

Abstract
Aflatoxin B1 (AFB) epoxide forms an unstable N7 guanine adduct in DNA. The adduct undergoes base-catalyzed ring opening to give a highly persistent formamidopyrimidine (FAPY) adduct which exists as a mixture of forms. Acid hydrolysis of the FAPY adduct gives the FAPY base which exists in two separable but interconvertible forms that have been assigned by various workers as functional, positional, or conformational isomers. Recently, this structural question became important when one of the two major FAPY species in DNA was found to be potently mutagenic and the other a block to replication [Smela, M. E.; Hamm, M. L.; Henderson, P. T.; Harris, C. M.; Harris, T. M.; Essigmann, J. M. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6655−6660]. NMR studies carried out on the AFB–FAPY bases and deoxynucleoside 3′,5′-dibutyrates now establish that the separable FAPY bases and nucleosides are diastereomeric N5 formyl derivatives involving axial asymmetry around the congested pyrimidine C5–N5 bond. Anomerization of the protected β-deoxyriboside was not observed, but in the absence of acyl protection, both anomerization and furanosyl → pyranosyl ring expansion occurred. In oligodeoxynucleotides, two equilibrating FAPY species, separable by HPLC, are assigned as anomers. The form normally present in duplex DNA is the mutagenic species. It has previously been assigned as the β anomer by NMR (Mao, H.; Deng, Z. W.; Wang, F.; Harris, T. M.; Stone, M. P. Biochemistry 1998, 37, 4374−4387). In single-stranded environments the dominant species is the α anomer; it is a block to replication.
Introduction
Aflatoxins are a group of toxic and carcinogenic fungal metabolites that frequently contaminate peanuts, corn, and other agricultural products; aflatoxin B1 (AFB, 1, Scheme 1) is the most potent of these mycotoxins.1 AFB is highly mutagenic with G → T transversions being the most frequent mutation observed in a wide variety of prokaryotic and eukaryotic systems.2−7 The mutagenicity is ascribed to the reaction of a highly electrophilic metabolite, the exo-8,9-epoxide (2), with DNA in which adduction occurs with high efficiency by attack of guanine N7 at the C8 position of the epoxide to give cationic adduct 3 (Scheme 1).8−15 However, it appears unlikely that 3 is the dominant contributor to the genotoxicity of AFB. Essigmann and co-workers have found 3 to be only moderately mutagenic in E. coli.16 Furthermore, although mainly G → T transversions were observed, nontargeted mutations of the 5′ neighboring base occurred a significant fraction of the time. Such nontargeted mutations have not been observed in living systems exposed to aflatoxin B1. NMR studies of oligodeoxynucleotide duplexes containing 3 have shown that the adduct exists with the aromatic portion of the aflatoxin moiety intercalated on the 5′ face of the guanine residue and that Watson–Crick base pairing is well maintained.17−19
Scheme 1.

Adduct 3 is chemically unstable and has a short half-life in vivo.12,20,21 Heat and/or acid treatment of 3 ruptures the glycosidic linkage to yield AFB–guanine (4) and an apurinic site in the DNA; this pathway occurs under physiological conditions, as well.21 Apurinic sites are noninformational and in E. coli induced for SOS response favor incorporation of adenine opposite the apurinic site, thereby leading to the observed G → T transversion.22,23 However, Essigmann was able to rule this out as the major source of mutation.16 The situation with apurinic sites is more complex in mammalian cells where a variety of mutations is observed.
Adduct 3 also decomposes by a second route involving base-catalyzed cleavage of the imidazole ring to give a formamidopyrimidine (FAPY) derivative.12,24 FAPY formation occurs even at physiological pH and represents a significant decomposition pathway for 3 in cellular DNA.25 AFB–FAPY adducts are more robust than the cationic precursor, requiring harsher conditions to achieve deglycosylation to give the AFB–FAPY base. The DNA lesions are highly persistent in vivo and, consequently, are thought to be the dominant lesion responsible for the genotoxicity of AFB.21,25−29
The structure of the AFB–FAPY lesion is not well understood. Multiple isomeric forms have been observed. Several studies have been carried out on the adducted base and on oligonucleotides, but the results fail to provide a complete picture of the structure. The NMR spectrum of the AFB–FAPY base shows that it exists as four species which can be temporarily separated by HPLC into two fractions, one much larger than the other, with each fraction containing a pair of inseparable species.12,24 Lin et al. assigned the major fraction as 6 and made a tentative assignment of the minor fraction as the cyclic precursor, 8-hydroxydihydroguanine 7 (entry 1, Scheme 2).12 More detailed investigation using NMR spectroscopy led Hertzog et al. to reassign the minor species as regioisomer 8 having the formyl group on N4. Both existed as a pair of geometrical isomers of the formamide (6-ab and 8-ab, entry 2, Scheme 2).24 The N4 assignment is in doubt when viewed in the light of other alkylated FAPY adducts that have been investigated.30−34 Multiple FAPY species exist in those cases as well, but in no case has an N4 formyl species been observed. We now show that the separable species are, in fact, atropisomers 6-A and 6-B (entry 3, Scheme 2).
Scheme 2.

Oligodeoxynucleotides containing the AFB–FAPY lesion also partition chromatographically into two fractions which gradually re-equilibrate. In the oligonucleotides studied thus far it has been observed that in the conversion of cation 3 to the FAPY lesions one FAPY species appears initially (generally, but not always, the earlier eluting), but as the reaction progresses a second species gradually appears and ultimately predominates over the first by a ratio of ∼2:1.35 When the oligodeoxynucleotide containing the mixture of FAPY species is combined with a complementary strand, NMR spectra indicate the presence of a complex mixture of species.36 However, after the sample has aged for several days only one species remains. Two-dimensional NMR studies have shown the latter to have the formyl group attached to N5, the aflatoxin moiety intercalated on the 5′ face of the formamidopyrimidine residue and the sugar to be the β-furanoside.36 It is noteworthy that in the duplex that was studied only one geometrical isomer of the formamide was observed, possibly due to stabilization by hydrogen bonding of the formyl group to the exocyclic amino group of the 3′ adenine.36 Overall, the structure closely parallels that of a duplex containing adduct 3.17 No structural studies have been reported on DNA containing other AFB–FAPY isomers.
Recently, Essigmann and co-workers examined the mutational properties in E. coli of the AFB–FAPY adduct introduced into a plasmid via a site specifically adducted 13-mer oligodeoxy-nucleotide.35 Both FAPY species were studied, the late eluting (major) one in pure form and the early eluting (minor) one as a mixture with the other. The late eluting species was found to be a complete block to replication, but the early eluting one was 6-fold more mutagenic than cationic adduct 3. The dominant mutation was a G → T transversion. These results are consistent with results in AFB-treated E. coli, which supports the contention that the FAPY adduct is the dominant mutagenic lesion produced by AFB. Thus, it becomes crucially important to understand the structures of the AFB–FAPY species present in DNA and the factors that control equilibria among them.
This paper describes structural studies of AFB–FAPY derivatives conducted with the adducted base, nucleoside, and oligodeoxynucleotides. The results provide insight into the structural basis of the differential processing that occurs during replication of DNA containing the two forms of the AFB–FAPY lesion. It should be noted that the species present in oligodeoxynucleotides termed “FAPY minor” and “FAPY major” in Essigmann's work are designated herein as FAPYs I and II, respectively, reflecting the order of formation in the base-catalyzed cleavage of 3.
Materials and Methods
Biological Hazards
Aflatoxin B1 and many of its derivatives are potently carcinogenic. Great care should be exercised to avoid personnel exposure. Crystalline material presents an inhalation hazard because the crystals develop electrostatic charge and cling to dust particles. The dust produced by scraping preparative TLC plates should be regarded as particularly hazardous.
Chemicals
Aflatoxin B1 was purchased from Aldrich Chemical Co. (Milwaukee, WI). Aflatoxin concentration was measured spectrophotometrically using an extinction coefficient of 2.18 × 104 M−1 cm−1 at 362 nm. 2,4,6-Triaminopyrimidine and sodium dithionite were purchased from Aldrich Chemical Co. (Milwaukee, WI). Na15NO2 was purchased from Isotec Inc. (Miamisburg, OH). Adenosine deaminase type IV (from bovine spleen suspension), bacterial nucleoside phosphorylase, and thymidine phosphorylase were purchased from Sigma Chemical Co. (St. Louis, MO). Dimethyldioxirane was synthesized as described by Murray and Jeyaraman37 and Adam et al.38,39 Solutions of dioxirane were stored over anhydrous MgSO4 at −20 °C and were used within 1 month of preparation. AFB epoxide (2),40 AFB–N7–guanine adduct 4,8 2′-deoxyguanosine 3′,5′-dibutyrate (9),15 AFB–FAPY base (6),12,24 and the analogous methyl-FAPY base34 were prepared by literature methods. Dimethyl sulfoxide-d6, methanol-d4, and D2O were obtained from Cambridge Isotope Laboratories, Woburn, MA.
Adduction of 2′-Deoxyguanosine 3′,5′-Dibutyrate with AFB Epoxide
AFB epoxide (2, 8 mg, 24 μmol) and 2′-deoxyguanosine 3′,5′-dibutyrate (9, 20 mg, 48 μmols) were dissolved in anhydrous CH2Cl2 (3 mL). The mixture was stirred in a capped vial for at least 6 h at room temperature. Separation of the product mixture by HPLC (Alltech C18 column, 250 mm × 10 mm, 0−67% CH3CN/0.3 M phosphate buffer, pH 7.0 gradient over 40 min (3.0 mL/min)) gave the N7 and O6 adducts 10 and 11 as the principal products, eluting at 33.9 and 37.6 min, respectively, plus an additional product of uncertain structure eluting at 34.5 min. Complete purification of 10 was not achieved by further chromatography. Instability in DMSO-d6 complicated the assignment of structure.
N7-AFB-2′-Deoxyguanosinium 3′,5′-Dibutyrate (10)
1H NMR (300 MHz, DMSO-d6) δ (aflatoxin signals) 6.96 (1H, d, J = 5.6 Hz, H6a), 6.70 (1H, s, 9-OH), 6.50 (1H, s, H5), 6.29 (1H, s, H8), 5.42 (1H, s, H9), 4.20 (1H, d, J = 5.6 Hz, H9a), 3.89 (3H, s, 4-OCH3), H2 and H3 signals obscured, (2′-deoxyguanosine signals) 8.60 (1H, s, H8), 6.32 (2H, br, 2-NH2), 6.05 (1H, m, H1′), 5.30 (1H, ∼d, H3′), 4.42 (2H, m, H5′). 4.32 (1H, m, H4′), 2.65 (1H, m, H2′), other H2′ signal obscured, (butyrate signals) 2.35 (4H, m, αH), 1.58 (4H, m, βH), 0.89 (6H, m, γH). HRMS FAB: calcd for C35H38N5O13 (M + H)+ 736.2466; found 736.2492.
O6-AFB-2′-Deoxyguanosine 3′,5′-Dibutyrate (11)
1H NMR (500 MHz, DMSO-d6) δ (aflatoxin signals) 6.75 (1H, d, J = 5.9 Hz, H6a), 6.65 (1H, s, H8), 6.60 (1H, s, H5), 6.18 (1H, s, 9-OH), 4.54 (1H, s, H9), 4.07 (1H, d, J = 5.9 Hz, H9a), 3.91 (3H, s, 4-OCH3), ∼3.30 (2H, m, AFB H3) ∼2.49 (2H, m, AFB H2), (2′-deoxyguanosine signals) 7.93 (1H, s, H8) 6.55 (2H, br, 2-NH2), 6.15 (1H, m, H1′), 5.30 (1H, ∼d, H3′), 4.26 and 4.17 (2H, m, H5′), 4.14 (1H, m, H4′), 2.97 (1H, m, H2′a), 2.41 (1H, m, H2′b), (butyrate signals) 2.33 and 2.27 (4H, m, αH), 1.57 and 1.48 (4H, m, βH) 0.90 and 0.83 (6H, m, γH). 13C NMR (DMSO-d6) δ (aflatoxin signals) 200.8 (C1), 34.9 (C2), 28.7 (C3), 177.5 (C3a), 103.0 (C3b), 161.9 (C4), 56.9 (OCH3), 91.0 (C5), 165.8 (C5a), 114.0 (C6a), 104.2 (C8), 76.6 (C9), 50.9 (C9a), 104.5 (C9b), 154.4 (C9c), 159.4 (C11), 116.0 (C11a), (2′-deoxyguanosine signals) 151.3 (C2), 154.6 (C4), 113.8 (C5), 157.9 (C6), 137.9 (C8), 82.7 (C1′), 35.1 (C2′), 74.4 (C3′), 81.3 (C4′), 63.4 (C5′), (butyrate signals) 172.7 and 172.3 (CO), 35.3 and 35.1 (αC), 17.9 and 17.8 (βC), 13.5 and 13.4 (γC). An HMBC correlation was seen between H8 of the AFB moiety and C6 of dGuo. HRMS FAB: calcd for C35H38N5O13 (M + H)+ 736.2466; found 736.2471.
Unidentified Product
1H NMR (DMSO-d6) δ (aflatoxin signals) 6.23 (1H, J = 6.6 Hz, d, H6a), 4.62 (1H, s, H8), 6.47 (1H, s, H5), 5.62 (1H, s, 9-OH), 4.13 (1H, s, H9), 3.75 (1H, J = 6.6 Hz, d, H9a), 3.89 (3H, s, 4-OCH3), H2 and H3 obscured, (2′-dGuo signals) 8.39 (1H, s, H8), 6.93 (2H, br, 2-NH2), 5.85 (1H, m, H1′), 5.32 (1H, d, H3′), 4.30 (2H, m, H5′), 4.18 (1H, m, H4′), 3.03 (1H, m, H2′), other H2′ signal obscured, (3,5′-butyrate signals) 2.30, 2.15 (4H, m, αH), 1.55, 1.42 (4H, m, βH), 0.88, 0.75 (6H, m, γH). HRMS FAB: calcd for C35H40N5O14 (M + H)+ 754.2571; found 754.2547.
Synthesis of 18O6-AFB-2′-Deoxyguanosine 3′,5′-Dibutyrate
18O-Labeled 2′-deoxyguanosine, prepared by enzymatic deamination of the 2′-deoxyriboside of 2,6-diaminopurine in 99% H218O,41 was converted to the dibutyrate and reacted with AFB epoxide as described above.
Synthesis of AFB–FAPY Deoxynucleoside Dibutyrate 13
AFB–FAPY dibutyrate 13 was prepared directly from the epoxide reaction mixture without isolation of the N7 adduct. Two HPLC fractions were collected, designated as FAPY A (major and first eluting) and FAPY B (minor and second eluting). 1H NMR data (obtained via COSY and TOCSY studies) (500 MHz, 9:1 MeOH-d4/DMSO-d6) δ 3.38−3.56 (m, 2H, AFB–H3), 2.58−2.64 (m, 2H, AFB–H2), 2.21−2.55 (m, 2H, sugar H2′, H2″), 5.2−5.25 (m, 1H, sugar H3′), 4.07−4.12 (m, 1H, sugar H4′), 4.11−4.18 (m, 2H, sugar H5′), 0.95 (t, 3H, butyrate CH3), 1.03 (t, 3H, butyrate CH3), 1.64 (m, 2H, butyrate β-CH2), 1.69 (m, 2H, butyrate β-CH2), 2.33 (m, 2H, butyrate α-CH2), 2.45 (m, 2H, butyrate α-CH2). Assignments for all other protons are shown in Table 1. 13C NMR data (obtained via HMQC and HMBC studies) are shown in Table 2. HRMS (FAB+): calcd for C35H40N5O14 (M + H)+ 754.2572; found for FAPY A 754.2559, FAPY B 754.2554.
Table 1.
| proton | 6-Aac | 6-Ab | 6-Ba | 6-Bb | 13-Aa | 13-Ab | 13-Ba | 13-Bb |
|---|---|---|---|---|---|---|---|---|
| Aflatoxin | ||||||||
| H5 | 6.39 | 6.39 | 6.40 | 6.40 | 6.48 | 6.48 | 6.63 | 6.63 |
| H6a | 6.49 | 6.41 | 6.46 | 6.49 | 6.48 | 6.48 | 6.44 | 6.44 |
| OHb | 6.08 | 6.08 | 6.11 | 6.13 | 6.11 | 6.13 | ||
| H8 | 6.27 | 5.69 | 6.04 | 5.48 | 6.44 | 5.83 | 6.45 | 5.80 |
| H9 | 4.93 | 5.08 | 4.82 | 4.84 | 5.03 | 5.12 | 5.09 | 5.18 |
| OMe | 3.92 | 3.92 | 3.94 | 3.94 | 4.03 | 4.03 | 4.03 | 4.03 |
| H9a | 3.94 | 3.94 | 3.92 | 3.88 | 4.00 | 4.03 | 4.01 | 4.00 |
| Pyrimidine | ||||||||
| NH1 | 9.65 | 9.49 | 9.91 | 9.80 | 9.90 | 9.78 | ||
| formyl | 7.59 | 8.29 | 7.52 | 8.22 | 7.74 | 8.47 | 7.66 | 8.36 |
| NH2b | 6.04c | 6.04c | 6.56 | 6.56 | 6.56 | 6.56 | ||
| NH4 | 6.06 | 6.06 | 6.29 | 6.16 | 6.02c | 6.01c | ||
| (J = 6.9 Hz) | ||||||||
| Deoxyribose | ||||||||
| H1′ | 5.94 | 5.90 | 5.96 | 5.95c | ||||
Spectra were recorded in DMSO-d6.
Spectra were recorded in MeOH-d4/DMSO-d6 (9:1) mixtures. Chemical shifts are in ppm relative to MeOD.
The capital letter indicates the major (A) and minor (B) chromatographically separable forms; the small letter indicates major (a) and minor (b) formyl isomers. Thus 6-Aa is the major formyl isomer of the major HPLC fraction.
Assignment uncertain.
Table 2.
13C Chemical Shifts of FAPY Nucleoside 13a
| carbon | FAPY 13-Aa | FAPY 13-Ab | FAPY 13-Ba | FAPY 13-Bb |
|---|---|---|---|---|
| Aflatoxin | ||||
| C6a | 114.2b | |||
| C8 | 95.4 | 102.9 | 95.8 | 103.6 |
| C5 | 92.1b | |||
| C9 | 75.4 | 75.2 | 76.3 | 76.1 |
| OMe | 57.3b | |||
| C9a | 55.0b | |||
| C2 | 35.9b | |||
| C3 | 30.1 | |||
| Pyrimidine | ||||
| CHO | 168.0 | 165.5 | 168.1 | 165.7 |
| C5 | 92.1 | 89.3 | 92.3 | 89.6 |
| Deoxyriboseb | ||||
| C1′ | 82.6b | |||
| C2′ | 37.3b | |||
| C3′ | 76.8b | |||
| C4′ | 82.7b | |||
| C5′ | 64.6b | |||
| Butyratesb | ||||
| CH3 | 13.8b | |||
| β-CH2 | 19.1b | |||
| α-CH2 | 36.5b | |||
Chemical shifts for 13C signals were determined from HMQC or HMBC 2-D spectra recorded in MeOH-d4/DMSO-d6 (9:1) mixtures at 288 K. Chemical shifts are in ppm (std: MeOH-d4). The chemical shifts are derived from two-dimensional experiments; their accuracy is subject to data point limitations, and signals separated by only a few Hz are not resolved from one another.
Only one set of signals seen for the four species. The values are listed for Aa which was present in the highest concentration.
Synthesis of [5-15N] Labeled AFB–FAPY Nucleoside 13
[7-15N] 2′-Deoxyguanosine was prepared according to a literature procedure42−44 and converted to the dibutyrate as described above. [5-15N]-Labeled AFB–FAPY nucleoside 13 was synthesized from [7-15N]-2′-deoxyguanosine 3′,5′-dibutyrate according to the procedure described above for the synthesis of the unlabeled 13.
Synthesis of [5-15N] AFB–FAPY Base 6
[5-15N]-Labeled AFB–FAPY nucleoside 13 (20 A260 units), dissolved in 2.0 mL of 0.1 M HCl containing 5% MeOH, was heated at 70 °C for 1 h. The reaction was cooled, neutralized with 2 mL of 0.1 M NaOH, and lyophilized. The sample was fractionated by HPLC (YMC-ODS-AQ column, 10 mm × 250 mm, 35% MeOH/H2O, 3.0 mL/min) to give two fractions 6-B and 6-A (retention times 15 and 18.6 min) in a ratio of 1:3.5. The samples were collected at −70 °C and lyophilized immediately to minimize re-equilibration.
Synthesis of Deprotected AFB–FAPY Nucleoside 15
Unprotected FAPY nucleoside 15 was prepared from N7-AFB-5′-O-(dimethoxytrityl)-2′-deoxyguanosine (14). AFB epoxide (5 mg, 15 μmol) and 5′-O-DMT-2′-deoxyguanosine (50 mg, 87 μmol) were dissolved in anhydrous THF (3 mL). The mixture was stirred for 2.5 h at room temperature. An aliquot was converted to the N7-guanine adduct to confirm formation of the cationic adduct. The reaction mixture was made basic with 8 mL of pH 9.5 buffer (15 mM Na2CO3/30 mM NaHCO3) and stirred overnight. Detritylation with 0.1 M HCl was monitored by TLC (95% CH2Cl2-MeOH with trace of triethylamine). After 15 min, the reaction was neutralized with dil NaOH and lyophilized to give crude 15 which was dissolved in 20 mL of 5% aqueous methanol and clarified by filtration through a 0.45 μm filter. HPLC indicated a complex mixture of isomeric species was present from which only deoxyribopyranoside 15 was isolated in pure form.
HPLC Studies of AFB–FAPY Oligonucleotides
HPLC experiments were performed on a Beckman Coulter system with a System Gold 126 solvent module and System Gold 168 diode array detector. 32 Karat v. 7.0 (Beckmann Coulter, Inc, Fullerton, CA) software was used for the acquisition, processing, and analysis of HPLC data. The diode array detector was configured to monitor both 254 and 360 nm wavelengths. Purification and analysis were performed using a YMC ODS-AQ C-18 column (250 mm × 4.6 mm i.d., 0.65 mL/min for analysis, and 250 mm × 10 mm i.d., 2.0 mL/min for purification). The mobile phase consisted of ammonium formate buffer (0.1 M, pH 8.0) and acetonitrile, with a gradient of 1% to 30% acetonitrile over 25 min and 30% to 80% over 5 min.
Preparation and Equilibration of a 13-mer FAPY Oligonucleotide
A solution of 1 μmol of 5′-d(CCTCTTCGAACTC) in 100 μL of phosphate buffer (10 mM sodium phosphate, 100 mM sodium chloride, pH 7.0) was mixed with 5 equiv of AFB epoxide dissolved in 100 μL of CH2Cl2. The resulting biphasic mixture was stirred at 5 °C for 30 min. After removal of the organic layer, the AFB–N7–Gua adducted oligonucleotide was purified from the aqueous layer by HPLC (retention time 20.8 min). The adducted oligonucleotide was lyophilized, redissolved in 500 mL of sodium carbonate solution (100 mM, pH 10), and placed in a 37 °C water bath to facilitate imidazole ring opening to give 5′-d(CCTCTTCAFBFAACTC) in which AFBF = 5. The reaction was followed by HPLC (FAPY I and II eluted at approximately 20 and 21 min, and the N7 adduct eluted just before FAPY II (20.8 min)). When it appeared that no further reaction was occurring, the FAPY-containing peaks were collected and lyophilized. The major peak, FAPY II, dissolved in H2O, was mixed with approximately 4 equiv of the complementary oligonucleotide, 5′-GAGTTCGAAGAGG-3′ in phosphate buffer (10 mM sodium phosphate, 100 mM sodium chloride, pH 7.0). The subsequent re-equilibration was monitored by HPLC; to ensure complete denaturation of duplex DNA, the column was heated to 50 °C.
pH Dependence of the Rate of Equilibration of FAPY Oligonucleotides I and II
FAPY I and II samples were brought to 90% or better purity by HPLC. Samples (10 μM) in sodium phosphate buffer (0.2 M) at pH 6.0, 7.0, and 8.0 were prepared in triplicate and allowed to equilibrate at room temperature. The equilibration was monitored by HPLC (Phenomenex Gemini C-18 column, 250 mm × 4.6 mm id., 5 μ, 110 Å) at a flow rate of 0.8 mL/min. The mobile phase consisted of ammonium formate buffer (0.1 M, pH 8.0) with an acetonitrile gradient of 1% to 30% over 25 min.
Results
Preparation of AFB–FAPY Base 6 and Nucleoside Dibutyrate 13
Protected AFB–FAPY nucleoside 13 was synthesized (Scheme 3), first without isotopic label and then with 15N at the N7 position of the starting 2′-deoxyguanosine. Deoxyguanosine was converted to 3′,5′-dibutyrate 9 which had sufficient lipophilicity to permit the reaction with AFB epoxide to be carried out in organic media. In dichloromethane, the reaction of AFB epoxide with 9 was complete within 6 h at room temperature giving a mixture of N7 adduct 10 and O6 adduct 11 plus an additional product of uncertain structure.
Scheme 3.

The structures of 10 and 11 were initially assigned by acid hydrolysis which with 10 gave N7-adducted guanine 4 and with 11 gave AFB-8,9-diol (12), both of which had been previously characterized. The structure assignments for 10 and 11 were confirmed by spectroscopic analysis. The NMR signals for the AFB H8 signals were shifted downfield for both 10 and 11 relative to epoxide 2, i.e., from δ 5.48 for 2 to δ 6.29 and 6.65 for 10 and 11, respectively. For 10, the purine H8 had moved from δ 7.88 to δ 8.60 reflecting the effect of positive charge on N7. For 11, assignment of the site of attachment as O6 was confirmed by examining 18O isotope shift effects in the 13C spectrum. Using 11 prepared from dGuo containing a 1:1 mixture of 16O and 18O at the O6 position, small differences (2−3 Hz) in chemical shifts were observed for the signals for guanine C6 and aflatoxin H8 with and without 18O such that the two signals appeared as doublets. Isotopic shifts were not detected for any other carbons. The unidentified product is tentatively assigned as involving reaction of the epoxide at C5 of guanine followed by reaction of OH− at C4. This assignment is based on the mass spectrum that shows the incorporation of a molecule of water and on the NMR signal for H8 of the aflatoxin residue being shifted upfield to 4.62 ppm, indicative of AFB attachment to carbon rather than to a heteroatom. Detailed NMR studies were precluded by instability of the adduct in DMSO-d6.
Cationic adduct 10 was converted to FAPY nucleoside dibutyrate 13 by overnight treatment with pH 9.5 buffer at ambient temperature. Nucleoside 13 was depurinated by heating at 70 °C in 0.1 M HCl to give FAPY base 6. AFB–FAPY nucleoside dibutyrate 13 and base 6 labeled with 15N at the N5 position were synthesized in the same manner starting with [7-15N] N7-AFB-2′-deoxyguanosine.
Characterization of FAPY Base 6
The 1H NMR spectrum of FAPY base 6 showed it to be four species. A preparative separation of 6 by reversed phase HPLC gave two fractions (∼8:1 ratio) which were collected at −70 °C and lyophilized for storage. The major fraction was designated 6-A, and the minor one, 6-B; 6-A eluted after 6-B. Each fraction contained two inseparable components. The HPLC separation of 6, the relative amounts of the two components, and the 1H spectra were consistent with previous reports for 6 prepared from AFB-adducted DNA.12,24 The lyophilized samples of 6-A and 6-B were redissolved in DMSO-d6, and 1H NMR spectra were immediately acquired. Repeat NMR analysis of 6-B over a period of 15 h showed that it rapidly equilibrated back to the original mixture of 6-A and 6-B with a half-life of ∼2.5 h (panel A of Figure 1).
Figure 1.

(Panel A) Conversion of AFB–FAPY base 6-B to an equilibrium mixture in which 6-A predominates. (Panel B) 1H NMR spectrum of the formyl region of the equilibrium mixture of 6 with 15N label at N5 showing structural assignments for the four diastereomers.
The 1H NMR spectra of 6-A and -B were very similar, each containing a pair of formyl signals, one near δ 8.2 and the other near δ 7.6. For 6-A, the signals appeared at δ 7.59 and 8.29; for 6-B, the signals appeared at δ 7.52 and 8.22. The four species are designated 6-Aa and -Ab and 6-Ba and -Bb, respectively. The AFB H8 signals for the four species followed the reverse trend; for 6-Aa and -Ab the H8 signals appeared at δ 6.27 and 5.69 and for 6-Ba and -Bb at δ 6.04 and 5.58. In NOESY spectra of 6, chemical exchange cross-peaks were observed for the formyl protons between 6-Aa and -Ab and between 6-Ba and -Bb but not between 6-A and -B. The 1H spectrum of labeled 6 (equilibrated mixture of the four isomers) showed 15N splitting of all four of the formyl signals into doublets with coupling constants of ∼17 Hz (Panel B in Figure 1) proving that the formyl group was attached at N5 in all species. Structure 8, proposed by Hertzog,24 could thus be eliminated.
Assignment of the stereochemical relationships among the isomers of 6 was not practicable by NMR spectroscopy due to the absence of nonexchangeable protons in the pyrimidine fragment. The prospects appeared much better with nucleoside 13.
Characterization of FAPY Nucleoside Dibutyrate 13
The 1H NMR spectrum of nucleoside dibutyrate 13 showed it also was a mixture of four species. HPLC separation of 13 gave fractions designated 13-A and -B in a ∼10:1 ratio. A typical separation is shown in the right insert in Figure 2. A subtle difference in the chromatographic separation of bases 6-A and -B and nucleosides 13-A and -B is that the major component elutes first with the nucleosides but last with the bases. For the nucleosides, the major component, 13-A, could be brought to a relative state of purity by reversed phase HPLC using a methanol–water gradient and was stable for many days in MeOH-d4/DMSO-d6. However, 13-A and -B interconverted during HPLC purification. On account of the low abundance of 13-B coupled with interconversion during chromatography, it was not possible to obtain pure 13-B in sufficient quantities for useful NMR studies.
Figure 2.

1H NMR spectrum (MeOH-d4/DMSO-d6) of AFB–FAPY nucleoside dibutyrate 13 showing structural assignments for the four diastereomers. (Left inset) the formyl region of an identical sample having 15N label at N5. (Right inset) HPLC separation of 13-A and 13-B.
The 1H NMR spectrum of the mixture of FAPY nucleoside species (Figure 2) showed strong similarities to the spectrum of the corresponding mixture of FAPY bases. As with 6, two of the formyl signals were associated with 13-A, and two, with 13-B. A sample of 13 with a 15N label at position N5 showed 1H–15N coupling for all four formyl groups (left insert in Figure 2) in the 1H NMR spectrum, and a 15N spectrum showed signals for the four species appearing in the range 130−134 ppm (data not shown).
Further NMR studies were carried out on unlabeled 13 using pure 13-A and mixtures of 13-A and -B in DMSO-d6, DMSO-d6/D2O (1:1), and MeOH-d4/DMSO-d6 (9:1). The studies included one-dimensional and COSY, TOCSY, HMQC, HMBC, NOESY, and ROESY two-dimensional spectra. The two stereoisomers comprising 13-A are designated 13-Aa and -Ab; the corresponding stereoisomers comprising 13-B are designated 13-Ba and -Bb. The formyl signals for 13-Aa and -Ab appeared at δ 7.74 and 8.47, respectively, and those for 13-Ba and Bb, at δ 7.66 and 8.36. Assigned 1H NMR resonances for 6 and 13 are listed in Table 1 and 13C resonances for 13 in Table 2; key connectivities derived from two-dimensional spectra are summarized in Figures 3 and 4.
Figure 3.

Important proton–proton and proton–carbon correlations seen for AFB–FAPY nucleosides 13-Aa, 13-Ab, 13-Ba, and 13-Bb. (Panel A) TOCSY results. Additional correlations were seen for pyrimidine 4-NH with H1′, H2′, and H2″. Correlation between H9 and H9a was not seen because the dihedral angle is close to 90°. (Panel B) HMBC results. Additional TOCSY and HMBC correlations were seen within the deoxyribose moiety.
Figure 4.

Significant NOESY correlations seen for FAPY nucleosides 13-Aa, 13-Ab, 13-Ba, and 13-Bb.
In one-dimensional spectra, resolved signals for all protons in the region of the aflatoxin–pyrimidine linkage were seen for mixtures of the four diastereomers, such that it was possible to make individual spectral assignments for each stereoisomer. The relative areas of the individual signals were the same as those for the formyl hydrogens. For each proton type, the separation between the signals for the four stereoisomers, i.e., chemical shift dispersion, was generally dependent on the proximity of that hydrogen to N5. Well resolved resonances were seen for the aflatoxin protons H8 and H9 sets. The dispersion of chemical shifts within the sets for aflatoxin H5, pyrimidine N4H, and deoxyribose H1 was somewhat less, and the chemical shift dispersion for the remaining proton sets on aflatoxin, the sugar fragments, and the butyrates was insufficient to permit reliable differential assignments. For the formyl protons, the pyrimidine C5 carbons, and the aflatoxin C8 carbons, the signals for 13-Ab and -Bb were similar as were those for 13-Aa and -Ba, but the two groups differed significantly from each other. The four species exhibited virtually identical carbon spectra (observed via HMBC and HMQC experiments) except for the carbon atoms near N5.
The four-bond coupling between the formyl proton and H8 of the AFB moiety could be observed for 13-Aa and -Ba (4J = ∼0.5 Hz) in one-dimensional spectra but 13-Ab and -Bb did not show this long-range coupling. TOCSY spectra showed correlations between the formyl proton and aflatoxin H8 in 13-Aa and -Ba but not in 13-Ab and -Bb. HMBC spectra showed a correlation between the formyl proton and aflatoxin C8 in 13-Aa and -Ba but not in 13-Ab or -Bb. Also, HMBC correlations of the formyl proton with pyrimidine C5 were observed in 13-Ab and -Bb but not in 13-Aa or -Ba. The chemical shift data are consistent with this pairing of stereoisomers (see Tables 1 and 2).
NOESY and ROESY spectra showed a strong cross-peak between the formyl proton and aflatoxin H8 in 13-Ab and -Bb but not in 13-Aa and -Ba. All four showed correlations of aflatoxin H8 with pyrimidine C5 and with the formyl carbon. They also showed correlation between aflatoxin H5 and the anomeric proton in 13-Aa and -Ab but none in 13-Ba or -Bb. NOESY and saturation transfer experiments with the observation of the formyl proton, aflatoxin H8, and aflatoxin H9 showed 13-Aa to be in exchange with -Ab and likewise 13-Ba with -Bb. However, there was no evidence of exchange on the NMR time scale between 13-A and -B. With a sample of pure 13-A, i.e., a mixture of 13-Aa and -Ab, correlations of H1′ with H2′, H2″, and H4′ were observed but not with H3′; a mixture of all four stereoisomers of 13 showed the correlations of H1′ with H2′, H2″, and H4′ but not with H3′.
For comparison, the NMR spectra of methyl-FAPY base derived from 7-methyl deoxyguanosine were examined. The achiral methyl-FAPY base exists as two separable but inter-converting forms with the formyl protons separated by ∼0.3 ppm.30,34 In 1:1 DMSO-d6/D2O, the 1H NMR spectrum contained singlets at δ 7.96 and 7.69 for the formyl protons of the two geometrical isomers of the formamide and singlets at δ 2.91 and 2.79 for the two methyl groups. The HMBC spectrum showed the same pattern of long-range coupling of the formyl protons with the methyl carbon and with C5 as in the case of 13; i.e., the upfield formyl proton correlated with the methyl carbon but not with C5, and the downfield formyl proton correlated with C5 but not the methyl carbon. Both stereoisomers showed long-range coupling of the methyl protons with the formyl carbon and with C5. Kadlubar et al. have assigned the geometrical isomers via NOE difference measurements.34 Our assignments are consistent with theirs. A noteworthy difference between the geometrical isomers of the methyl-FAPY base and 6 and 13 is that the isomers of the methyl-FAPY base can be separated by HPLC, whereas those of 6 and 13 cannot. It has been proposed that the geometrical isomer of methyl-FAPY having the carbonyl oxygen proximal to the pyrimidine ring is stabilized by hydrogen bonding with the proton on N4. We suspect that the formyl isomers of the AFB–FAPY species interconvert more rapidly due to an inability to achieve sufficient planarity to form this hydrogen bond. However, our failure to resolve the geometrical isomers of 6 and 13 may only reflect smaller differences in HPLC retention times.
Structural Relationships among the Four Stereoisomers of 6 and 13
The 1H spectra of the mixtures of stereoisomers of 6 and 13 showed strong similarity to each other. In each case the four formyl and H8 signals were separated into two pairs of similar signals which were separated from each other by more than 0.5 ppm. The two fractions, A and B, resulting from HPLC fractionation of 6 and 13 each contained one upfield and one downfield formyl signal. A similar situation existed with H8. The four formyl proton chemical shifts are in the sequence Ab > Bb >> Aa > Ba for both 6 and 13. The four H8 signals for 6 and 13 are in the sequence Ba ≈ Aa >> Ab ≈ Bb. The correlation of H8 signals with the formyl signals was made on the basis of relative intensities. For the formyl signals of 6 the intensities are Aa > Ab > Ba > Bb; for 13 they are Aa > Ab > Ba ≈ Bb. In comparing the spectra of the stereoisomers of 6 with those of 13 the aflatoxin derived signals show strong similarities, ruling out the remote possibility that there might be structural differences in this portion of 6 and 13. For both 6 and 13, NOESY spectra revealed chemical exchange between the Aa and Ab isomers and between the Ba and Bb isomers but not between the A and B forms. It can be concluded that the stereochemical relationships among the four stereoisomers of 6 and 13 are the same. Consequently, the stereochemical relationships among the stereoisomers of 13 can safely be used to establish the relationships among the stereoisomers of 6.
Based on the extensive NMR studies, 13-Ab and -Bb can be assigned as the E isomers, and 13-Aa and -Ba, as the Z (Figure 5) and likewise for the geometric isomers of 6.
Figure 5.
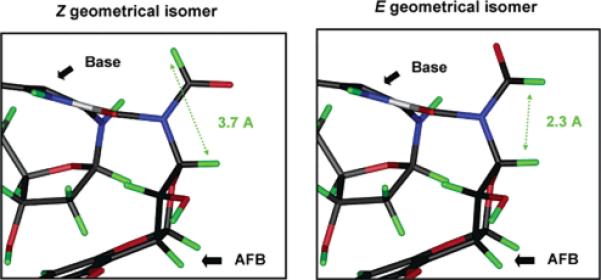
Molecular models showing the geometric isomers of the formyl groups in AFB–FAPY base 6 and nucleoside 13. (Left panel) the preferred Z isomer showing the distance (3.7 Å) between the formyl proton and aflatoxin H8. (Right panel) E isomer. The close proximity (2.3 Å) of the formyl proton and aflatoxin H8 allows a strong NOE to be observed.
Atropisomers at the Pyrimidine C5–N5 Bond
The steric bulk of the AFB substituent on N5 of the pyrimidine plus the presence of substituents on carbons 4 and 6 creates high rotational barriers in 6 and 13. The resulting stereogenic bond creates diastereomers on account of the chirality of the aflatoxin moiety. With the Aa/Ab and Ba/Bb pairs of stereoisomers assigned as geometrical isomers of the formamide, the Aa/Ba and Ab/Bb pairs must be the C5–N5 atropisomers.
Molecular models of 13 (Figure 6) suggest that smaller differences are to be expected between the NMR spectra of atropisomers than between those of the geometrical isomers; this has been borne out by the proton spectra (see Table 1). TOCSY and HMBC correlations were essentially identical for each pair of atropisomers of 13 even though there were major differences between pairs of geometrical isomers (Figure 3). However, a significant difference between the atropisomers was seen in the NOESY spectrum of 13 where the Aa and Ab stereoisomers showed correlation between H5 of the AFB moiety and the anomeric proton, whereas Ba and Bb did not (Figure 6). Models reveal that the Ra rotamer at C5–N5 can assume a conformation in which H5 can make a close enough approach to the anomeric proton (∼2.8 Å) to produce an NOE, whereas the Sa rotamer cannot. On this basis, the stereoisomers Aa and Ab of 13 are assigned as Ra, and Ba and Bb, as Sa. The stereoisomers of 6 can be assigned accordingly.
Figure 6.
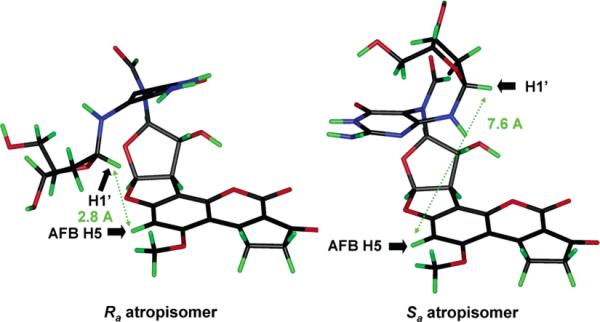
Molecular models of two rotamers of 13 (butyrate esters not shown). (Left) Ra configuration at C5–N.5 The close proximity (2.8 Å) of AFB H5 to H1′ leads to the observed NOE (shown by the arrow). (Right) Sa configuration at C5–N5. This orientation places AFB H5 too distant (7.8 Å) from the anomeric proton for an NOE to be observed.
Unprotected AFB–FAPY Deoxyriboside
The possibility of deoxyribose isomerization was examined using AFB–FAPY dGuo lacking acyl protection of the hydroxyl groups on the deoxyribose. Unprotected FAPY nucleoside was prepared from 5′-dimethoxytrityl-AFB–FAPY dGuo (14, Scheme 4). HPLC analysis of the crude deprotected deoxynucleoside following treatment of 14 with dilute acid showed a complex mixture of components from which only the major product was isolated in pure form and in sufficient quantity to permit structural characterization. The compound was assigned as pyranoside 15 on the basis of an HMBC correlation of H5′ and H5″ with the anomeric carbon. NMR methodology used with 13 revealed 15 had the Ra configuration at the C5–N5 bond and was the expected mixture of E and Z geometrical isomers of the formamide. Ring expansion of deoxyribose is consistent with the well-known preference of ribose and deoxyribose derivatives to assume β-pyranose structures; it is assumed that the mixture also contained atropisomers of α- and β-furanosides and the imine intermediate (Scheme 4). In contrast, protected FAPY nucleosides 13-A and 13-B showed no evidence of anomerization; only the β anomer was detected in the 2-D NMR studies.
Scheme 4.

Equilibration Studies on AFB–FAPY in Oligonucleotides
Equilibration of AFB–FAPY species in DNA was examined using the 13-mer oligodeoxynucleotide 5′-d(CCTCTTCAFBFAACTC) in which AFBF = 5; this was the construct employed by the Essigmann group in their mutagenesis studies.35 The adducted oligodeoxynucleotide can be resolved into two components by reversed phase HPLC.
Only FAPY I was observed at early time points in the hydrolysis of the cationic adduct, but as the reaction progressed FAPY II appeared and eventually became the dominant species in a final ratio of 2−3:1. FAPY I eluted ahead of FAPY II in this sequence. This is consistent with observations reported by Essigmann. Re-equilibration of individual samples of I and II led to the same final mixture. The rate of equilibration between I and II was found to be pH dependent between pH 6 and pH 8, increasing rapidly with decreasing pH (Figure 7).
Figure 7.

Interconversion of single-stranded FAPY oligonucleotides I and II as a function of pH. Samples were dissolved in 0.2 M sodium phosphate buffer at the designated pH, and equilibration at ambient temperature was followed by HPLC.
Form I was the more stable species in double-stranded DNA. Freshly prepared II was combined with the complementary 13-mer at neutral pH, and aliquots of the mixture were analyzed by HPLC over a period of 6 h (Figure 8). During this time the composition of the duplex changed to one in which I was the only FAPY species that could be detected. Reversion of II to I in the presence of the complementary strand also showed acid catalysis. Whereas a facile equilibration had been observed near neutrality, a duplex containing II was found to be stable for many days at pH 9.
Figure 8.

At time 0, FAPY I and II (5′-d(CCTCTTCAFBFAACTC)) were allowed to equilibrate. Addition of excess complementary strand DNA (C) shifted the equilibrium in favor of FAPY I over 6 h. Only I and II exhibited absorption at 360 nm.
AFB–FAPY equilibria were examined in other DNA sequences. The 10-mer 5′-d(CTATAFBFATTCA) was of particular interest being the oligodeoxynucleotide for which the structure had been determined by Mao et al.36 It showed the same dependence of the equilibrium ratio of FAPY forms I and II on hybridization state as had been observed with the 13-mer. Several additional sequences were also examined. We conclude that the observed stabilization of FAPY form I in double-stranded DNA is a general phenomenon.
Discussion
Structures of AFB–FAPY Base 6 and Nucleoside 13
When considering the nature of the biologically active species formed by alkaline cleavage of the guanine N7 adduct of AFB epoxide, an examination of the literature quickly revealed that the first question to be resolved was the site of attachment(s) of the formyl group in the two separable forms of the FAPYs. Structure 8, assigned to the minor form by Hertzog et al.,24 is inconsistent with the structures that have been assigned more recently for other FAPY adducts. However, it is not out of the question that the bulkiness or structural complexity of the aflatoxin residue leads to a different partitioning between the N5 and N4 formyl products arising from ring opening of N7 adduct 3. To explore this question we chose to synthesize the unknown AFB–FAPY dGuo. The only known AFB–FAPY species, base 6, had been prepared by isolation from adducted DNA.
AFB epoxide reacts well with dGuo in double-stranded DNA where intercalation holds 2 in an orientation optimal for attack by N7, but aqueous reactions with guanine and guanine nucleosides give very low yields due to competition for the epoxide by water. This problem was circumvented by carrying out the adduction reaction in an organic medium using the 3′,5′-dibutyrate derivative (9) of dGuo to obtain solubility. Adduction at N7 gave cationic adduct 10 in satisfactory yield but also two unexpected adducts: O6 adduct 11 plus one of uncertain structure.
The identification of O6 adduct 11 as a significant product of adduction of dGuo dibutyrate by epoxide 2 raised the question of whether this adduct arises in epoxide-treated DNA. Such an adduct would be expected to be highly mutagenic. Adduct 11 might have escaped detection in previous examinations of the reaction of 2 with DNA where workup involved depurination under acidic conditions which would have hydrolyzed the ether linkage in 11 to give dGuo and AFB diol. However, a cursory search for 11 in epoxide-treated calf thymus DNA indicated that 11, if present at all, was below the level of detection. The unidentified adduct was unstable, which made it impossible to acquire HMBC and other spectra needed to fully characterize it. However, on the basis of limited NMR and mass spectra, it appears possible that it is a C5 adduct arising by addition of the AFB moiety at C5 of dGuo and hydroxide at C4.
Although it should be possible to assign the site of formylation in AFB–FAPY bases and nucleosides by two-dimensional NMR methods, in particular by HMBC spectra, we chose to use 15N substitution, the method having been successfully employed by Humphreys et al. for assignment of the structure of the isomers of methyl-FAPY derived from N7-methyl guanosine.33 Labeling the amino group with 15N at the 5 position would lead to 15N–1H coupling in 1H spectra for formyl groups residing on N5, but the coupling would not be seen if the formyl group was on N4. When this experiment was performed all formyl species in 6 and 13 showed ∼17 Hz coupling to 15N5; hence there is only one regioisomer of AFB–FAPY.
Two types of stereoisomerism are available to FAPY base 6: geometrical isomerism of the formamide moiety and atropisomerism involving hindered rotation around the pyrimidine C5–N5 bond. The observation of four species in the 1H NMR spectrum shows that both types of isomerization are present. With the aid of extensive two-dimensional NMR studies of 6 and nucleoside 13, it was possible to assign structures to both the geometric isomers (Figure 5) and the atropisomers (Figure 6), the latter being separable, the former not.
The Ra atropisomer is strongly favored over the Sa in equilibrium mixtures of both 6 and 13; for both atropisomers there is a preference for the formyl group to adopt the Z geometry. It is noteworthy that NMR studies have shown that the aflatoxin moiety of the FAPY adduct intercalates on the 5′ face of the pyrimidine ring in double-stranded DNA.36 This orientation represents an Ra absolute configuration for the C5–N5 bond there also. The implication of this observation is that the aflatoxin moiety has an inherent preference for stacking on the 5′ face of the pyrimidine ring. This preference can be expected to remain true even for single-stranded oligonucleotides such as those present at the replication fork.
The fact that the C5–N5 atropisomers of 13 and of 6 can be separated by chromatography whereas the formamide geometrical isomers cannot is an indication that the rotation around the C5–N5 bond is slower than that around the amide linkage. In general, the other FAPY adducts that have been examined by NMR have been FAPY bases with achiral substituents, and thus restricted rotation around the C5–N5 bond would create enantiomers rather than diastereomers. Tomasz et al. have identified C5–N5 rotamers with the FAPY nucleoside derived from 7-ethyl guanosine that were in slow exchange on the NMR time scale but were unable to resolve them by chromatography.31 However, they found evidence for atropisomerism with the FAPY adduct derived from mitomycin C.31
Anomers of the Deoxyribosyl Moiety
In addition to isomerization of base 6, nucleoside 13 can potentially undergo anomerization of the deoxyribose. Ordinary nucleosides do not readily anomerize; however, due to the presence of a proton on N4 of FAPY nucleosides, ring opening of the deoxyribose (or ribose) followed by reclosure can occur converting the β anomer to the R. Several examples have been reported of anomerization of FAPY nucleosides.31,45−47
The possibility was examined that 13 might have been inseparable mixtures of α and β anomers, i.e., a total of eight species rather than only four. Our inability to obtain samples of 13-B completely free of 13-A coupled with the fact that both 13-A and 13-B were mixtures of formamide geometrical isomers made it difficult to study this question. The α anomer would be expected to show a strong NOESY cross-peak between H1′ and H3′. The mixture of diastereomers showed no evidence of an NOE between H1′ and H3′. Thus, we conclude that β → α anomerization had not occurred.
Others have reported mixed results on the issue of anomerization in protected FAPY nucleosides. Tomasz et al. found the FAPY derived from the 3′,5′-diacetyl 7-ethyl derivative of guanosine was stable31 but the FAPY 3′,5′-diacetate derived from the 7-methyl derivative of deoxyguanosine underwent anomerization.31 Also, the FAPY derived from 7-ethylguanosine but lacking protection of the hydroxyl groups of the ribosyl fragment underwent anomerization and ring expansion to the ribopyranoside.31 Burgdorf and Carell reported that the FAPY derived from 3′,5′-silyl-protected deoxyguanosine was stable in DMSO for 6 h at 60 °C in DMSO but underwent slow anomerization in water–acetonitrile mixtures.48 Uncertainty exists as to the relationship of the experimental conditions employed in the studies with these other FAPY adducts to the conditions used in the present work. Consequently, it was deemed prudent to examine the issue of anomerization using the unprotected AFB–FAPY deoxyriboside (15).
Unprotected FAPY nucleoside 15 was prepared by acid cleavage of 5′-trityl derivative 14; it underwent spontaneous anomerization and deoxyribosyl ring enlargement to give a complex mixture of FAPY species. The dominant component of this mixture was isolated in pure form and shown by NMR studies to be the mixture of E/Z geometrical isomers of the Ra diastereomer of 15 in which the β-furanose ring has been converted to a β-pyranose. In view of the studies by Tomasz et al. the unidentified products are presumed to include the α- and β-furanosides.31 In our view, unprotected FAPY nucleoside 15 is a better model for FAPY nucleosides in DNA than FAPY dibutyrate 13; i.e., the influence on anomerization of the free 3′ and 5′ hydroxyl groups in 15 is more similar to that of the phosphate esters in DNA than that of the much more electron-withdrawing acyl groups in 13. Anomerization of the deoxyribose as well as geometrical isomerization of the formamide and atropisomerization at C5–N5 can be predicted to occur in DNA.
Equilibrium Studies of AFB–FAPY in Oligonucleotides
Oligonucleotides containing AFB–FAPY exist in two equilibrating but chromatographically separable forms designated FAPYs I and II. In single-stranded DNA, AFB–FAPY exists as a mixture of FAPYs I and II in which FAPY II predominates; FAPY I is the exclusive product in double-stranded DNA. There are other examples of adducts whose structures depend on whether they are in single- or double-stranded DNA. For example, the malondialdehyde adduct of deoxyguanosine exists in the cyclic (pyrimidopurinone) form in single-stranded DNA but opens to the extended (oxopropenyl) form when paired with deoxycytidine in double-stranded DNA.49,50 Likewise, the N2 adduct of acrolein with deoxyguanosine is cyclic in single-stranded DNA but open in duplex DNA.51−53 The structure of a DNA duplex containing FAPY I has been deduced by two-dimensional NMR.36 In this structure, the AFB moiety was found to be intercalated on the 5′ face of the lesion creating the Ra configuration at the C5–N5 bond. Furthermore, the deoxyribose was β and the formamide Z. No evidence for the E formamide was found. Exclusive existence of the Z geometry for the formamide is somewhat surprising but has been ascribed to hydrogen bonding between the carbonyl group and the exocyclic amino group of the adenine on the 3′ side of the adducted base.36 However, the possibility cannot be excluded that even in the absence of hydrogen bonding the E/Z equilibrium mixture strongly favored the Z form and the E form was below the level of detection since the ratio of Z to E forms was ∼6:1 for the Ra atropisomer of 13.
The structure of FAPY I in single-stranded DNA is not known but can be expected to be similar to that of nucleoside 13 and to the duplex where the Ra and Z configurations are preferred. AFB–FAPY I is the direct product in hydrolytic reactions carried out with N7 adduct 3 in single-stranded oligodeoxy-nucleotides. Both the cationic and the FAPY adducts show a strong preference for intercalation on the 5′ face of the pyrimidine moiety. Consequently, FAPY I is being formed from 3 with minimal reorganization. Enhanced stabilization of the Ra atropisomer may result from stacking interactions with the 5′ neighboring base.
Only limited information is currently available on the structure of FAPY II. The observation that equilibration is an acid-catalyzed process provides strong support for our contention that FAPY II is the anomer of FAPY I. There are a number of precedents for acid-catalyzed anomerization of glycosylamines.54 Our assignment of FAPY II as the α anomer is fully in accord with Greenberg's report that the FAPY adducts derived from deoxyadenosine and deoxyguanosine readily anomerize in DNA.46,55 Greenberg has employed Endonuclease IV, which specifically cleaves α anomers in double-stranded DNA, to probe the anomeric configuration of unsubstituted FAPY adducts.47 However, this strategy was unsuccessful with AFB–FAPY oligonucleotides, perhaps because the bulky AFB moiety renders the α anomer a poor substrate for the enzyme.
It appears that β anomers are inherently more stable than α in duplex DNA. Greenberg's studies show β anomers of the unsubstituted FAPYs are strongly favored in double-stranded DNA.47 Also, Germann et al. have compared the stabilities of duplexes containing an α-deoxyadenosine versus the normal β-deoxyadenosine and found the Tm of the latter was significantly higher.56
The major difference therefore between the structure of the FAPY adduct in single- and double-stranded DNA is that the deoxyribose moiety exists as a 2−3:1 mixture of α and β anomers in a single-stranded environment but is exclusively β in duplex. Our protected nucleoside experiments did not address this issue because anomeric equilibria were not observed with the dibutyrate 13. However, Tomasz et al. have observed a ∼2:1 ratio of α and β anomers with the unprotected FAPY riboside derived from N7-ethylguanosine.31 Thus the ratio of FAPYs I and II observed with the 13-mer oligodeoxynucleotide is comparable to that observed with nucleosides.
Biochemical Implications
The studies of AFB–FAPY adducts in the context of the base, nucleosides, and oligonucleotides now make it possible to interpret the divergent response of the two forms of the lesion in replication experiments. The underlying chemistry had been obscured by uncertainty as to whether the isomers being observed are regioisomers or stereoisomers. Also, the effects of duplex versus single-strand environments on isomer equilibria were not well understood. First, it should be noted that several studies of the AFB–FAPY35,57 and related FAPY lesions32,58 in oligonucleotides have presumed the two equilibrating but chromatogaphically separable species to be C5–N5 rotamers, whereas the present work leads to the conclusion that they are anomers. Second, the terms “FAPY major” and “FAPY minor” used to describe the two AFB–FAPY species have had different definitions in different publications. In the mutagenesis studies the AFB–FAPY lesion was introduced using a single-stranded oligonucleotide;35 under the conditions of those experiments the species referred to as “FAPY major” can now be identified as FAPY II. The NMR studies were carried out with the AFB–FAPY lesion imbedded in a duplex. Under those conditions the exclusive AFB–FAPY lesion is FAPY I (referred to as “FAPY minor” in ref 35).36 It is not unexpected that α-linked FAPY II would be a more serious impediment to polymerase bypass than FAPY I, which is β-linked, and could even be a total block to replication. Germann et al. have found α-deoxyadenosine incorporated into DNA to be a significant block to replication in a bacterial system.56 It is not yet known whether mammalian polymerases are more successful than bacterial polymerases in bypassing AFB–FAPY II. However, it should be emphasized that on the basis of our results it is unlikely that significant amounts of FAPY II will be present in genomic DNA.
Smela et al. have pointed out that the persistence in vivo of the AFB–FAPY lesion coupled with its high mutagenic potential make it a dangerous lesion.35 However, the lesion may be even more dangerous than they recognized. Whereas they regarded it as being the minor form of AFB–FAPY, it will, in fact, be essentially the only one present in genomic DNA. During replication, the adducted DNA will be in a single-stranded form in the replication complex. However, the period during which it will be in this form is probably too brief for anomerization to occur to any significant extent; under the conditions of our equilibration studies the half-life for conversion of FAPY I to FAPY II at pH 7 is more than 16 h. Furthermore, the conformational constraints imposed by the replication complex may make it impossible for anomerization of the deoxyribose to occur.
A study by Alekseyev et al. of the repair of AFB–FAPY adducts has shown the lesion to be efficiently repaired by a nucleotide excision pathway in E. coli and that base excision repair, which is the primary pathway for repair of unsubstituted FAPY adducts, does not participate significantly.59 One unresolved issue in that study was the stereoisomer status of the FAPY adduct. Whereas the mutagenesis studies had been carried out employing site specifically modified single-stranded M13 vectors, the repair studies employed randomly modified double-stranded pSP72 vectors. Therefore, the AFB–FAPY adducts present in the repair studies can be expected to have been the naturally occurring FAPY I in contrast to the mutagenesis studies having been conducted with a mixture of FAPYs I and II.
There are many compounds, naturally occurring and synthetic, which react with DNA primarily at the N7 of guanine and therefore have the potential to lead to FAPYs. With the exception of the unsubstituted FAPYs of dGuo and dAdo arising from oxidative damage to DNA and, to a lesser extent, methyl FAPY, little is known about the occurrence, chemistry, and biology of these adducts. It will be interesting to see if the structural and biological behavior we have observed for the AFB–FAPY adduct has generality or is unique to aflatoxin B1.
Supplementary Material
Acknowledgment
This research was generously supported by the U.S. Public Health Service via Project Grants ES-03755 and CA-55678, Center Grants ES-00267 and CA-68485, and Instrumentation Grant RR-05805. We thank Dr. Donald Stec for assistance acquiring NMR spectra.
Footnotes
Supporting Information Available: Experimental procedures for synthesis and isolation of 13, isolation of 15, and NMR studies; tables of 1H and 13C chemical shifts for 15; 1H NMR and UV spectra of 6-A, 6-B, 10, 11, 13-Aab, 13-Bab, and 15; 13C spectra of 11 showing aflatoxin C8 and C6 with 6-16O and 6-18O; 1H–15N COSY and 15N INEPT spectra of 13 and complete ref 53. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Busby WF, Jr., Wogan GN. In: Chemical Carcinogens. 2nd ed. Searle C, editor. American Chemical Society; Washington, DC: 1984. pp. 945–1136. [Google Scholar]
- 2.Foster PL, Eisenstadt E, Miller JH. Proc. Natl. Acad. Sci. U.S.A. 1983;80:2695–2698. doi: 10.1073/pnas.80.9.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMahon G, Davis EF, Huber LJ, Kim YS, Wogan GN. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1104–1108. doi: 10.1073/pnas.87.3.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollstein M, Sidransky D, Vogelstein B, Harris CC. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 5.Chang Y-J, Mathews C, Mangold K, Marien K, Hendricks J, Bailey G. Mol. Carcinog. 1991;4:112–119. doi: 10.1002/mc.2940040206. [DOI] [PubMed] [Google Scholar]
- 6.Levy DD, Groopman JD, Lim SE, Seidman MM, Kraemer KH. Cancer Res. 1992;52:5668–5673. [PubMed] [Google Scholar]
- 7.Trottier Y, Waithe WI, Anderson A. Mol. Carcinog. 1992;6:140–147. doi: 10.1002/mc.2940060209. [DOI] [PubMed] [Google Scholar]
- 8.Coles BF, Welch AM, Hertzog PJ, Smith JRL, Garner RC. Carcinogenesis. 1980;1:79–90. doi: 10.1093/carcin/1.1.79. [DOI] [PubMed] [Google Scholar]
- 9.Garner RC, Miller EC, Miller JA, Garner JV, Hanson RS. Biochem. Biophys. Res. Commun. 1971;45:774–780. doi: 10.1016/0006-291x(71)90484-0. [DOI] [PubMed] [Google Scholar]
- 10.Swenson DH, Lin JK, Miller EC, Miller JA. Cancer Res. 1977;37:172–181. [PubMed] [Google Scholar]
- 11.Martin CN, Garner RC. Nature. 1977;267:863–865. doi: 10.1038/267863a0. [DOI] [PubMed] [Google Scholar]
- 12.Lin J-K, Miller JA, Miller EC. Cancer Res. 1977;37:4430–4438. [PubMed] [Google Scholar]
- 13.Essigmann JM, Croy RG, Nadzan AM, Busby WF, Jr., Reinhold VN, Buchi G, Wogan GN. Proc. Natl. Acad. Sci. U.S.A. 1977;74:1870–1874. doi: 10.1073/pnas.74.5.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croy RG, Essigmann JM, Reinhold VN, Wogan GN. Proc. Natl. Acad. Sci. U.S.A. 1978;75:1745–1749. doi: 10.1073/pnas.75.4.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buchi G, Fowler KW, Nadzan AM. J. Am. Chem. Soc. 1982;104:544–547. [Google Scholar]
- 16.Bailey EA, Iyer R, Stone MP, Harris TM, Essigmann JM. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1535–1539. doi: 10.1073/pnas.93.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopalakrishnan S, Harris TM, Stone MP. Biochemistry. 1990;29:10438–10488. doi: 10.1021/bi00498a002. [DOI] [PubMed] [Google Scholar]
- 18.Giri I, Jenkins MD, Schnetz-Boutaud NC, Stone MP. Chem. Res. Toxicol. 2002;15:638–47. doi: 10.1021/tx010187n. [DOI] [PubMed] [Google Scholar]
- 19.Jones WR, Johnston DS, Stone MP. Chem. Res. Toxicol. 1998;11:873–881. doi: 10.1021/tx980047m. [DOI] [PubMed] [Google Scholar]
- 20.Wang TV, Cerutti P. Biochemistry. 1980;19:1692–1698. doi: 10.1021/bi00549a027. [DOI] [PubMed] [Google Scholar]
- 21.Groopman JD, Croy RG, Wogan GN. Proc. Natl. Acad. Sci. U.S.A. 1981;78:5445–5449. doi: 10.1073/pnas.78.9.5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeb LA, Preston BD. Annu. Rev. Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 23.Lawrence CW, Borden A, Banerjee SK, LeClerc JE. Nucleic Acids Res. 1990;18:2153–2157. doi: 10.1093/nar/18.8.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hertzog PJ, Smith JRL, Garner RC. Carcinogenesis. 1982;3:723–725. doi: 10.1093/carcin/3.6.723. [DOI] [PubMed] [Google Scholar]
- 25.Croy RG, Wogan GN. Cancer Res. 1981;41:197–203. [PubMed] [Google Scholar]
- 26.Essigmann JM, Croy RG, Bennett RA, Wogan GN. Drug Metab. Rev. 1982;13:581–602. doi: 10.3109/03602538209011088. [DOI] [PubMed] [Google Scholar]
- 27.Kaden DA, Call KM, Leong P-M, Komives EA, Thilly WG. Cancer Res. 1987;47:1993–2001. [PubMed] [Google Scholar]
- 28.Leadon SA, Snowden MM. Mol. Cell. Biol. 1988;8:5331–5338. doi: 10.1128/mcb.8.12.5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ball RW, Wilson DW, Coulombe RA. J. Cancer Res. 1990;50:4918–4922. [PubMed] [Google Scholar]
- 30.Boiteux S, Belleney J, Roques BP, Laval J. Nucleic Acids Res. 1984;12:5429–5439. doi: 10.1093/nar/12.13.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomasz M, Lipman R, Lee MS, Verdine GL, Nakanishi K. Biochemistry. 1987;26:2010–2027. doi: 10.1021/bi00381a034. [DOI] [PubMed] [Google Scholar]
- 32.Gopalakrishnan S, Patel DJ. J. Am. Chem. Soc. 1993;115:9321–9322. [Google Scholar]
- 33.Humphreys WG, Guengerich FP. Chem. Res. Toxicol. 1991;4:632–636. doi: 10.1021/tx00024a005. [DOI] [PubMed] [Google Scholar]
- 34.Kadlubar FF, Beranek DT, Weis CC, Evans FE, Cox R, Irving CC. Carcinogenesis. 1984;5:587–592. doi: 10.1093/carcin/5.5.587. [DOI] [PubMed] [Google Scholar]
- 35.Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM. Proc. Natl. Acad. Sci. U.S.A. 2002;99:6655–6660. doi: 10.1073/pnas.102167699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao H, Deng ZW, Wang F, Harris TM, Stone MP. Biochemistry. 1998;37:4374–4387. doi: 10.1021/bi9718292. [DOI] [PubMed] [Google Scholar]
- 37.Murray RW, Jeyaraman R. J. Org. Chem. 1985;50:2847–2853. [Google Scholar]
- 38.Adam W, Bialas J, Hadjiarapoglou L. Chem. Ber. 1991;124:2377–2370. [Google Scholar]
- 39.Adam W, Chan YY, Cremer D, Gauss J, Scheutzow D, Schindler M. J. Org. Chem. 1987;52:2800–2803. [Google Scholar]
- 40.Baertschi S, Raney KD, Stone MP, Harris TM. J. Am. Chem. Soc. 1988;110:7929–7931. [Google Scholar]
- 41.Baer H-P, Drummond GI, Duncan EL. Mol. Pharm. 1966;2:67–76. [PubMed] [Google Scholar]
- 42.Massefski W, Jr, Redfield A, Das Sarma U, Bannerji A, Roy S. J. Am. Chem. Soc. 1990;112:5350–5351. [Google Scholar]
- 43.Gaffney BL, Kung PP, Jones RA. J. Am. Chem. Soc. 1990;112:6748–6749. [Google Scholar]
- 44.Barrio MDCG, Scopes DIC, Holtwick JB, Leonard NJ. Proc. Natl. Acad. Sci. U.S.A. 1981;78:3986–3988. doi: 10.1073/pnas.78.7.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haraguchi K, Delaney MO, Wiederholt CJ, Sambandam A, Hantosi Z, Greenberg MM. Nucleic Acids Res. Suppl. 2001:129–130. [PubMed] [Google Scholar]
- 46.Haraguchi K, Greenberg MM. J. Am. Chem. Soc. 2001;123:8636–8637. doi: 10.1021/ja0160952. [DOI] [PubMed] [Google Scholar]
- 47.Patro JN, Haraguchi K, Delaney MO, Greenberg MM. Biochemistry. 2004;43:13397–13403. doi: 10.1021/bi049035s. [DOI] [PubMed] [Google Scholar]
- 48.Burgdorf LT, Carell T. Chemistry. 2002;8:293–301. doi: 10.1002/1521-3765(20020104)8:1<293::aid-chem293>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 49.Schnetz-Boutaud NC, Saleh S, Marnett LJ, Stone MP. Biochemistry. 2001;40:15638–15639. doi: 10.1021/bi011242u. [DOI] [PubMed] [Google Scholar]
- 50.Mao H, Schnetz-Boutaud NC, Weisenseel JP, Marnett LJ, Stone MP. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6615–6620. doi: 10.1073/pnas.96.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de los Santos C, Zaliznyak T, Johnson F. J. Biol. Chem. 2001;276:9077–9082. doi: 10.1074/jbc.M009028200. [DOI] [PubMed] [Google Scholar]
- 52.Kim HY, Voehler M, Harris TM, Stone MP. J. Am. Chem. Soc. 2002;124:9324–9325. doi: 10.1021/ja020333r. [DOI] [PubMed] [Google Scholar]
- 53.Cho YJ, et al. J. Am. Chem. Soc. 2005;127:17686–17696. doi: 10.1021/ja053897e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Capon B. Chem. Rev. 1969;69:407–498. [Google Scholar]
- 55.Greenberg MM, Hantosi Z, Wiederholt CJ, Rithner CD. Biochemistry. 2001;40:15856–15861. doi: 10.1021/bi011490q. [DOI] [PubMed] [Google Scholar]
- 56.Aramini JM, Cleaver SH, Pon RT, Cunningham RP, Germann MW. J. Mol. Biol. 2004;338:77–91. doi: 10.1016/j.jmb.2004.02.035. [DOI] [PubMed] [Google Scholar]
- 57.Giri I, Stone MP. Biopolymers. 2002;65:190–201. doi: 10.1002/bip.10222. [DOI] [PubMed] [Google Scholar]
- 58.Gopalakrishnan S, Liu X, Patel DJ. Biochemistry. 1992;31:10790–10801. doi: 10.1021/bi00159a021. [DOI] [PubMed] [Google Scholar]
- 59.Alekseyev YO, Hamm ML, Essigmann JM. Carcinogenesis. 2004;25:1045–1051. doi: 10.1093/carcin/bgh098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.