

Abstract
We have previously shown that mice with a CNS restricted knock-out of the integrin β1 subunit gene (Itgb1-CNSko mice) have defects in the formation of lamina and folia in the cerebral and cerebellar cortices that are caused by disruption of the cortical marginal zones. Cortical structures in postnatal and adult Itgb1-CNSko animals are also reduced in size, but the mechanism that causes the size defect has remained unclear. We now demonstrate that proliferation of granule cell precursors (GCPs) is severely affected in the developing cerebellum of Itgb1-CNSko mice. In the absence of β1 expression, GCPs lose contact with laminin in the meningeal basement membrane, cease proliferating, and differentiate prematurely. In vitro studies provide evidence thatβ1 integrins act at least in part cell autonomously in GCPs to regulate their proliferation. Previous studies have shown that sonic hedgehog (Shh)-induced GCP proliferation is potentiated by the integrin ligand laminin. We show that Shh directly binds to laminin and that laminin–Shh induced cell proliferation is dependent on β1 integrin expression in GCPs. Taken together, these data are consistent with a model in which β1 integrin expression in GCPs is required to recruit a laminin–Shh complex to the surface of GCPs and to subsequently modulate the activity of signaling pathways that regulate proliferation.
Keywords: integrin, laminin, sonic hedgehog, proliferation, granule cell precursor
Introduction
Cerebellar granule cells are excellent models to analyze the mechanisms that control neurogenesis in the CNS. Molecular markers that distinguish different stages of granule cell differentiation are available (Kuhar et al., 1993; Diaz et al., 2002; Zhao et al., 2002), and the cells undergo sequential steps of development in spatially well defined regions. Granule cell precursors (GCPs) are generated in the rhombic lip and move across the developing cerebellar surface to form the external granule cell layer (EGL). GCPs in the outer part of the EGL proliferate and generate postmitotic granule cells that are displaced to more internal parts of the EGL, where they start to migrate along Bergmann glial fibers through the Purkinje cell layer (PCL) to form the internal granule cell layer (IGL) (Herrup and Kuemerle, 1997; Hatten, 1999).
Several lines of evidence suggest that extracellular matrix (ECM) proteins are instrumental in regulating GCP proliferation and differentiation. First, proliferating GCPs are in contact with laminin (LN) and proteoglycans. These molecules strongly enhance the ability of sonic hedgehog (Shh), a mitogen for GCPs, to induce GCP proliferation (Dahmane and Ruiz-i-Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999; Pons et al., 2001; Rubin et al., 2002). Second, differentiating GCPs are in contact with vitronectin (VN), and VN can induce GCP differentiation (Pons et al., 2001). Finally, collagen IV (CoIV) regulates the extension of granule cell axons (Bhatt et al., 2000). The Discoidin domain receptor 1 (DDR-1) mediates effects of CoIV on axon extension (Bhatt et al., 2000), but the receptors that mediate effects of other ECM components are not known. Good candidates are β1 integrins that bind to LN and VN and cooperate with growth factor receptors to stimulate cell proliferation in many tissues (Giancotti and Ruoslahti, 1999). β1 integrins also regulate the distribution of ECM components around cells (Schwarzbauer, 1999). Integrins may therefore affect signaling pathways that regulate GCP proliferation and differentiation, as well as the distribution and local concentration of morphogens such as Shh by organizing the ECM (Bellaiche et al., 1998; The et al., 1999; Pons and Marti, 2000).
To determine the function of β1 integrins in the regulation of proliferation and/or differentiation of GCP, we have analyzed the cerebellum of mice with a CNS restricted knock-out of the β1 integrin subunit gene (Itgb1CNSko mice). These mice were generated by crossing Itgb1flox mice with nestin-Cre mice. Our previous studies have shown that in Itgb1-CNSko mice the lamination and foliation of cortical structures is perturbed, because of defects in the cortical marginal zone, where basement membrane deposition is affected, radial glial fibers do not form endfeet, and the Cajal Retzius cell layer is disrupted (Graus-Porta et al., 2001). However, Itgb1CNSko mice have additional defects that cannot easily be explained by defects in the cortical marginal zones. Notably, the cerebellar cortex of adult mice is strongly reduced in size. We demonstrate that the defect in cerebellar size is caused by defects in GCP proliferation, and we provide evidence that β1 integrins, LN, and Shh cooperate to regulate GCP proliferation.
Materials and Methods
Mouse lines and analysis of Cre activity. The mouse lines have been described: Itgb1+/- (Stephens et al., 1995), Itgb1flox (Graus-Porta et al., 2001), nestin-Cre (Trouche et al., 1999), Rosa26lacZflox (Mao et al., 2001). Protocols for genotyping and to analyze Cre activity by X-gal staining have been described (Graus-Porta et al., 2001).
Histology and immunohistochemistry. Brains were fixed overnight at 4°C in Carnoy (60% ethanol, 30% chloroform, and 10% acetic acid), embedded in paraffin, sectioned at 8 μm, and stained with hematoxylin and eosin (Graus-Porta et al., 2001). For BrdU labeling (Sigma, St. Louis, MO), mice were injected intraperitoneally with 100 μg BrdU/gm body weight and killed 1 hr later. Immunohistochemistry with anti-BrdU antibodies (BD Biosciences, San Diego, CA) was performed as described (Graus-Porta et al., 2001). For staining with Math-1 antibodies (provided by E. Johnson, University of Texas Southwestern Medical Center, Dallas, TX), brains were frozen, embedded in OCT compound (VWR, West Chester, PA), and 12 μm cryosections were prepared, and fixed in 4% paraformaldehyde for 5 min. For staining with antibodies to p27–Kip1 (BD Biosciences), TAG-1 (provided by E. Stoeckli, University of Zurich, Zurich, Switzerland), calbindin (Sigma), GABAA receptor α6 subunit (Chemicon, Temecula, CA), integrin α7 subunit (provided by U. Mayer, University of Manchester, Manchester, UK), NeuN (Chemicon), P-CREB (BD Biosciences), and LN-1/2 (provided by L. Sorokin, University of Uppsala, Uppsala, Sweden), Carnoy-fixed paraffin sections were used. In experiments with anti-p27–Kip1 antibody, sections were microwaved three times for 3 min in 10 mm citrate buffer, pH 6.0, before blocking. For LN chain-specific antibody staining, postnatal day 10 brains were frozen in OCT and sectioned at 8 μm in a cryostat. Antibodies to LNα1, β1 (provided by D. Abrahamson, University of Kansas Medical Center, Kansas City, KA), LNα2 (Alexis Biochemicals, San Diego, CA), LNα3, α4, β2 (provided by R. Timpl and T. Sasaki, Max Planck Institute, Martinsried, Germany), LNα5 (Miner et al., 1997), and LNα1 (Chemicon) were used to stain unfixed sections after blocking in 10% normal goat serum in PBS. Caspase-3 antibody (PharMingen, San Diego, CA) was used on vibratome sections treated with acetone for 30 sec at –20°C. For detection, we used secondary antibodies coupled to fluorescence dyes (Jackson ImmunoResearch, West Grove, PA) and either conventional or deconvolution fluorescence microscopy.
Cerebellar cell cultures. Cells were purified from P4–5 cerebella using a modification of the procedure described by Hatten et al. (1998) Cerebellar pieces were incubated at 37°C for 5–10 min in buffer consisting of Earle's balanced salt solution (Invitrogen, Carlsbad, CA), supplemented with 0.1% glucose, 1% trypsin, 0.1% DNase (Worthington, Lakewood, NJ), 1 mm MgSO4,and6mm NaOH. The buffer was replaced with BME (Invitrogen) containing 0.05% DNase and 0.25% glucose. The tissue was triturated using pipettes of decreasing bore size to obtain a single cell suspension. Cells were harvested by centrifugation at room temperature (RT), resuspended in EBSS containing 0.1% glucose, the cell suspension was passed through a cell strainer (BD Biosciences), recentrifuged, and resuspended in serum-free medium consisting of Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen), glutamine, 20 mm KCl and penicillin–streptomycin. Where indicated, cells were further purified by Percoll gradient centrifugation (Hatten et al., 1998). Cells (2 × 105/well) were plated onto glass coverslips coated with 10 μg/ml poly-l-lysine (PLL) or coated with 10 μg/ml PLL and 10 μg/ml of either LN or VN (Sigma). Cells were cultured for3din serum-free medium in a humidified atmosphere at 37°C with 5% CO2. Where indicated, 1–3 μg/ml of Shh were added directly after plating the cells, or 50 ng/ml EGF or IGF (Sigma). For preincubation experiments, 1–3 μg/ml Shh in Neurobasal medium was added onto the coated glass plates and incubated for 2 hr at 37°C. Coated glass plates were washed three times with Neurobasal medium, cells were plated and cultured without additional Shh for 3 d. Four hours before fixation, cultures were treated with 50 ng/ml BrdU. To test cell adhesion, cells were plated in 24 well plates coated with either 10 μg/ml PLL, LN, or VN, cultured for 1 d and imaged. The recombinant Shh used in this study was a purified autocleaved 19 kDa N-terminal fragment (Pons et al., 2001). The Shh antibody for function blocking studies was obtained from the Developmental Studies Hybridoma Bank.
For immunostainings of cerebellar cultures, cells were fixed in 4% PFA treated with 4 N HCl for 10 min to denature DNA, neutralized for 5 min with 0.1 m sodium borate buffer, pH 8.5, and washed three times in PBS. Cells were blocked for 1 hr at RT in 10% NGS, 0.1% Triton X-100 in PBS (PBS+), and incubated with primary antibodies for 1 hr at RT in PBS+. After washing in PBS, sections were incubated with secondary antibody (Vector Laboratories, Burlingame, CA) for 1 hr at RT in PBS+. Colometric detection using the ABC Elite Kit (Vector Laboratories) was performed according to the manufacturer's instructions. Primary antibodies were anti-BrdU and optionally anti-GFAP (Dako, Glostrup, Denmark). Secondary antibodies were either biotinylated (Vector Laboratories) or coupled to fluorescence dyes (Jackson ImmunoResearch). Quantification was performed by imaging six randomly selected fields with Nomarski optics. The number of total and BrdU-positive cells was counted (∼200–1000 cells per field). In general, three or more experiments were performed. For the preincubation with VN, two experiments were performed. Statistical significance was evaluated by Student's t test.
ELISA assays. Nunc PolySorb 96 well plates (Nalgen, Rochester, NY) were coated with varying concentrations of LN-1, CoIV, or fibronectin (all Sigma) in PBS for 2 hr at 37°C. Wells were blocked for 2 hr with 4 mg/ml BSA (Sigma) in PBS, rinsed with PBS, and incubated overnight at 4°C with 1 μg/ml Shh in PBS containing 4 mg/ml BSA. Wells were incubated with antibodies to Shh for 1 hr at 37°C and rinsed with PBS, followed by incubation for an additional hour with secondary antibodies that was coupled to peroxidase (Amersham Biosciences, Buckinghamshire, UK). The wells were rinsed with PBS and water, and 100 μl peroxidase substrate (3, 3′, 5, 5′-tetramethylbenzidine) was added. The color reaction was stopped after a 30 min incubation at room temperature by adding 50 μl 1 N sulfuric acid, and the absorbance was measured at 450 nm. The percentage of binding relative to direct immobilization of 1 μg/ml Shh was determined. The experiments were performed at least three times in duplicates, and the mean and SD were determined.
Results
Expression of β1 integrins in the cerebellum
To analyze the function of β1 integrins in the cerebellum, we first examined the expression pattern of the Itgb1 gene by in situ hybridization of sagittal sections from wild-type mice. At embryonic day 12.5 (E12.5) and E14, Itgb1 was expressed throughout the cerebellar anlage (Fig. 1A) (data not shown). At postnatal day 14 (P14), Itgb1 was still widely expressed, including expression in premigratory and postmigratory granule cells, and in Purkinje cells. Hybridization with sense-control probes confirmed that the observed signals were specific (Fig. 1A). We also analyzed the distribution of integrin α subunits in the developing cerebellum. Previous studies have shown that GCPs and differentiating granule cells in the EGL express the integrin α6β1, an LN receptor (Pons et al., 2001). We could demonstrate that these cells expressed in addition the integrin α7 subunit that heterodimerizes with β1 to form a second LN receptor (Fig. 1B). Interestingly, expression of both the α6 and α7 subunits is downregulated during migration and terminal differentiation of granule cells, as is evident by the lack of expression in the molecular layer and the IGL (Fig. 1B) (Pons et al., 2001). Thus, GCPs and granule cells at early stages of their differentiation in the EGL, but not their differentiated descendants in the IGL, coexpress at least two LN receptors.
Figure 1.

Integrin expression in the cerebellum. A, In situ hybridization was performed with an Itgb1-specific probe on sagittal sections. In wild-type mice, Itgb1 was expressed throughout the cerebellar primordium (CPR), choroid plexus (CP), and mesencephalon (MS) at E14, and in the external granule cell layer (EGL), the internal granule cell layer (IGL), and the Purkinje cell layer (PCL) at P14. No signal was detected with the sense control probe (control). B, Left panel, Proliferating GCPs in the EGL were visualized by staining sections of mice injected with BrdU with antibodies to BrdU. Middle panel, Immunostaining for the integrin α7 subunit (red). Sagittal sections from a P2 wild-type cerebellum revealed thatα7 expression was restricted to the EGL. A single folium is shown. Inset panel shows the same areas stained with DAPI to reveal nuclei. Left panel, Expression of the integrin α7 subunit was not detectable in the EGL of Itgb1-CNSko mice. Scale bars: A, 200 μm; B, 50 μm.
Gene inactivation by Cre—Lox-mediated gene ablation
To address Itgb1 function in cerebellar development, we wanted to inactivate its expression, by crossing a mouse line carrying an Itgb1flox allele (Graus-Porta et al., 2001) with a suitable Cre mouse line. We have previously characterized a nestin-Cre mouse line that induces Cre-mediated recombination in the precursors of neurons and glia in the CNS (Tronche et al., 1999; Graus-Porta et al., 2001). This mouse line may be suitable to analyze cerebellar development, but a detailed characterization of the Cre recombination pattern in different cell types of the cerebellum has not been described. We therefore crossed nestin-Cre mice with a reporter mouse line that carries a Rosa26lacZflox transgene. In this mouse line, LacZ expression is activated by Cre recombination (Mao et al., 1999). LacZ activity was detectable at E10.5 within the neural tube, including the rhombic lip (Fig. 2A). In sagittal sections of E12.5 (data not shown) and E15.5 embryos (Fig. 2B), recombination was evident throughout the cerebellar anlage. At P7, all cells in the EGL, IGL, and the PCL were LacZ-positive (Fig. 2B). In contrast, no recombination was evident in the cerebellar meninges (Fig. 1C) and blood vessels (data not shown). We conclude that the nestin-Cre allele induces efficient recombination in the cerebellum in the precursors of neurons and glia starting at or before E10.5.
Figure 2.
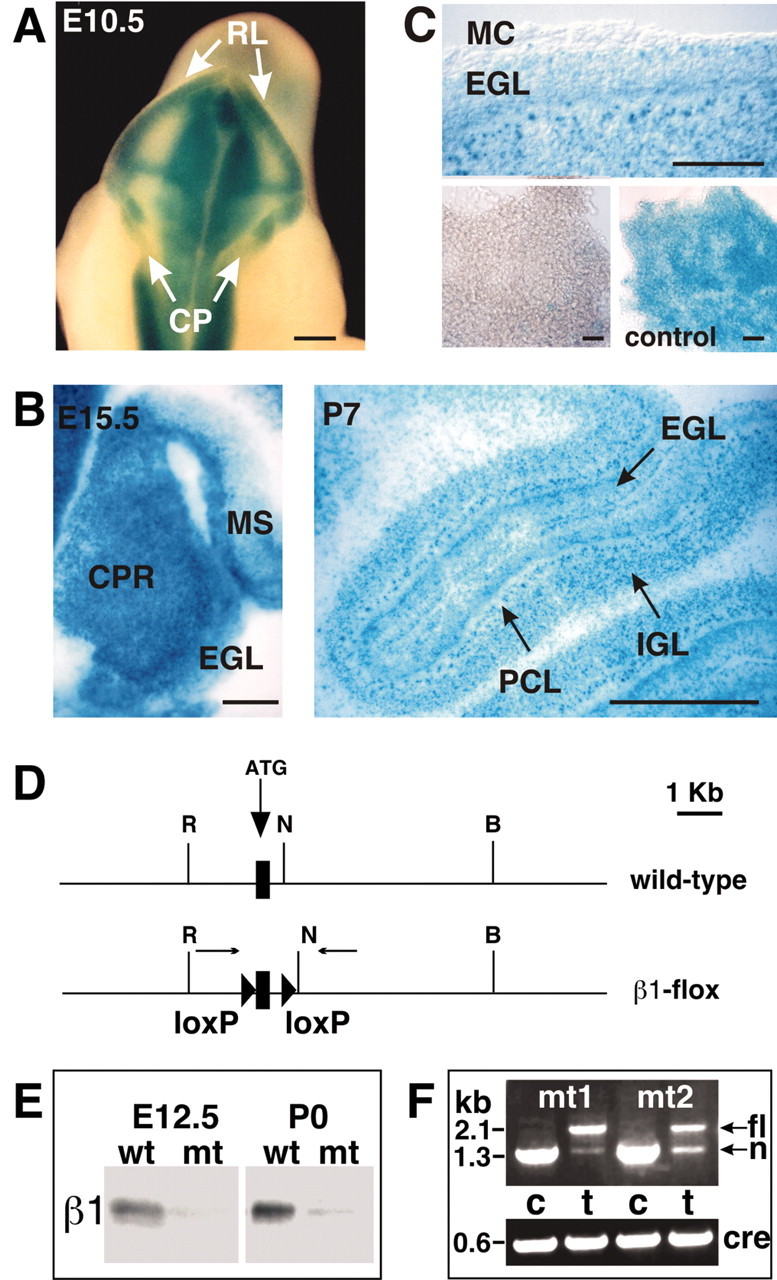
Cre recombination pattern and inactivation of the Itgb1 gene in the cerebellum. A–C, Offspring of intercrosses between Rosa26lacZflox mice and nestin-Cre mice were analyzed for LacZ expression to determine the Cre recombination pattern in the cerebellum. A, At E10.5, recombination was evident in whole-mount staining in the rhombic lip (RL), but not in the developing choroid plexus (CP). B, In histological sections of E15.5 embryos, recombination was apparent throughout the cerebellar primordium (CPR) including the forming EGL. Recombination also occurred in the mesencephalon (MS). At P7, recombination was evident in all cell layers of the cerebellum, including the EGL, IGL, and PCL. Note that the P7 sections were cut thinner than the E15.5 sections, revealing the nuclear localization of LacZ. C, The top panel shows a detail of a sagittal P7 section, revealing that the meningeal cell layer (MC) was not recombined. The bottom panels show meningeal cell layers dissected from P7 mice. Meninges were spread out on a coverslip and photographed. Nestin-Cre did not induce recombination in the meninges (bottom left panel). In control mice that carried a fully recombined Rosa26lacZflox locus, the meninges were LacZ-positive (bottom right panel, control). D, Diagram of the Itgb1 wild-type and Itgb1flox alleles. The first coding exon (rectangle), the lox P sites (triangles), the primers used for analyzing Cre-mediated recombination (arrows), and several restriction endonuclease cleavage sites are indicated (R = EcoRI; N = NheI; B = BamHI). E, The neural tube from E12.5 mice and the cerebellar anlage from P0 mice were dissected. Protein extracts were analyzed by Western blotting. β1 protein was expressed in wild-type mice, but very low amounts in the mutants. Low amounts of β1 protein in the mutants were expected because of β1 expression in blood vessels and meninges. F, DNA was prepared from the cerebellum (c) and tail (t) of two mutant mice at P0 and analyzed by PCR for Cre-mediated recombination. In mouse tail DNA the 2.1 kb Itgb1flox allele (fl) and the 1.3 kb Itgb1null allele (n) were detectable. In the cerebellar DNA only a 1.3 kb band indicative of both the Itgb1null allele and the recombined Itgb1flox allele was apparent. Primers against Cre were used to confirm the presence of the Cre transgene. Scale bars, 200 μm.
We next crossed the Itgb1+/-nestin-Cre mice with Itgb1flox/flox mice (Fig. 2D). To confirm that the Itgb1 gene was effectively inactivated in the mutant offspring (referred to as Itgb1-CNSko mice), we dissected the neural tube and cerebellar anlage from wild-type and mutant mice at E12.5 and P0, respectively. Extracts were prepared, and the β1 protein levels were determined by Western blot (Fig. 2E). The β1 protein was detectable in wild-type mice, but it was almost absent in the mutants. Trace amounts of β1 protein were evident after longer exposure of the blots. This was expected because the Itgb1 gene is expressed in blood vessels and meninges that contaminated our samples, but did not express Cre (Fig. 2C). We conclude that the nestin-Cre transgene induces efficient recombination of the Itgb1flox allele in developing neurons and glia at or before E12.5. Recombination was confirmed by PCR analysis (Fig. 2F).
We also analyzed the expression of integrin subunits by immunohistochemistry. Unfortunately, none of several available β1-specific antibodies stained neural tissue of wild-type mice reproducibly (data not shown). However, the integrin β1 subunit is an exclusive partner for several integrin α subunits, and heterodimerization of the α and β1 subunits is essential for integrin cell surface expression (Hemler, 1999). We therefore analyzed the expression of the integrin α7 subunit that is expressed in the EGL in wild-type mice (Fig. 1B). As expected, α7 expression was not detected in the mutants (Fig. 1B), demonstrating that inactivation of the β1 subunit leads to the inactivation of the α7β1 heterodimer, and likely of other αβ1 heterodimers as well.
β1 integrins are critical for GCP proliferation
We have previously shown that Itgb1-CNSko mice have defects in the glial fiber network, in the assembly–remodeling of meningeal basement membranes, and in the formation of the Cajal Retzius cell layer. As a consequence, the laminar organization of cortical structures is affected, and cerebellar folia fuse (Graus-Porta et al., 2001). The cerebellum of adult Itgb1CNSko mice is also reduced in size, as evident from the decreased depth of the folia and the irregular appearance of the IGL (Fig. 3A,B), and granule cells accumulate in ectopic positions along the fusion lines of adjacent cerebellar folia (Fig. 3C,D). We now addressed whether defects in cerebellar size were caused by a developmental defect such as impaired expansion of the GCP pool in the absence of β1 integrins. We therefore performed cell counts of dissociated cerebellar cortices at varying postnatal ages during the growth phase of the cerebellar cortex. Cell numbers were comparable between wild-type and mutant mice at P0, but they were reduced in the mutants by P5 and P7 (Fig. 3G). The reduction in total cell numbers was likely a consequence of a reduction in the number of granule cells, because the IGL is clearly reduced in the adult mutant cerebellum, and granule cells are by far the most abundant cell population in the cerebellum (Altman and Bayer, 1997).
Figure 3.
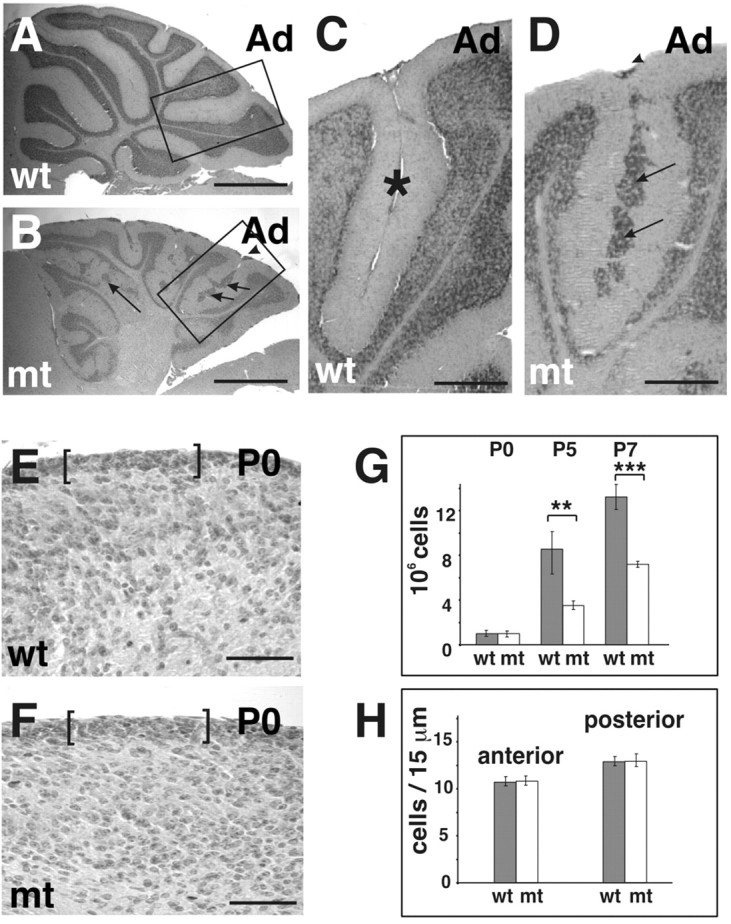
Cerebellar defects in Itgb1- CNSko mice. A, B, Sagittal sections of adult (Ad) animals were stained with hematoxylin–eosin. The cerebellar cortex of wild-type (wt) mice showed a regular foliation pattern. The cerebellar cortex of the mutant (mt) mice lacked fissures and the depth of folia was reduced, but a rudimentary internal foliation pattern was discernible from the shape of the IGL. Note that the overall thickness of the IGL was drastically reduced. Granule cell ectopia remained along the fusion lines of folia (arrows) and on the surface of the cerebellum (arrowhead). C, D, Higher magnification view of the areas indicated in A and B. In wild-type mice, adjacent cerebellar folia were separated by fissures (asterisks). In mutants, adjacent folia fused and granule cell ectopia formed at the cerebellar surface (arrowhead) and along the fusion line (arrows). E, F, Part of a midsagittal sections through the central lobe of the cerebellum of a wild-type and mutant mouse at P0. The EGL is indicated by brackets. G, The cerebellum was dissected from animals of the indicated age and dissociated. The total cell number was determined. For each age, cell counts were performed for three wild-type and three mutant animals. Error bars indicate the SD. A Student's t test was performed (***p < 0.001; **p < 0.01). Scale bars: A, B, 600 μm; C, D, 100 μm; F, G, 50 μm. H, Cell numbers in the EGL were determined both in the anterior and posterior aspect of the central lobe. Cells were counted on two adjacent midsagittal sections for two wild-type and two mutant mice. The mean and SD were determined.
To address that granule cells were predominantly affected and to determine the mechanism that caused the reduction in cell number, we analyzed whether the EGL formed normally in Itgb1-CNSko mice before birth and whether the postnatal defects were caused by changes in cell proliferation or cell death of cells within the EGL. Cell counts in histological sections confirmed that the EGL contained normal numbers of granule cells in the Itgb1-CNSko at P0, providing evidence that the generation of the initial GCP pool was not affected (Fig. 3E–H). However, BrdU pulse labeling demonstrated a proliferative defect in GCPs in the EGL by P2 (Fig. 4C–E), particularly within the forming folia, which were fused in the mutant (Fig. 4B,E). The defect was most prominent within the fastest growing folia where the proliferation rate is presumably highest. Therefore, the EGL of the folia where the primary (pf) and secondary fissure (sf) would form showed a 50–70% reduction in proliferation. In contrast, little defect was observed at this time in the slower growing EGL of the folium where the prepyramidal fissure (ppf) would form (Fig. 4C).
Figure 4.

Defective granule cell precursor proliferation. A, B, Sagittal sections of P2 animals were stained with hematoxylin–eosin. The folia in the mutant mice lacked fissures, and adjacent EGLs within the folia were fused. Note that in the mutants the EGL in some folia was thinner than in the wild-type mice. Boxed areas indicate folia that would be separated by the primary fissure (pf), prepyramidal fissure (ppf), and secondary fissure (sf). C–I, Mice were injected with BrdU and killed 1 hr later. Sagittal cerebellar sections were analyzed for BrdU incorporation. C, The number of proliferating GCPs was determined at P2 by counting BrdU-positive cells in the EGL of the folia indicated in A and B. Two sections were counted per animal (2 wild-type and 2 mutant mice). Error bars indicate the SD. A Student's t test was performed (***p < 0.001). D–I, Double staining for BrdU (green) and LN (red). D, E, Example of a P2 folium. At P2, proliferation was greatly diminished in the mutant cerebellum, in particular within the developing folia. Note that LN was incorporated into the folium in the wild-type cerebellum but was absent in the mutant. Scattered BrdU-positive cells in deeper layers of the cerebellum (arrows) were probably glial cells (WM, white matter). Arrowheads indicate LN immunostaining around blood vessels. F, G, At P7, BrdU-negative ectopia formed in the fused folia (arrows) in the mutant, in areas where GCPs in wild-type mice were in contact with ECM components. The EGL–PCL boundary is outlined. H, I, BrdU labeling at P14 revealed strong proliferative defects at the cerebellar surface (dashed line), and almost complete absence of proliferation within the folia (asterisks) of the mutants. The IGL–PC border is outlined (dotted line). At this stage the basement membrane component LN was also absent from the cerebellar surface (dashed line) in the mutant. J, The number of apoptotic cells was determined at P7 by counting caspase 3-positive cells in the EGL of the folia indicated in A and B. Five to seven sections were counted each in three wild-type and two mutant mice. Error bars indicate the SD. K, L, Caspase 3 staining (red) at P7. The PCL–IGL boundary (dotted line) and the cerebellar surface (dashed line) is outlined. Arrows point to caspase 3-positive cells. Scale bars: A, B, 100 μm; D–I, 40 μm; K, L, 60 μm.
The proliferative defects were more pronounced by P7. At this time point, the EGL in wild-type mice contained several layers of proliferating cells (Fig. 4F). In the mutants, the proliferation zone was multiple cell layers deep, but nonproliferating cells also accumulated within this zone. This was most evident within the fused folia, where islands of nonproliferating cells were intermixed with proliferating cells (Fig. 4G). By P14, GCPs in wild-type mice were still proliferating, albeit in reduced number when compared with earlier developmental ages, and proliferating cells were confined to one or two cell layers (Fig. 4H). In the mutants, GCP proliferation had ceased almost completely within all folia and at the cerebellar surface (Fig. 4I).
We also analyzed apoptosis in the cerebellum of wild-type and Itgb1-CNSko mice by Tunel and Caspase 3 staining (Fig. 4J–L). There was no difference in the number of apoptotic cells, providing evidence that cell death did not contribute to the defect in granule cell numbers in Itgb1-CNSko mice.
The onset of the proliferative defects correlated with defects in the deposition of ECM components such as LN, collagen IV, and entactin into meningeal basement membranes. Accordingly, proliferative defects were first evident within the folia where ECM was absent from the onset of their formation (Fig. 4, compare D, E, and F,G) (data not shown). At the cerebellar surface, where defects in the formation of the basement membrane were evident only starting at P7, a similar correlation between decreased cell proliferation and depletion of ECM components was observed (Fig. 4I). These data provide evidence that ECM has an important role in maintaining granule cell precursor proliferation in vivo.
We next investigated which particular LN chains and, by inference, which LN trimers are normally present in the meningeal basement membrane during this period of GCP proliferation. Immunostaining of sections of wild-type P7 and P10 brains with a panel of LN chain-specific antibodies revealed the presence of LNs α1, α2, α4, α5, β1, and β2 in the meningeal basement membrane of the cerebellum (Fig. 5) (data not shown). These chains can associate to form LNs-1, -2, -3, -4, -10, and -11, which are all likely ligands for β1 integrins (Cologonato and Yurchenko, 2000).
Figure 5.

Expression of LN subunits in the cerebellum. Sagittal cerebellar sections were stained with antibodies directed against different LN subunits as indicated in the panels. A, Part of a cerebellum at P10 is shown. B–F, High-magnification view of cerebellar folia at P7. The arrows point to the meningeal basement membranes adjacent to the EGL. The arrowheads point to blood vessels. Scale bars: A, 400 μm; B–F, 50 μm.
We conclude that β1 integrins are critical for the expansion of the GCP pool within the EGL during postnatal ages. Although the EGL formed normally in the Itgb1-CNSko mice before birth, we cannot exclude that β1 integrins may have additional functions in GCPs before birth. Small amounts of β1 protein may have remained early after Cre-mediated recombination that could have escaped detection.
Upregulation of p27–Kip1 and premature GCP differentiation
In the absence of β1 integrins, nonproliferating cells accumulate within the proliferation zone of the EGL (Fig. 4G), suggesting that self-renewing GCPs exited the cell cycle prematurely, leading to a depletion of the precursor pool within the EGL and to granule cell differentiation. To test this hypothesis, we analyzed whether the expression of cell cycle regulators was affected and whether the nonproliferating cells had acquired characteristics of differentiated granule cells.
We first analyzed the expression of p27–Kip1, a negative regulator of cyclin-dependent kinases and of the cell cycle in GCPs (Huard et al., 1999; Miyazawa et al., 2000). In wild-type mice, p27–Kip1 is expressed in granule cells in the IGL and in differentiating but not proliferating cells in the EGL (Fig. 6A,B). In the Itgb1-CNSko mice, expression of p27–Kip1 was upregulated in ectopically located BrdU-negative cells within the fused proliferative zone of the EGL (Fig. 6B,C). We next analyzed the expression pattern of markers for granule cell differentiation. These included Math-1, which is specifically expressed in proliferating GCPs, and TAG-1, a marker for early differentiating granule cells (Fig. 6A) (Furley et al., 1990; Helms and Johnson, 1998). Ectopically localized BrdU-negative, p27–Kip1-positive cells within the proliferative zone of the EGL did not express Math-1, in contrast to the proliferating cells (Fig. 6D). Moreover, the nonproliferating cells were TAG-1-positive (Fig. 6B,D). They also started to express at subsequent ages markers for differentiated postmigratory granule cells such as P-CREB, NeuN, and GABAARα6 (Fig. 6E) (data not shown) (Mullen et al., 1992; Thompson and Stephenson, 1996; Pons et al., 2001). Collectively, these data strongly indicate that in the absence of β1 integrins a cell cycle exit checkpoint is deregulated in normally self-renewing GCPs within the EGL, leading to their premature differentiation.
Figure 6.
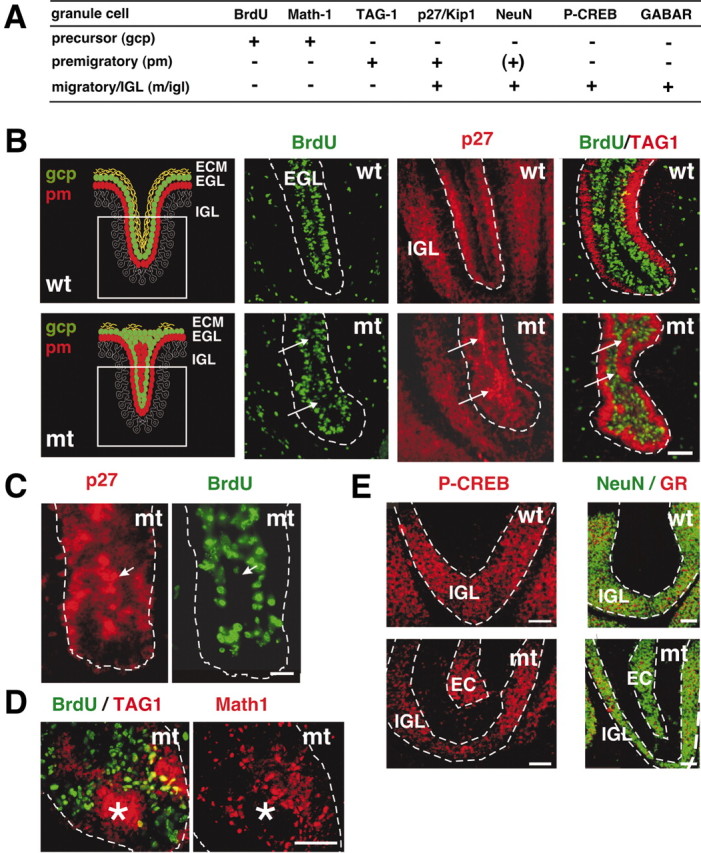
Premature differentiation of granule cells. The expression of molecular markers was analyzed to determine the differentiation state of BrdU-negative granule cells within the proliferation zone of the EGL. A, Summary of the expression patterns of molecular markers during granule cell differentiation [gcp, GCPs in the EGL; pm, premigratory granule cells in the EGL; m/igl, migrating cells and cells in the IGL; (–), no expression; (+), expression]. B–D, Analysis of P7 wild-type and mutant cerebella (sagittal sections). Mice were injected with BrdU and killed 1 hr later. B, The left panels show a schematic illustration of the differentiation states of granule cells in the EGL of a wild-type (top panel) and mutant (bottom panel) folium at P7. The boxed area indicates the region shown in the subsequent immunostainings. Immunostaining for BrdU (green) and p27–Kip1 (red) on adjacent sections: in wild-type, postmitotic but not BrdU-positive cells expressed p27–Kip1. In the mutant, BrdU-negative ectopia formed in the fused folia (arrows) in areas in which GCPs in wild-type mice are in contact with ECM components (Fig. 4 D,E). p27–Kip1 was expressed in the BrdU-negative ectopia in the mutants (arrows). Double staining for BrdU (green) and TAG-1 (red): GCPs that ceased to proliferate started to differentiate in ectopic locations (arrows). C, D, Higher magnification of a tip of a folium in the mutant. C, Staining for p27–Kip1 (red) and BrdU (green): granule cells that ceased to proliferate upregulated p27–Kip1 (arrow). D, Double staining for BrdU (green) and TAG-1 (red) or Math-1 (red) on an adjacent section at P7: the BrdU-negative ectopia expressed TAG-1, but not Math-1 (asterisk). Thus, the cells lost their GCP character in an ectopic location and initiated differentiation. The EGL is outlined. E, Staining for P-CREB (red), or NeuN (green) and GABARα6 (GR; red) in sections from P14 animals: in wild-type mice P-CREB, NeuN, and GABARα6 were expressed only in the IGL. In contrast, these proteins were expressed in mutant mice in nonproliferating cells in ectopic positions (EC) in the EGL. Scale bars, 40 μm.
β1 integrins act cell autonomously to regulate GCP proliferation in vitro
Shh and LN cooperate in vitro to induce GCP proliferation by an unknown mechanism (Pons et al., 2001). Because GCPs and granule cells during early stages of their differentiation express the LN receptors α6β1 and α7β1 (Fig. 1) (Pons et al., 2001), the proliferative defects described here may be a consequence of defects in LN–Shh-dependent signaling pathways. To test this hypothesis, we compared the proliferative response of cultured wild-type and Itgb1-deficient GCPs to Shh on different ECM substrates.
We first determined whether β1 integrins are essential to mediate interactions of GCPs and differentiating granule cells with LN. We therefore cultured cells from wild-type and Itgb1-CNSko mice on different substrates. For these experiments, LN-1 or the control substrate VN or PLL were coated directly onto plastic. We chose the LN-1 isoform because it is expressed in the cerebellum (Fig. 5), and readily available in purified form. Both wild-type and Itgb1-deficient cells adhered to and spread equally well on PLL, and did not adhere to VN. Wild-type cerebellar cells also attached to and spread on LN-1. In contrast, Itgb1-deficient cells only loosely adhered to LN-1 and formed instead clusters on top of glial cells (Fig. 7A) (data not shown). We therefore conclude that by deleting β1 integrins interactions of granule cells with LN isoforms such as LN-1 are abolished, suggesting that these cells do not express any other LN receptors, such as dystrolycan (Henry and Campbell, 1999).
Figure 7.
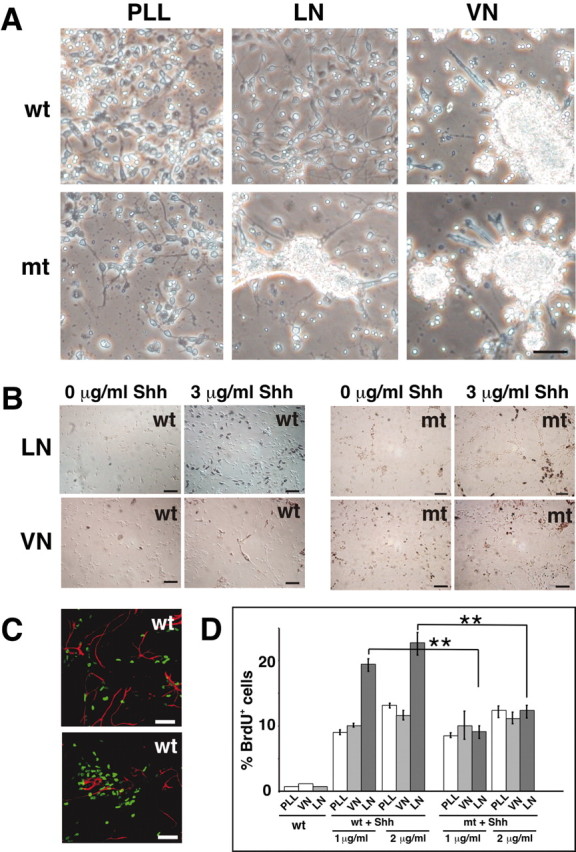
Effects of Shh and ECM on granule cell precursor proliferation. A, Wild-type and Itgb1-deficient cells were plated in plastic tissue culture wells coated with PLL, LN-1, or VN. Phase-contrast images of cells after 1 d in culture. Note that wild-type granule cells adhered to and spread on PLL and LN-1, but not on VN, and that adhesion to LN-1 was abolished in Itgb1-deficient cells. B, Cerebellar cells were plated on PLL–LN-1 or PLL–VN and cultured for 3 d in the absence or presence of Shh. Before fixation, cultures were pulsed with BrdU for 4 hr and stained with an anti-BrdU antibody (brown color). Both wild-type (wt) and Itgb1-deficient (mt) cells were plated and compared. C, Immunostaining for GFAP (red) and BrdU (green) showing that the proliferating cells are not GFAP-positive glial cells. D, Quantification of BrdU-positive wild-type and mutant cells cultured in the presence or absence of Shh (1 and 2 μg/ml) on ECM substrates. Analysis was performed by counting total cells and BrdU-immunostained cells. BrdU-positive cells were expressed as a percentage of the total cell number. A minimum of 1000 cells was counted per experiment. A Student's t test was performed (**p ≤ 0.01). Scale bars, 40 μm.
We next wanted to determine the proliferative response of GCPs to LN-1 and Shh in vitro. To ensure that potential differences in the proliferative response of wild-type and Itgb1-deficient cells were not caused by defects in cell adhesion or spreading, we plated cells on surfaces that had been precoated with PLL and were subsequently coated with LN or VN. Both wild-type and Itgb1-deficient cells attached to and spread equally well on these mixed substrates (Fig. 7B). Subsequent to plating, cells were cultured for 3 d in the presence or absence of Shh, pulse-labeled with BrdU, and immunostained with anti-BrdU antibodies to quantify the number of proliferating cells (Fig. 7B,D). Cultures were also stained with anti-GFAP antibodies to exclude that BrdU-incorporating cells were glial cells (Fig. 7C). In cultures of wild-type cells, <0.5% of the GCPs incorporate BrdU on any substrate in the absence of Shh. As previously demonstrated, addition of Shh to these cultures increased the proliferative response significantly, an effect that was further stimulated when cells were plated on LN-1 (Fig. 7B,D) (Pons et al., 2001). In contrast, we observed that in Itgb1-deficient cells the stimulatory effect of LN-1 on Shh induced proliferation was impaired. Whereas the basic proliferative response to Shh was maintained, the effect of LN-1 to potentiate Shh-promoted GCP proliferation was abolished, and Itgb1-deficient cells on LN-1 proliferated at comparable levels to wild-type cells on PLL and VN (Fig. 7B,D).
Because the initial in vitro experiments were performed with mixed cerebellar cultures that contain additional cell types, such as glia (Fig. 7C), we wanted to exclude that Shh and LN-1 may have affected GCPs secondarily, for example by inducing the release of mitogens from glia. We therefore cultured granule cells and their precursors that had been purified to near homogeneity by Percoll gradient centrifugation (Hatten et al., 1998). Staining of the purified cells with antibodies to GFAP confirmed that the cultures were essentially glia-free (data not shown). Under these culture conditions, Shh also induced proliferation of GCPs. Importantly, this response was still greatly enhanced by LN-1 (Fig. 8A), providing strong evidence that β1 integrins act cell autonomously in GCPs to regulate proliferation.
Figure 8.

β1 integrins and LN-1 cooperate with Shh but not with EGF or IGF to regulate GCP proliferation. A, Granule cells were purified by Percoll gradient centrifugation, plated on the indicated substrates and, where indicated, were treated with 1 μg/ml Shh. Quantification was performed in three independent experiments as described in Figure 7D. The mean and SD were determined, and a Student's t test was performed (***p < 0.001). B, Cells were cultured on LN-1 in the presence of 1 μg/ml Shh. Either no antibody (–) was added, 50 μg/ml antibody to Shh (α-Shh), or 50μg/ml control IgG. The quantification was performed as described in Figure 7D. C, Cells were cultured on PLL or PLL/LN-1 for 3 d in the presence of 50 ng/ml EGF or IGF. The cells were labeled for the final 6 hr of culturing with BrdU, and the percentage of BrdU-positive cells was determined. The results show the mean and SD from three different experiments.
To confirm that the Shh preparation used here was not contaminated with other growth factors we added simultaneously Shh and antibodies to Shh to wild-type cells cultured on LN-1 substrates (Fig. 8B). As expected, GCP proliferation was blocked by the antibody to Shh, but not by a control antibody.
Taken together, these data show that β1 integrins are essential to mediate the cooperative effect of Shh and LN-1 on GCP proliferation, but are not required for the basic proliferative response of GCPs to Shh. It is striking that although both wild-type and Itgb1-deficient cells equally well attached to and spread on mixed PLL–LN-1 substrates (because of the presence of PLL), only wild-type cells react to the presence of LN-1. This strongly suggests that cell attachment and spreading, and close proximity to LN-1 alone is not sufficient to induce proliferation, but that β1 integrins in GCPs have active roles in mediating responses to Shh–LN-1.
LN and β1 integrins are not required for EGF and IGF signaling in GCPs
β1 integrins regulate cell proliferation in many cells (Giancotti and Ruoslahti, 1999). Therefore, β1 integrins may be required for maintaining the basic proliferative capacity of different cell types, including GCPs, rather than affecting specifically responses to Shh. We therefore analyzed whether proliferative responses of GCPs to mitogens such as EGF and IGF were dependent on β1 integrins, and modulated by LN-1. Both growth factors induced cell proliferation to a similar extent in wild-type and Itg-deficient cells, and the mitogenic response was not potentiated by LN-1 (Fig. 8C). These data demonstrate that β1 integrins and LN-1 are not generally required for GCP proliferation, but specifically for proliferation in response to Shh–LN-1.
Shh binds to LN-1
Shh has been shown to bind to the ECM component VN (Pons and Marti, 2000). We therefore reasoned that Shh may also bind to LN-1 and that β1 integrins may help to recruit Shh–LN-1 complexes to the surface of GCPs. This may subsequently lead to the activation of signaling pathways via integrins and Shh receptors. We therefore tested whether LN-1 and Shh can bind to each other. For this purpose we performed ELISA assays with purified Shh and LN-1. To perform the experiments, we raised an antibody against the N-terminal signaling fragment of Shh, which was expressed in and purified from Escherichia coli cells as a fusion protein with GST. The antibody was affinity purified, and specifically detected Shh, but not LN in Western blots with purified proteins (data not shown). We next coated increasing concentrations of LN-1 or control substrates such as BSA or fibronectin onto 96 well plates, blocked nonspecific binding sites with BSA, incubated the substrates with 1 μg/ml Shh, followed by extensive washing and immunodetection of bound Shh. Shh bound efficiently to LN-1 in a dose-dependent manner, demonstrating that Shh and LN-1 directly bind to each other (Fig. 9A). No binding was observed to control substrates such as BSA or fibronectin (data not shown).
Figure 9.

Shh binds to LN-1. A, Increasing amounts of LN-1 were coated on 96 well plates, blocked with BSA, and 1 μg/ml Shh was added (+Shh) or omitted (–Shh). The plates were washed with PBS, and bound Shh was detected by ELISA assays. The values were normalized against 1 μg/ml Shh that was directly bound to plastic (100%). The experiment was performed three times, and the mean and SD were determined. B, Dishes were precoated with ECM substrates, incubated with the indicated amounts of Shh, and subsequently washed. Wild-type cells were plated onto the substrates and cultured for 3 d without further addition of Shh. The cells were labeled for the final 4 hr of culturing with BrdU, and the number of BrdU-positive cells was quantified as described in the legend to Figure 7. Two independent representative experiments are shown.
We next analyzed interactions between Shh and LN-1 in a cell biological assay, by analyzing proliferative responses to preformed Shh–LN-1 complexes. Therefore, we coated coverslips first with PLL and LN or VN. We next washed the coverslips thoroughly and added Shh. After an additional incubation step, the substrates were washed to remove unbound Shh. Cells were added and cultured for 3 d without further addition of Shh. There was only a slight increase in the number of BrdU-positive cells in cultures plated on PLL–Shh or VN–Shh. In contrast, we observed a strong increase in the number of BrdU-positive cells on LN-1–Shh (Fig. 9B). These data show that Shh that is bound to LN-1 promotes GCP proliferation, suggesting that presentation of Shh by LN-1 could be important to induce GCP proliferation. However, it is unlikely that direct binding is the only mechanism, because VN, which has no positive effect on GCP proliferation (Fig. 7B,D), also binds to Shh (Pons and Marti, 2000). Because GCPs express receptors that mediate interactions with LN-1 but not VN (Fig. 7A), the results rather are consistent with a model in which integrin-mediated recruitment or retainment of Shh–LN-1 complexes is important, followed by the activation of integrin—Shh-activated signaling pathways.
Discussion
We provide evidence that β1 integrins, their LN ligands, and Shh cooperate to regulate GCP proliferation in the EGL. It has previously been shown that LN and Shh cooperate to regulate GCP proliferation in the EGL (Pons et al., 2001). However, the mechanism of this cooperation has not been addressed. We show that GCPs in the EGL express LN receptors of the β1 integrin family, and that the cells proliferate in close apposition to basement membranes containing LN isoforms. Using a conditional mutant, we demonstrate that in the absence of β1 integrins, interactions with LN are perturbed. Furthermore, the rate of GCP proliferation is impaired. Detailed analysis of the proliferative defects shows that upregulation of the cell cycle inhibitor p27–Kip1 and premature differentiation of Itgb1-deficient GCPs lead to a reduction in the final number of mature granule cells, and a reduced size of the cerebellum. We also demonstrate that LN-1 and Shh bind to each other and that the cooperative effect on GCP proliferation is dependent on β1 integrin expression in GCPs. The data are consistent with a model in which β1 integrins act at two sequential steps. First, they help to recruit LN–Shh complexes to the surface of GCPs. Second, they subsequently affect more directly signaling pathways that are important for GCP proliferation.
Cell autonomous functions for β1 integrins
When β1 integrins are inactivated in neurons and glia using nestin-Cre, GCP proliferation is drastically impaired, but not abolished. This suggests that β1 integrins, although not essential for cell proliferation, are critical for the efficient expansion of the GCP pool in the EGL. Accordingly, proliferative defects are most obvious during peak times of GCP proliferation, i.e., in the most rapidly growing cerebellar folia where the rate of cell proliferation is highest. Several lines of evidence suggest that β1 integrins act cell autonomously in GCPs to regulate cell proliferation. First, GCPs express the LN receptors integrin α6β1 and α7β1. Second, proliferating GCPs in the cerebellum in vivo are in close apposition to basement membranes that contain LNs. Third, LN-1 and Shh cooperate in vitro to induce proliferation of highly purified GCPs. Fourth, the effect of LN-1 and Shh on cell proliferation in vitro is dependent on β1 integrin expression in GCPs.
Previous studies have shown that β1 integrins act cell autonomously within epidermal stem cells to regulate their proliferation (Caroll et al., 1995; Brakebusch et al., 2000; Raghavan et al., 2000). Epidermal stem cells express higher integrin levels than their differentiated descendants, and ectopic overexpression of integrins leads to increased cell proliferation (Carroll et al., 1995). This suggests that the levels of cell surface integrins determine the proliferative capacity of these cells. We have shown that granule cells in the EGL express the integrin α7 subunit that forms heterodimers with β1 (Hemler, 1999). The expression of α7 is downregulated during granule cell migration, suggesting that GCPs, similarly to stem cells in the skin, regulate the cell surface levels of integrin receptors. We have not observed proliferative defects in the cerebellum of α7-deficient mice (S. Blaess and U. Mueller, unpublished observations). This may be explained by the fact that GCPs also express the integrin α6β1 (Pons et al., 2001), and the two receptors could have redundant functions. Mice lacking α6 die at birth (Georges-Labouesse et al., 1996), thereby preventing the analysis of α6–α7 double knockout mice. Further studies, for example with mice carrying floxed α6 alleles, will be necessary to address the function of specific αβ1 heterodimers during GCP proliferation.
Integrins, LN, and Shh
An important finding of our studies is that β1 integrins, their LN ligands, and Shh cooperate to regulate GCP proliferation. Shh is the most potent promoter of GCP proliferation that has been described, and its ability to stimulate GCP proliferation is regulated by proteoglycans, LN, and SDF1-α by unknown mechanisms (Klein et al., 2001; Pons et al., 2001; Rubin et al., 2002). Our data provide the first insights into the mechanisms by which LN modulates Shh function. We not only confirm that the proliferative responses to Shh are potentiated by LN-1, but also show that Shh binds to LN-1 and that expression of β1 integrins in GCPs is required for LN-1 to enhance GCP proliferation. These data are consistent with a model where β1 integrins help to recruit Shh–LN-1 complexes to the cell surface of GCPs, increasing the apparent concentration of Shh close to the Patched–Smoothened receptor complex. However, binding to LN-1 alone cannot be sufficient, because Shh also binds to VN, but VN appears to induce differentiation (Pons and Marti, 2000; Pons et al., 2001). The results rather are consistent with a model in which β1 integrins not only help to recruit Shh–LN-1 complexes to the cell surface, but also regulate the activity of cellular second messenger systems more directly.
Our data show that several additional LN isoforms are expressed in the cerebellum. Studies with genetically modified mice have shown that different LN isoforms have distinct functions. For example, mutations in the LN β2 subunit gene lead to defects in the kidney. The LN β1 subunit, although coexpressed with β2 cannot substitute for its function (Noakes et al., 1995). Intriguingly, the LN-10 and -11 isoforms are only expressed during development of the cerebellum, but not in adult animals (J. Miner, unpublished observation). It will therefore be important to test in the future to what extent different LN isoforms affect Shh function in specific ways.
In vitro studies have provided strong evidence for cooperative signaling between integrins and growth factor receptors of the tyrosine kinase family, where integrins enhance the ability of these kinases to activate downstream signaling targets like MAP kinases (Giancotti and Ruoslahti, 1999). Recent evidence suggests that Shh regulates cell proliferation in different ways from classic mitogenic growth factors. First, Shh keeps cells proliferating, but it does not induce quiescent cells to reenter the cell cycle. Second, stimulation of GCP proliferation by Shh is independent of MAP kinases (Kenney and Rowitch, 2000). Because Shh activates gene expression, it may maintain a gene expression program required for cell proliferation, such as the expression of the cell cycle regulator cyclin D1 (Kenney and Rowitch, 2000). GCPs express cyclin-dependent kinases (cdks), cyclin D1, and cyclin D2. In contrast, postmitotic granule cells express p27–Kip1, a cdk inhibitor. Cell proliferation in the EGL is enhanced in p27–Kip1-deficient mice and decreased in cyclin D2-deficient mice (Huard et al., 1999; Miyazawa et al., 2000). Likewise, expression of cyclin D1 is suppressed in cells that are not anchored to the ECM, and anchorage to the ECM is necessary to down-regulate p27–Kip1 (Fang et al., 1996; Zhu et al., 1996). This may provide an explanation for the proliferative defect observed in GCPs, in which Shh and β1 integrins may fulfill a dual role in regulating the cell cycle. First, they could cooperate to maintain cyclin D1 and D2 expression. Second, they could prevent the expression of the cell cycle inhibitor p27–Kip1, an interpretation that is consistent with our finding that p27–Kip1 is upregulated in the Itg-deficient GCPs. This dual control would ensure a tight regulation of cell proliferation for generating a precise number of granule cells at the appropriate time during development.
β1 integrins, basement membrane assembly, and cell proliferation
The data presented here provide compelling evidence that integrins act cell autonomously within GCPs to regulate their proliferation. The cell autonomous requirement is consistent with a signaling function for β1 integrins in GCP, where they may act at least in part by modulating Shh-dependent signaling pathways. However, the data leave open the possibility that integrins and their ECM ligands contribute to the regulation of GCP proliferation in other ways as well. Integrins are key regulators of basement membrane assembly, and meningeal basement membrane assembly is defective in Itgb1-CNSko mice (Graus-Porta et al., 2001). The spatially restricted assembly of basement membranes in the proliferation zone of the EGL could influence the proliferative behavior of GCPs in several ways. First, GCPs are not only in contact with basement membranes, but also with other GCPs. This could generate asymmetry in the cellular cytoskeleton, affecting cell polarity and orientation of the cleavage spindle during cell division. Second, signaling molecules that promote GCP proliferation and differentiation in the EGL have been suggested to be secreted from meningeal cells, the precursors themselves, and from Purkinje cells (Barakat et al., 1981; Hatten, 1985; Gao et al., 1991; Smeyne et al., 1995; Klein et al., 2001). Cell–ECM interactions could help to recruit secreted signaling molecules locally into the meningeal basement membranes. Shh has been shown to be concentrated at basement membranes, and other signaling molecules such as wnt-wingless, fibroblast growth factors, and bone morphogenetic proteins interact with ECM components and heparan sulfate proteoglycans (Baeg and Perrimon, 2000). Their distribution and signaling function could be affected in the Itgb1-CNSko mice. Interestingly, proliferating cells in other tissues such as skin and bone marrow are situated in close proximity to basement membranes. Thus, cell–ECM interaction may play a fundamental role in maintaining the proliferative capacity of self-renewing precursors and stem cells in many tissues.
Footnotes
This work was supported by the Novartis Research Foundation (U.M.) and the National Institutes of Health (U.M., J.M.). The generation of the Itgb1flox mice was initiated under United States National Institutes of Health Grant NS19090 (L.F.R.). S.B. was supported by the Roche Research Foundation; D.G. by the Swiss Foundation for Research on Muscular Diseases. L.F.R. is an investigator of the Howard Hughes Medical Institute. We thank E. Stoeckli, L. Sorokin, U. Mayer, D. Abrahamson, R. Timpl, T. Sasaki, and J. E. Johnson for antibodies; S. Orkin for Rosa26lacZflox mice; C. Damsky for the mouse line carrying an Itgb1null allele; R. Klein for nestin-Cre mice; members of the laboratory for helpful discussions; D. Monard, A. Matus, W. Krek, A. Kralli, E. Stoeckli, A. Joyner, and M. Zervas for comments.
Correspondence should be addressed to Ulrich Müller, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92073. E-mail: umueller@scripps.edu.
DOI:10.1523/JNEUROSCI.5241-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243402-11$15.00/0
S.B. and D.G.-P. contributed equally to this work.
References
- Altman J, Bayer SA (1997) Development of the cerebellar system in relation to its evolution, structure, and function. New York: CRC.
- Baeg GH, Perrimon N (2000) Functional binding of secreted molecules to heparan sulfate proteoglycans in Drosophila Curr Opin Cell Biol 12: 575–580. [DOI] [PubMed] [Google Scholar]
- Barakat I, Wittendorp-Rechenmann W, Rechenmann RV, Sensenbrenner M (1981) Influence of meningeal cells on the proliferation of neuroblasts in culture. Dev Neurosci 4: 363–372. [DOI] [PubMed] [Google Scholar]
- Bellaiche Y, The I, Perrimon N (1998) Tout-velu is a Drosophila homologue of the putative tumour suppressor EXT-1 and is needed for Hh diffusion. Nature 394: 85–88. [DOI] [PubMed] [Google Scholar]
- Bhatt RS, Tomoda T, Fang Y, Hatten ME (2000) Discoidin domain receptor 1 functions in axon extension of cerebellar granule neurons. Genes Dev 14: 2216–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakebusch C, Grose R, Quondamatteo F, Ramirez A, Jorcano JL, Pirro A, Svensson M, Herken R, Sasaki T, Timpl R, Werner S, Fassler R (2000) Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J 19: 3990–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JM, Romero MR, Watt FM (1995) Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell 83: 957–968. [DOI] [PubMed] [Google Scholar]
- Colognato H, Yurchenco PD (2000) Form and function: the laminin family of heterotrimers. Dev Dyn 218: 213–234. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz-i-Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126: 3089–3100. [DOI] [PubMed] [Google Scholar]
- Diaz E, Ge Y, Yang YH, Loh KC, Serafini TA, Okazaki Y, Hayashizaki Y, Speed TP, Ngai J, Scheiffele P (2002) Molecular analysis of gene expression in the developing pontocerebellar projection system. Neuron 36: 417–434. [DOI] [PubMed] [Google Scholar]
- Fang F, Orend G, Watanabe N, Hunter T, Ruoslahti E (1996) Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science 271: 499–502. [DOI] [PubMed] [Google Scholar]
- Furley AJ, Morton SB, Manalo D, Karagogeos D, Dodd J, Jessell TM (1990) The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell 61: 157–170. [DOI] [PubMed] [Google Scholar]
- Gao WO, Heintz N, Hatten ME (1991) Cerebellar granule cell neurogenesis is regulated by cell-cell interactions in vitro. Neuron 6: 705–715. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M (1996) Absence of integrin alpha 6 leads to epidermolysis bullosa and neonatal death in mice. Nat Genet 13: 370–373. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E (1999) Integrin signaling. Science 285: 1028–1032. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U (2001) Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367–379. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A, Lewis P, McMahon AP, Linde A (2001) The whereabouts of a morphogen: direct evidence for short- and graded long-range activity of hedgehog signaling peptides. Dev Biol 236: 364–386. [DOI] [PubMed] [Google Scholar]
- Hatten ME (1985) Neuronal regulation of astroglial morphology and proliferation in vitro. J Cell Biol 100: 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME (1999) Central nervous system neuronal migration. Annu Rev Neurosci 22: 511–539. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Gao WQ, Morrison MA, Mason CA (1998) The cerebellum: purification and coculture of identified cell populations. In: Culturing nerve cells (Banker G, Goslin K, eds). Cambridge, Massachusetts: MIT.
- Helms AW, Johnson JE (1998) Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development 125: 919–928. [DOI] [PubMed] [Google Scholar]
- Hemler ME (1999) Integrins. In: Guidebook to the Extracellular Matris, Anchor and Adhesion Proteins (Kreis T, Vale R, eds), pp 196–212. New York: Oxford University Press.
- Henry MD, Campbell KP (1999) Dystroglycan inside and out. Curr Opin Cell Biol 11: 602–607. [DOI] [PubMed] [Google Scholar]
- Herrup K, Kuemerle B (1997) The compartmentalization of the cerebellum. Annu Rev Neurosci 20: 61–90. [DOI] [PubMed] [Google Scholar]
- Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME (1999) Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development 126: 1927–1935. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH (2000) Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 20: 9055–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Rubin JB, Gibson HD, DeHaan EN, Alvarez-Hernandez X, Segal RA, Luster AD (2001) SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development 128: 1971–1981. [DOI] [PubMed] [Google Scholar]
- Kuhar SG, Feng L, Vidan S, Ross ME, Hatten ME, Heintz N (1993) Changing patterns of gene expression define four stages of cerebellar granule neuron differentiation. Development 117: 97–104. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH (1999) Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci USA 96: 5037–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner J, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR (1997) THe laminin alpha chains: expression, developmental transitions, and chromosomal locations of alpha 1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel alpha 3 isoform. J Cell Biol 137: 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y (2000) A role for p27–Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci 20: 5756–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM (1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116: 201–211. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP (1995) The renal glomerulus of mice lacking s-laminin/laminin beta 2: nephrosis despite molecular compensation by laminin beta 1. Nat Genet 10: 400–406. [DOI] [PubMed] [Google Scholar]
- Pons S, Marti E (2000) Shh synergizes with the extracellular matrix protein vitronectin to induce spinal motor neuron differentiation. Development 127: 333–342. [DOI] [PubMed] [Google Scholar]
- Pons S, Trejo JL, Martinez-Morales JR, Marti E (2001) Vitronectin regulates Sonic hedgehog activity during cerebellum development through CREB phosphorylation. Development 128: 1481–1492. [DOI] [PubMed] [Google Scholar]
- Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E (2000) Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol 150: 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JB, Choi Y, Segal RA (2002) Cerebellar proteoglycans regulate sonic hedgehog responses during development. Development 129: 2223–2232. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer J (1999) Basement membranes: Putting up the barriers. Curr Biol 9: R242–244. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Chu T, Lewin A, Bian F, S SC, Kunsch C, Lira SA, Oberdick J (1995) Local control of granule cell generation by cerebellar Purkinje cells. Mol Cell Neurosci 6: 230–251. [DOI] [PubMed] [Google Scholar]
- Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH (1995) Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev 9: 1883–1895. [DOI] [PubMed] [Google Scholar]
- The I, Bellaiche Y, Perrimon N (1999) Hedgehog movement is regulated through tout velu-dependent synthesis of a heparan sulfate proteoglycan. Mol Cell 4: 633–639. [DOI] [PubMed] [Google Scholar]
- Thompson CL, Stephenson FA (1994) GABAA receptor subtypes expressed in cerebellar granule cells: a developmental study. J Neurochem 62: 2037–2044. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99–103. [DOI] [PubMed] [Google Scholar]
- Wallace VA (1999) Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9: 445–448. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22: 103–114. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Kho A, Kenney AM, Yuk DI, Kohane I, Rowitch DH (2002) Identification of genes expressed with temporal-spatial restriction to developing cerebellar neuron precursors by a functional genomic approach. Proc Natl Acad Sci USA 99: 5704–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Ohtsubo M, Bohmer RM, Roberts JM, Assoian RK (1996) Adhesion-dependent cell cycle progression linked to the expression of cyclin D1, activation of cyclin E-cdk2, and phosphorylation of the retinoblastoma protein. J Cell Biol 133: 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]