Gertler FB. Neuron. 1999;22:313–325. doi: 10.1016/s0896-6273(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Hall A. Rho GTPases. J. Biol. Chem. 1998;273:20685–20688. doi: 10.1074/jbc.273.33.20685. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Insall RH. Curr. Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, Pollard TD. Proc. Natl. Acad. Sci. USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. EMBO J. 1995;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Pages C, Kedersha NL, Fazikas L, Medley Q, Debant A, Streuli M. EMBO J. 1995;14:2827–2838. doi: 10.1002/j.1460-2075.1995.tb07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HJ, Ming G, He Z, Lehmann M, McKerracher L, Tessier-Lavigne M, Poo M-M. Science. 1998;281:1515–1518. doi: 10.1126/science.281.5382.1515. [DOI] [PubMed] [Google Scholar]
- Suetsugu S, Miki H, Takenawa T. EMBO J. 1998;17:6516–6526. doi: 10.1093/emboj/17.22.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills Z, Bateman J, Korey C, Comer A, Van Vactor D. Neuron. 1999a;22:301–312. doi: 10.1016/s0896-6273(00)81091-0. [DOI] [PubMed] [Google Scholar]
- Wills Z, Marr L, Zinn K, Goodman CS, Van Vactor D. Neuron. 1999b;22:291–299. doi: 10.1016/s0896-6273(00)81090-9. [DOI] [PubMed] [Google Scholar]