

Abstract
Autism is a severe disorder that involves both genetic and environmental factors. Expression profiling of the superior temporal gyrus of six autistic subjects and matched controls revealed increased transcript levels of many immune system related genes. We also noticed changes in transcripts related to cell communication, differentiation, cell cycle regulation and chaperone systems. Critical expression changes were confirmed by qPCR (BCL6, CHI3L1, CYR61, IFI16, IFITM3, MAP2K3, PTDSR, RFX4, SPP1, RELN, NOTCH2, RIT1, SFN, GADD45B, HSPA6, HSPB8 and SERPINH1). Overall, these expression patterns appear to be more associated with the late recovery phase of autoimmune brain disorders, than with the innate immune response characteristic of neurodegenerative diseases. Moreover, a variance-based analysis revealed much greater transcript variability in brains from autistic subjects compared to the control group, suggesting that these genes may represent autism susceptibility genes and should be assessed in follow-up genetic studies.
Keywords: DNA microarray, gene expression, transcriptome, autism, qPCR, post mortem, temporal cortex
INTRODUCTION
Autism spectrum disorder (ASD) is a life long pervasive developmental disorder first manifesting itself before age 3. ASD is diagnosed on the basis of several behavioral dysfunctions: impaired social interaction, impaired communication, restricted and repetitive interests and activities (Lord et al., 2000). Neuropathological studies of postmortem brains from subjects with autism have revealed abnormalities in neuronal organization of the cerebral cortex and reduced number of Purkinje cells in the cerebellum, suggesting that altered neuronal maturation and/or defective cortical organization may play a role in the development of ASD (Bauman and Kemper, 2003). Recent publications suggest a link between the volume of the superior temporal gyrus (STG) and language development in autistic children (Bigler et al., 2007). The superior temporal gyrus is involved in auditory processing, including language, and has also been implicated as a critical structure in social cognition (Baron-Cohen et al., 1999).
The exact etiology of autism is unknown, although it is believed to result from a complex combination of genetic, environmental, and immunological factors (Persico and Bourgeron, 2006). A number of genes have been recently identified as promising candidates, including Reelin (RELN) (Persico et al., 2001), serotonin transporter (5HTT) and engrailed 2 (EN2) and MET (Campbell et al., 2006). In addition to genetic factors, environmental factors appear to contribute significantly to the risk of developing the disease: prenatal rubella infections, anticonvulsants, antiemetics taken during pregnancy, perinatal hypoxia, and postnatal infections have all been identified as putative contributors to ASD (Baird et al., 2003). In particular, increased occurrence of maternal immune abnormalities during early pregnancy and greater incidence of familial autoimmunity suggest that some of the non-genetic ASD predisposing factors may act through altering the response of the materno-fetal immune system (Fatemi et al., 2002b; Shi et al., 2003; Smith et al., 2007). This view is also supported by recent findings of dysregulated production of antibodies, cytokines, and immune cells in autistic patients (Ashwood et al., 2006), which could be either caused by, or the result of altered fetal neurodevelopment.
In order to better understand the molecular changes associated with ASD, we assessed the transcriptome of the temporal cortex of postmortem brains from autistic subjects and compared it to matched healthy controls. This assessment was performed using oligonucleotide DNA microarrays on six autistic-control pairs. While the sample size is limited by the availability of high-quality RNA from postmortem subjects with ASD, this sample size is sufficient to uncover robust and relatively uniform changes that may be characteristic of the majority of subjects. Our study revealed a dramatic increase in the expression of immune system-related genes. Furthermore, transcripts of genes involved in cell communication, differentiation, cell cycle regulation and cell death were also profoundly affected. Many of the genes altered in the temporal cortex of autistic subjects are part of the cytokine signaling/regulatory pathway, suggesting that a dysreactive immune process is a critical driver of the observed ASD-related transcriptome profile.
MATERIALS AND METHODS
1. Human brain samples
The Autism Tissue Program (ATP) (http://www.brainbank.org/) database was extensively reviewed and the resulting tissue request for frozen samples of superior temporal gyrus (STG) was approved by ATP. Initial review and subsequent analysis of more than 30 sample pairs revealed that 8 pairs of autistic (AUT) – control (CONT) subjects had satisfactory RNA quality (RIN>7.0), assessed by Agilent BioAnalyzer (Santa Clara, CA) to be included in the further study. A secondary, in-depth review of the clinical data eliminated two additional subject pairs, resulting in the 6 final pairs presented in this study (Table 1). The control and experimental samples showed no statistically significant difference in mean age, post-mortem interval (PMI), RNA integrity number (RIN) and present calls/5′:3′ integrity ratios by Affymetrix GeneChip Operating Software (GCOS) (Santa Clara, CA). Brain tissue recovery, guided by an institutionally approved informed consent procedure, is coordinated nationally by the ATP and the NIMH/NINDS Harvard Brain Tissue Resource Center.
Table 1.
Autistic and control subjects.
| CONTROL SAMPLES | ||||||||
|---|---|---|---|---|---|---|---|---|
| Diagnosis | ATP ID# | SEX | AGE | PMI | RIN | % P | 5′:3′ | Cause of death |
| Control | UMB1377 | F | 5 | 20.0 | 7.8 | 41.9% | 1.98 | Drowning |
| Control | UMB1407 | F | 9 | 20.0 | 7.6 | 45.5% | 1.59 | Asthma |
| Control | UMB1185 | M | 4 | 17.0 | 7.4 | 51.8% | 1.41 | Drowning |
| Control | B4211 | M | 30 | 23.0 | 7.4 | 51.7% | 1.07 | Cardiac arrhythmia |
| Control | B5873 | M | 28 | 23.0 | 7.6 | 49.3% | 1.06 | Unknown |
| Control | B3829 | M | 22 | 24.0 | 7.1 | 53.6% | 1.07 | Central hepatic laceration |
| MEAN | 16.3 | 21.2 | 7.5 | 49.0% | 1.36 | |||
| AUTISM SAMPLES | ||||||||
| Diagnosis | ATP ID# | SEX | AGE | PMI | RIN | % P | 5′:3′ | Cause of death |
| Autism | UMB4671 | F | 4 | 13.0 | 7.5 | 50.6% | 1.00 | Trauma |
| Autism | UMB1174 | F | 7 | 14.0 | 8.0 | 45.0% | 1.54 | Sudden death, seizure |
| Autism | UMB144 | M | 10 | 22.0 | 7.5 | 45.9% | 1.58 | Drawning |
| Autism | B5173 | M | 30 | 20.0 | 7.2 | 50.4% | 1.55 | Gastrointestinal hemorrage |
| Autism | B5000 | M | 27 | 8.3 | 7.2 | 54.0% | 1.45 | Drowning |
| Autism | B5144 | M | 20 | 23.7 | 7.4 | 53.9% | 1.04 | Trauma |
| MEAN | 16.3 | 16.8 | 7.5 | 50.0% | 1.36 | |||
Autism Tissue Program identifier is depicted by ATP ID#. The age, postmortem interval (PMI), Bioanalyzer 2100 RNA integrity number (RIN), present call (%P) and Affymetrix 5′:3′ GAPDH ratio were not significantly different across the two groups (two-tailed t-test = 1.00, 0.17, 0.91, 0.65 and 0.99, respectively).
2. Sample preparation and hybridization
Brain material was homogenized and total RNA isolated using Trizol® reagent (Invitrogen, Carlsbad, CA), with RNA quality assessed via analysis on an Agilent 2100 Bioanalyzer. Only samples with an RIN>7.0 were considered for further analysis. The samples were primed with a standard T7-oligo(dT) primer and cDNA synthesis was performed using five μg of total RNA according to the Affymetrix® manufacturer’s protocol. Amplified antisense RNA (aRNA) was produced using in vitro transcription directed by T7 polymerase. Fifteen micrograms of the purified and fragmented aRNA were hybridized to Affymetrix Human Genome 133 plus 2 microarrays. Image segmentation analysis and generation of DAT files was performed using Microarray Suite 5.0 (MAS5).
3. Microarray data analysis
All microarrays had exceptional quality based on present calls and 5′:3′ GAPDH integrity ratios calculated by GCOS (Table 1). Segmented images were normalized and log2 transformed using GC-robust multi-array analysis (GC-RMA) (Wu et al., 2004), with GC-RMA normalized expression levels utilized for all of our subsequent analyses. All microarray data will be made publicly available at the time of publication.
A. Identification of differentially expressed genes
The data were subjected to four types of analysis: 1) magnitude assessment by calculating the average log ratio (ALR=MeanAUT-MeanCONT) for the RMA-generated log2 values; 2) a Student’s pairwise two-tailed t-test between the RMA intensities of AUT and CONT samples (p1); 3) a Student’s groupwise two-tailed t-test between the RMA intensities of AUT and CONT sample groups (p2); 4) a Student’s pairwise two-tailed t-test using the difference in each gene’s relative rank within the AUT and CONT sample groups (p3). Genes were considered differentially expressed between AUT and CONT samples if they reported AUT-CONT absolute ALR >1 (corresponds to a 2-fold change) and a statistical significance of p<0.05 in at least 2 probability measurements. This strategy allowed us to eliminate small expression changes that may be statistically significant, which are a major source of type I errors.
We felt that such a multifaceted analysis strategy was essential to maximize the true discovery and minimize the putative confounds arising from a limited sample size and cohort diversity. For example, pairwise analysis (while giving up power by reducing the degrees of freedom in statistical tests) allows meaningful comparisons across an age range and inclusion of subjects with different genders. In addition, relative rank analysis is independent of the normalization method applied to the microarray dataset.
Two-way hierarchical clustering of the data was performed using GenePattern software (Subramanian et al., 2005). This clustering was performed on log2 transformed GC-RMA normalized expression levels using row (gene) centering and Pearson correlation.
B. Enrichment detection by literature search
Automated enrichment detection tools, while powerful for initial assessment of data, suffer from low specificity and a limited knowledge base. These programs are not tissue-type specific and also fail to assess the cell-type specificity of the observed changes. Thus, once we identified differentially expressed genes between the AUT and CONT samples, we attempted to classify them into common biological functions based on a comprehensive search of NCBI-listed published literature.
C. Enrichment detection using GSEA
The goal of this analysis was to uncover gene group enrichments in the whole dataset using an unbiased approach. The method derives its power by focusing on gene sets, that is, groups of genes that share common biological function. Gene set analysis was performed using Gene Set Enrichment Analysis (GSEA) software version 2 with the entire microarray probe set collapsed to a gene symbol and gene pathways generated from Biocarta (converted from probe set list with manual curation). Statistical significance was calculated using paired Student’s t test. False discovery rate threshold in GSEA was set at q<0.05.
D. Identification of genes showing increased variance
Finally, we performed an exploratory, variance-based assessment of the dataset. At the heart of this analysis lies an assumption that not all diseased subjects are equal with respect to the genetic and environmental liabilities which predisposed them to the disease; thus they may display a significantly greater variability in their overall gene expression pattern. In contrast, we expect that the control subjects will show comparable expression within their group; thus most genes will have a relatively low variance. If this hypothesis holds true, one would expect that the number of genes that would show standard deviation SDAUT-SDCONT>2 would greatly outnumber the ones with SDCONT-SDAUT>2.
4. qPCR verification of data
cDNA synthesis was performed using two independent reverse transcription reactions for each sample with High Capacity cDNA Archive Kit® (Applied Biosystems). For each 100 μl reaction, we used 700 ng of the same total RNA used for microarray analysis. Priming was performed with random hexamers. For each sample, amplified product differences were measured with 4 independent replicates using SYBR Green chemistry-based detection (Mimmack et al., 2004). β-actin was used as the endogenous reference gene since 1) it has been established as a stable reference gene in the literature (Chen et al., 2001), 2) it did not display significant variation in gene expression between autistic and control samples in the microarray studies and 3) it has been established as a stable reference gene in our previous studies of the human postmortem brain (Arion et al., 2006; Arion et al., 2007). The efficiency for each primer set was assessed prior to qPCR measurements, and a primer set was considered valid if its efficiency was >80%. The qPCR reactions were carried out on an ABI Prism 7300 thermal cycler (Applied Biosystems Inc.), quantified using ABI Prism 7300 SDS software (with the auto baseline and auto threshold detection options selected) and statistically analyzed using a Student’s one-tailed paired t-test in Microsoft Excel.
RESULTS
Differentially expressed transcripts in autistic superior temporal gyrus
Six autistic and six matched control brain tissue samples were assayed for differential gene expression. Approximately 38,000 genes represented by 54,000 probe sets were interrogated by GeneChip Human Genome U133 Plus 2.0 arrays (Affymetrix Inc, Santa Clara, CA). The investigated control and autistic brains showed comparable characteristics (Table 1). Based on our four statistical criteria we identified 152 differentially expressed gene products (Supplemental Material 1). Of these 130 showed increased expression, while 22 reported decreased levels in the brains of autistics subjects. A two-way clustering (genes x samples) of the expression levels of these gene probes resulted in separation of the samples in two distinct classes, with all but one brain from an autistic subject clustering together (Figure 1).
Figure 1. Two-way hierarchical clustering of expression data.

Hierarchical clustering was performed on log2-transformed expression level of 152 differentially expressed genes which demonstrated |ALR|>1 and p<0.05 in three distinct statistical analyses (groupwise, pairwise and rank-based). Samples were clustered vertically, gene probes were clustered horizontally. Genes are denoted by Affymetrix probes and NCBI gene symbols. Each colored square represents a normalized gene expression level, color coded for increase (red) or decrease (blue). Color intensity is proportional to magnitude of change. The clustering resulted in a near-perfect separation of samples into two discrete groups corresponding to diagnosis (vertical dendrogram, red for AUT and blue for CONT).
Importantly, an additional 69 genes showed |ALR AUT- CONT |>1 with significance in 2/3 statistical analyses, generally due to the lower statistical power of pairwise t-tests. These include multiple genes that have been associated with brain pathology, suggesting that this additional dataset may also contain important leads related to the pathophysiology of the disease. Finally, 66 additional genes showed significance by only one of three of the statistical methods.
Real time quantitative PCR (qPCR) validation of microarray data
To validate the microarray findings we selected 20 genes for qPCR analysis. The selected genes reported ALRAUT-CONT values ranging between −1.3 (2.4-fold decrease in AUT) to 3.7 (12.9-fold increase in AUT). Ten of the chosen genes showed statistical significance in all three microarray statistical analyses [heat shock 70kDa protein 6 (HSPA6); serpin peptidase inhibitor, clade H (SERPINH1); chitinase 3-like 1 (CHI3L1); growth arrest and DNA-damage-inducible, beta (GADD45B); cysteine-rich, angiogenic inducer, 61 (CYR61); phosphatidylserine receptor (PTDSR); interferon induced transmembrane protein 3 (IFITM3); aquaporin 4 (AQP4), heat shock 22kDa protein 8 (HSPB8) and transporter 1 ATP-binding cassette, sub-family B (TAP1)], seven reported significance in 2 of 3 assessments [secreted phosphoprotein 1 (SPP1); dystrobrevin, alpha (DTNA); regulatory factor X, 4 (RFX4); B-cell CLL/lymphoma 6 (BCL6); interferon gamma-inducible protein 16 (IFI16); Ras-like without CAAX 1 (RIT1) and growth arrest-specific 7 (GAS7)], and three genes showed expression difference in only in one of the statistical assessments [stratifin (SFN); mitogen-activated protein kinase kinase (MAP2K3) and Notch homolog 2 (NOTCH2)]. For all the tested genes the expression differences reported by qPCR agreed with the directionality revealed by the microarray data. The overall qPCR ΔΔ Ct and the microarray ALR for these gene transcripts were strongly correlated (Pearson r = 0.89, p<0.003) (Figure 2). Furthermore, the qPCR findings reached both pair-wise and group-wise statistical significance for 16 out of the 20 tested genes (80%). Interestingly, the success of validation did not depend on the strength of the statistical evidence: three out the four transcripts that did not reach statistical significance in qPCR reported initial significance in the microarray experiment by all three statistical analyses. In contrast, the differential expression of all three transcripts that showed significance in only one out of the three statistical assessments of the microarrays data were successfully validated by qPCR.
Figure 2. Data validation by qPCR.
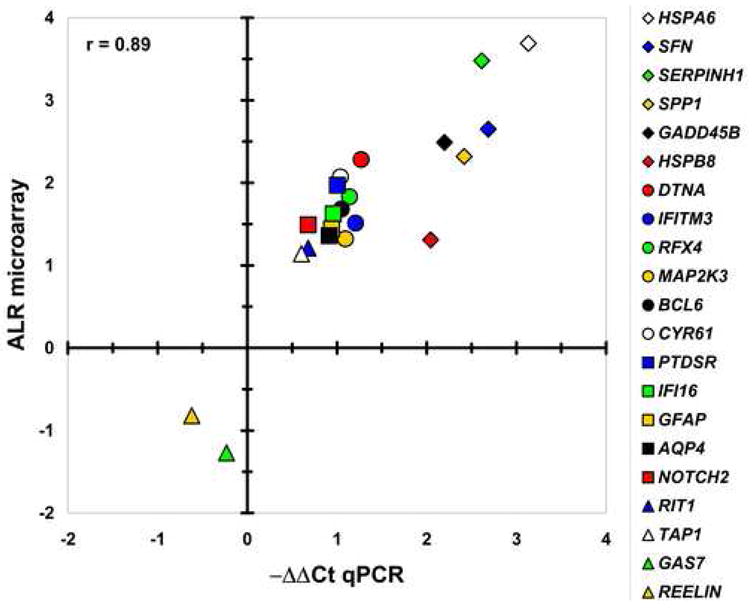
Differential expression of 22 genes was validated by qPCR. X-axis represents -ΔΔCt measured by qPCR, Y-axis denotes DNA microarray reported ALR. Each symbol represents a single gene. Note that the qPCR and microarray data were highly correlated (r=0.89; p<0.001), with agreement in the directionality of change for all investigated transcripts.
Furthermore, we tested by qPCR the expression of two additional genes with potential relation to autism: reelin (RELN) and glial fibrillary acidic protein (GFAP). Although these genes did not meet our inclusion criteria for differentially expressed genes in the microarray experiments, the data were suggestive of a biologically relevant expression change (RELN ALR=−0.82, p1=0.01, p2=0.036, p3= 0.042; GFAP ALR=1.44, p1=0.094, p2=0.095, p3= 0.12). Indeed, the follow-up qPCR assessment revealed a statistically significant differential expression between the AUT and CONT samples for both of these genes (RELN ΔΔ Ct = −0.62, p<0.05 and GFAP ΔΔ Ct = 0.94, p<0.05). This is in agreement with previous reports that biologically relevant expression differences between samples often do not reach significance in DNA microarray datasets (type II errors ) (Mirnics and Pevsner, 2004; Mirnics et al., 2006).
Classification of most changed genes according to function
Knowledge based classification
These classifications were performed on a selected gene set that is differentially expressed between AUT and CONT subjects; based on the success of our qPCR validation, we decided to perform this analysis using transcripts that both reported an |ALR>1| and that reached p<0.05 in at least 2/3 statistical significance comparisons. Of 221 such transcripts, 186 had increased expression in AUT compared to CONT, while only 35 genes showed reduced expression in the AUT samples. We subjected these transcripts to an extensive literature search and observed that 72 out of 193 (37.3%) annotated and differentially expressed transcripts were either immune system related or cytokine responsive transcripts (Supplemental Material 2). Following this first classification, we were able to more precisely sub-classify these 72 annotated genes into three major functional subcategories, which overlap to a different degree; 1) cell communication and motility, 2) cell fate and differentiation, and 3) chaperones (Figure 3). The deregulation of these gene pathways might indicate that the profound molecular differences observed in the temporal cortex of autistic subjects possibly originate from an inability to attenuate a cytokine activation signal.
Figure 3. Autistic samples show altered expression of transcripts involved in cell communication and differentiation.
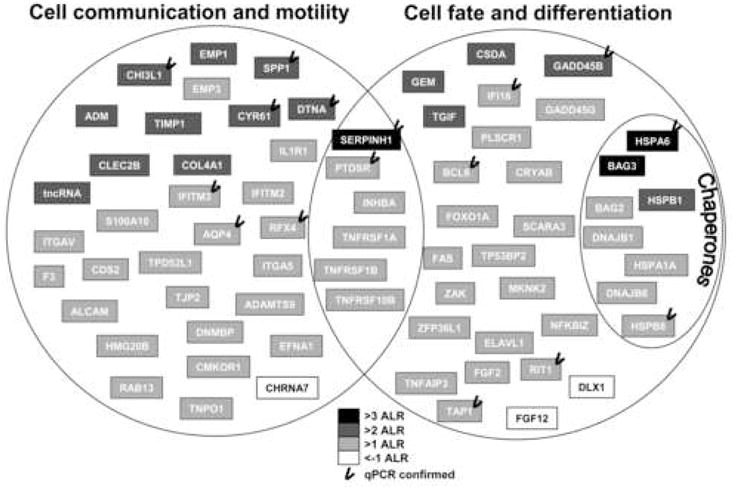
Differentially expressed genes were functionally classified based on literature search (for references, see Supplemental Material 2). We observed a strong overrepresentation of differentially expressed transcripts mediating cell communication and motility, cell fate and differentiation, and chaperones. The magnitude of the gene expression change is coded by cell shading, and checkmarks denote successful qPCR validation of differential expression. Note that the transcript inductions (grey boxes) greatly outnumbered transcript repressions (white boxes).
Furthermore, we observed reduced transcript levels for several genes involved in neuronal differentiation and outgrowth (FGF12, MYT1L, and GAS7), which is suggestive of altered neuronal maturation and connectivity in the assessed autistic individuals.
Finally, our dataset is consistent with previously published findings (Fatemi et al., 2002a; Yip et al., 2007). In addition to the above discussed RELN and GFAP expression changes, glutamic acid decarboxylase 1 (GAD67) and 2 (GAD65) levels were also reduced in our microarray dataset (GAD67 ALR=−0.55, p1=0.046, p2=0.017, p3=0.019; GAD65 ALR= −0.63, p1=0.026, p2=0.010, p3=0.017). Although the statistical significance of these findings were less compelling than the immune system related changes, they still very likely represent a core feature of autism.
Classification using pre-defined gene classes
While the literature-based classification of most changed genes is a powerful tool for in-depth classification of a subset of transcripts that form a network, it also suffers from unavoidable subjectivity. To circumvent this, we elected to perform GSEA, which identifies functional pathways in which gene expression changes are clustered, using the whole, unfiltered dataset with predefined functional classes of genes based on BioCarta. In essence, the genes in the AUT and CONT group were ranked according to their expression level. Then, an enrichment score (ES) and normalized enrichment score (NES – which is an ES normalized for a gene set size) were calculated in order to measure how much each gene set is overrepresented in the ranked list of genes. Two statistical parameters were calculated; a nominal p value for each gene set, estimating the statistical significance of the NES, and a q value, estimating the probability for NES to represent false discovery for a gene set. Using p< 0.01 and q < 0.05 (e.g FDR<5%), we identified 31 BioCarta gene sets that were differentially expressed between AUT and CONT samples (Table 2 and Supplemental Material 3). Interestingly, 19 out of the 31 gene sets were involved in immune system function. More specifically, these groups were related to antigen-specific immune response (TOLL, TNFR2, HIVNEF, DC, IL2R pathway), inflammation (NFKB, IL1R, INFLAM, GSK3, P38MAPK, IL6, NTHI, and TH1TH2 pathway), cell death (NFKB, TNFR2, P38MAPK, TID, 41BB, CASPASE, and FAS pathway), autoimmune diseases (NFKB, TOB1, FAS pathway), migration (MCALPAIN pathway) and targeting of the immune response to specific cells (NKT pathway). Thus, the data obtained using a pre-defined gene set were strongly supportive of our findings that resulted from a knowledge-based assessment. Additionally, the analysis also revealed a systemic transcript disturbance of the MET pathway, providing further support to the previously published findings (Campbell et al., 2007)
Table 2.
Gene pathways with altered expression in autism
| Gene Set | Genes | NES | p-val | q-val |
|---|---|---|---|---|
| NFKBPATHWAY | 22 | 2.10 | 0.0000 | 0.0000 |
| IL1RPATHWAY | 30 | 2.05 | 0.0000 | 0.0000 |
| TOLLPATHWAY | 32 | 2.05 | 0.0000 | 0.0000 |
| NKTPATHWAY | 25 | 2.00 | 0.0000 | 0.0000 |
| INFLAMPATHWAY | 28 | 1.96 | 0.0000 | 0.0000 |
| GSK3PATHWAY | 25 | 1.91 | 0.0000 | 0.0020 |
| TOB1PATHWAY | 16 | 1.87 | 0.0000 | 0.0040 |
| TNFR2PATHWAY | 17 | 1.86 | 0.0000 | 0.0040 |
| CARDIACEGFPATHWAY | 16 | 1.86 | 0.0000 | 0.0040 |
| P38MAPKPATHWAY | 37 | 1.84 | 0.0030 | 0.0050 |
| TIDPATHWAY | 17 | 1.79 | 0.0000 | 0.0120 |
| G1PATHWAY | 24 | 1.76 | 0.0000 | 0.0200 |
| HIVNEFPATHWAY | 54 | 1.73 | 0.0010 | 0.0290 |
| 4-1BBPATHWAY | 16 | 1.73 | 0.0020 | 0.0270 |
| MCALPAINPATHWAY | 23 | 1.70 | 0.0050 | 0.0340 |
| P53HYPOXIAPATHWAY | 18 | 1.69 | 0.0050 | 0.0350 |
| METPATHWAY | 36 | 1.68 | 0.0030 | 0.0360 |
| DEATHPATHWAY | 32 | 1.67 | 0.0060 | 0.0370 |
| ATMPATHWAY | 18 | 1.66 | 0.0160 | 0.0390 |
| IL6PATHWAY | 20 | 1.66 | 0.0130 | 0.0400 |
| RELAPATHWAY | 15 | 1.65 | 0.0060 | 0.0400 |
| ALKPATHWAY | 32 | 1.65 | 0.0060 | 0.0380 |
| NTHIPATHWAY | 20 | 1.65 | 0.0110 | 0.0410 |
| CASPASEPATHWAY | 21 | 1.65 | 0.0100 | 0.0390 |
| DCPATHWAY | 20 | 1.64 | 0.0140 | 0.0400 |
| AKTPATHWAY | 16 | 1.64 | 0.0070 | 0.0400 |
| ECMPATHWAY | 20 | 1.63 | 0.0130 | 0.0400 |
| RACCYCDPATHWAY | 21 | 1.63 | 0.0220 | 0.0400 |
| IL2RBPATHWAY | 33 | 1.61 | 0.0000 | 0.0450 |
| TH1TH2PATHWAY | 16 | 1.61 | 0.0110 | 0.0450 |
| FASPATHWAY | 26 | 1.60 | 0.0190 | 0.0470 |
Microarray gene expression data, subjected to functional pathway analysis with GSEA using BioCarta pathways, identified 31 differentially expressed groups of genes between AUT and CONT samples. The gene pathways are ranked by a net enrichment score (NES). The number of genes in each pathway is depicted in the Genes column. All the differentially expressed pathways reported a q-value based false discovery rate of less than 5% (q<0.05). Note that most of differentially expressed pathways are involved in immune response (shaded).
Variability within subject groups
Finally, to maximize the information that can be obtained from our data set, we performed an exploratory, variance-based assessment of all the microarray-represented transcripts. Autistic subjects display significant phenotypic variability which could be due to an intricate interplay of genetic and environmental factors. Thus, we hypothesized that this phenotypic diversity is due to subject-to-subject variability in gene expression. To test this, we calculated the intra-cohort standard deviation (SD) of normalized expression levels for each gene of the dataset. We identified 62 transcripts that showed higher variability in the brains of AUT subjects (SD(AUT)-SD(CONT)>2) (Table 3). In contrast, only 1 transcript showed higher variability in the CONT brains (SDCONT-SDAUT>2), suggesting that the molecular diversity among autistic subjects greatly exceeds the variability seen in the control cohort. Again, many of the genes showing a higher variability in the autistic cohort were related to the immune system and cytokine signaling, suggesting that the increased diversity present in autistic brains is due to a dysregulation of the immune response.
Table 3.
Autistic samples show increased transcriptome variability
| # | GENE NAME | SYMBOL | SDAUT | SDCONT | SDAUT — SDCONT |
|---|---|---|---|---|---|
| 1 | serpin peptidase inhibitor A3 (alpha-1 antiproteinase) | SERPINA3 | 4.21 | 0.88 | 3.33 |
| 2 | hypothetical protein FLJ10847 | FLJ10847 | 3.33 | 0.04 | 3.29 |
| 3 | stratifin* | SFN | 3.25 | 0.06 | 3.19 |
| 4 | secreted frizzled-related protein 2* | SFRP2 | 3.44 | 0.29 | 3.15 |
| 5 | secretory leukocyte peptidase inhibitor | SLPI | 3.13 | 0.07 | 3.06 |
| 6 | stratifin* | SFN | 3.37 | 0.33 | 3.04 |
| 7 | scavenger receptor class A, member 5* | SCARA5 | 3.70 | 0.66 | 3.03 |
| 8 | heat shock 70kDa protein 6* | HSPA6 | 3.27 | 0.38 | 2.89 |
| 9 | secreted frizzled-related protein 2* | SFRP2 | 3.60 | 0.71 | 2.88 |
| 10 | CD44 molecule* | CD44 | 2.88 | 0.01 | 2.87 |
| 11 | heat shock 70kDa protein 6* | HSPA6 | 3.69 | 0.94 | 2.75 |
| 12 | scavenger receptor class A, member 5* | SCARA5 | 3.16 | 0.47 | 2.69 |
| 13 | CD44 molecule* | CD44 | 3.32 | 0.67 | 2.65 |
| 14 | neuronal PAS domain protein 4 | NPAS4 | 3.75 | 1.16 | 2.59 |
| 15 | CD44 molecule* | CD44 | 2.72 | 0.15 | 2.58 |
| 16 | CD44 molecule* | CD44 | 2.68 | 0.11 | 2.57 |
| 17 | guanylate binding protein 2, interferon-inducible | GBP2 | 2.64 | 0.07 | 2.57 |
| 18 | chemokine (C-C motif) ligand 19 | CCL19 | 3.07 | 0.51 | 2.56 |
| 19 | family with sequence similarity 20, member A* | FAM20A | 2.57 | 0.01 | 2.55 |
| 20 | leptin receptor* | LEPR | 2.55 | 0.02 | 2.53 |
| 21 | solute carrier family 26 (sulfate transporter), member 2* | SLC26A2 | 2.97 | 0.45 | 2.52 |
| 22 | sine oculis homeobox homolog 2 | SIX2 | 2.57 | 0.08 | 2.50 |
| 23 | serpin peptidase inhibitor, clade H, member 1 | SERPINH1 | 2.81 | 0.37 | 2.43 |
| 24 | family with sequence similarity 20, member A* | FAM20A | 2.51 | 0.09 | 2.42 |
| 25 | chitinase 3-like 1 (cartilage glycoprotein-39) | CHI3L1 | 2.82 | 0.41 | 2.41 |
| 26 | --- | --- | 2.38 | 0.01 | 2.38 |
| 27 | thrombospondin, type I, domain containing 4* | THSD4 | 2.86 | 0.48 | 2.38 |
| 28 | superoxide dismutase 2, mitochondrial* | SOD2 | 2.43 | 0.07 | 2.35 |
| 29 | transmembrane protein 30B | TMEM30B | 2.37 | 0.03 | 2.34 |
| 30 | sine oculis homeobox homolog 1 | SIX1 | 2.40 | 0.06 | 2.34 |
| 31 | leptin receptor* | LEPR | 2.34 | 0.01 | 2.33 |
| 32 | CD44 molecule* | CD44 | 2.40 | 0.08 | 2.32 |
| 33 | bone morphogenetic protein 5 | BMP5 | 2.34 | 0.02 | 2.32 |
| 34 | thrombospondin, type I, domain containing 4* | THSD4 | 2.74 | 0.43 | 2.31 |
| 35 | chemokine (C-X-C motif) ligand 10 | CXCL10 | 2.31 | 0.02 | 2.30 |
| 36 | Integrin, beta-like 1 (with EGF-like repeat domains) | ITGBL1 | 2.30 | 0.02 | 2.28 |
| 37 | superoxide dismutase 2, mitochondrial* | SOD2 | 2.76 | 0.48 | 2.28 |
| 38 | interleukin 6 (interferon, beta 2) | IL6 | 2.29 | 0.02 | 2.26 |
| 39 | ADAMTS-like 3 | ADAMTSL3 | 2.46 | 0.20 | 2.26 |
| 40 | actin, gamma 2, smooth muscle, enteric | ACTG2 | 2.93 | 0.68 | 2.25 |
| 41 | myosin, heavy polypeptide 11, smooth muscle* | MYH11 | 2.69 | 0.44 | 2.25 |
| 42 | FLJ45224 protein | FLJ45224 | 2.27 | 0.04 | 2.24 |
| 43 | myosin, heavy polypeptide 11, smooth muscle* | MYH11 | 2.24 | 0.03 | 2.20 |
| 44 | platelet-derived growth factor receptor-like | PDGFRL | 2.23 | 0.03 | 2.20 |
| 45 | ras-related associated with diabetes | RRAD | 2.24 | 0.07 | 2.17 |
| 46 | bone morphogenetic protein 4 | BMP4 | 2.16 | 0.01 | 2.15 |
| 47 | secreted frizzled-related protein 4 | SFRP4 | 2.16 | 0.03 | 2.13 |
| 48 | solute carrier family 26 (sulfate transporter), member 2* | SLC26A2 | 2.94 | 0.83 | 2.11 |
| 49 | rabaptin, RAB GTPase binding effector protein 1 | RABEP1 | 2.12 | 0.02 | 2.11 |
| 50 | basonuclin 2* | BNC2 | 2.11 | 0.01 | 2.10 |
| 51 | leptin receptor* | LEPR | 2.14 | 0.04 | 2.10 |
| 52 | basonuclin 2* | BNC2 | 2.12 | 0.03 | 2.10 |
| 53 | selectin E (endothelial adhesion molecule 1) | SELE | 2.17 | 0.08 | 2.09 |
| 54 | chloride intracellular channel 6 | CLIC6 | 2.34 | 0.25 | 2.08 |
| 55 | ATP-binding cassette, sub-family G, member 2 | ABCG2 | 2.42 | 0.35 | 2.08 |
| 56 | aquaporin 3 | AQP3 | 2.24 | 0.17 | 2.07 |
| 57 | keratin 18 | KRT18 | 2.09 | 0.02 | 2.07 |
| 58 | microsomal glutathione S-transferase 1 | MGST1 | 2.06 | 0.02 | 2.04 |
| 59 | chemokine (C-C motif) ligand 4 | CCL4 | 2.38 | 0.36 | 2.03 |
| 60 | leptin receptor* | LEPR | 2.35 | 0.35 | 2.00 |
| 61 | regulator of G-protein signalling 16 | RGS16 | 2.14 | 0.14 | 2.00 |
| 62 | BCL2-associated athanogene 3 | BAG3 | 2.55 | 0.55 | 2.00 |
| 63 | CDNA FLJ34964 fis, clone NTONG2004095 | --- | 0.03 | 3.03 | −3.00 |
Variance-based analysis was derived from differences in standard deviation (SD) within the CONT and AUT sample groups. Asterisk denotes multiple probesets against the same genes that obtained similar results. Transcripts involved in immune-cytokine responses are highlighted in grey. In the entire microarray dataset we identified 62 transcripts with high expression variability (SDAUT — SDCONT >2) in AUT subjects and only 1 in the CONT subjects. These data suggest significant transcriptome heterogeneity within the diseased subjects.
DISCUSSION
The results of our study suggest that 1) in autism, transcript induction events greatly outnumbers transcript repression processes; 2) the neocortical transcriptome of autistic individuals is characterized by a strong immune response; 3) the transcription of genes related to cell communication, differentiation and cell cycle regulation is altered, putatively in an immune system-dependent manner, and 4) transcriptome variability is increased among autistic subjects, as compared to matched controls. Furthermore, our study also provides additional support for previously reported involvement of MET, GAD1, GFAP, RELN and other genes in the pathophysiology of autism. While the findings were obtained on a limited sample size, the statistical power, together with the previously reported postmortem data by other investigators suggest that the observed gene expression changes are likely to be critically related to the pathophysiology seen in the brain of the majority of ASD patients.
There have been three autism DNA microarray studies performed previously on human material. The first study by Purcell et al (2001) analyzed postmortem cerebellar tissue from subjects with autism and reported an upregulation of glutamate related transcripts. Unfortunately, the results from their and our studies are almost impossible to meaningfully compare, as different experimental designs, array platforms, brain regions, statistical approaches and control strategies were employed. Perhaps not surprisingly, in our study we did not observe a cortical glutamatergic transcript upregulation, rather, we observed a moderate/mild decrease in three glutamatergic transcripts (glutamate receptors GRID1/GluR δ-1 with ALR = −0.25, p<0.05 and GRIK2/GluR6 with ALR = −0.75, p<0.05 and glutamate transporter SLC1A1 with ALR = −0.44, p<0.05). Thus, the two datasets are neither convergent nor divergent, but simply very different, and the disparity can be possibly explained by the differential regulation of the glutamate system in the cerebellum and temporal cortex. Nevertheless, both studies report altered expression of glutamate system genes in the brain of autistic subjects, putatively suggesting a wide, region-specific and complex dysregulation of the glutamatergic network. This notion is further strengthened by the recent discovery that GRIK2/GluR6 may represent an autism susceptibility gene (Jamain et al., 2002; Dutta et al., 2007).
The other two DNA microarray studies focused on gene expression changes in the blood of patients with autism (Nishimura et al., 2007). The study by Nishimura and coworkers was successful in identifying peripheral biomarkers of the disease based on the expression of members of the FMR1-CYFIP1-JAKMIP1-GPR155 pathway. These distinct transcriptome profiles were able to correctly subclassify autism based on genetic etiology (e.g. 15q11-q13 duplication vs fragile X mutation). In the other report, Gregg et al point toward an abnormal activation of NK cells and/or CD8+ cytotoxic T cells in autism (Gregg et al., 2007), and we may only speculate that these findings could be a peripheral manifestation of the same immune processes that we are witnessing in the temporal cortex. Nevertheless, the transcriptome profiles in blood cells and brain in autism are bound to differ: peripherally expressed genes may show a co-regulation with the genes responsible for CNS pathophysiology, thus representing extremely valuable disease markers even if they are not causally involved in the brain-related pathophysiological events. In contrast, gene expression changes in the brains of subjects with autism may speak of the most critical aspects of the CNS pathophysiology, yet they may have no diagnostic value if they are not expressed or modulated in accessible, peripheral tissue.
It is noteworthy that our findings are consistent with many other studies that employed more classical experimental methods and focused on single genes or specific pathways. For example, glutamic acid decarboxylase transcripts (GAD1 and GAD2) were reduced in the temporal cortex of our autistic cohort; similar reductions of these two critical GABAergic enzymes were previously reported by Yip et al (2007) in cerebellar Purkinje cells. Furthermore, increased GFAP expression was observed in the cerebellum of postmortem autistic brains by several studies (Purcell et al., 2001; Vargas et al., 2005), while decreased RELN gene expression was previously found in cerebellar cortex of autistic individuals (Fatemi et al., 2001). Finally, we previously reported decreased transcription of MET, an established autism susceptibility gene (Campbell et al., 2006), which was recently confirmed by showing parallel decreases in MET protein levels and increases in transcripts of genes encoding other members of the MET pathway (Campbell et al., 2007).
The most prominent expression changes in our dataset are clearly related to neuroimmune disturbances in the cortical tissue of autistic subjects. The idea of brain inflammatory changes in autism is not novel; epidemiological, (DeLong et al., 1981; Yamashita et al., 2003; Libbey et al., 2005) serological studies (Vargas et al., 2005; Ashwood et al., 2006) and postmortem studies (Pardo et al., 2005; Vargas et al., 2005; Korkmaz et al., 2006) over the last 10 years have provided compelling evidence that immune system response is an essential contributor to the pathophysiology of this disorder (Ashwood et al. 2006). Finally, converging post-mortem assessments and measurements of cytokines in the CSF of autistic children (Vargas et al., 2005), may indicating an ongoing immunological process involving multiple brain regions.
Altered immune system genes are often observed across various brain disorders, albeit there are notable differences between the observed transcriptome patterns. The majority of neuroimmune genes found activated in the autistic brains overlap with mouse genes that are activated during the late recovery or “repair” phase in experimental autoimmune encephalomyelitis (Baranzini et al., 2005). This suggests a presence of an innate immune response in autism. However, the altered IL2RB, TH1TH2, and FAS pathways suggest a simultaneously occurring, T cell-mediated acquired immune response. Based on these combined findings we propose that the expression pattern in the autistic brains resembles a late stage autoimmune event rather than an acute autoimmune response or a non-specific immune activation seen in neurodegenerative diseases. Furthermore, the presence of an acquired immune component could conceivably point toward a potential viral trigger for an early-onset chronic autoimmune process leading to altered neurodevelopment and to persistent immune activation in the brain. Interestingly, recently obtained gene expression signatures of subjects with schizophrenia (Arion et al., 2007) show a partial, but important overlap with the altered neuroimmune genes found here in autism. These commonly observed immune changes may represent a long-lasting consequence of a shared, early life immune challenge, perhaps occurring at different developmental stages and thus affecting different brain regions, or yielding distinct clinical phenotypes due to different underlying premorbid genetic backgrounds. Furthermore, the comprehensive understanding of the disease process will require a precise identification of the cell populations that are the primary targets of these complex pathological processes, and various in vivo and in vitro experimental models will be essential for obtaining such information (Zirlinger and Anderson, 2003; de Ledesma et al., 2006; Sabatini et al., 2007).
Finally, the increased molecular variability within autistic subjects indicates that this disease is quite heterogenous at the molecular level, suggesting that we are seeing interplay between environment and genetics that gives rise to a unique gene expression pattern in the brain of each subject. Thus, the gene expression signatures of the disease are likely to sort along a continuum, just like the clinical symptoms of autism spectrum disorders do.
Supplementary Material
Supplemental Material 1. Differentially expressed transcripts between the AUT and CONT samples. Abbreviations: PROBE – Affymetrix probe identifier; SYMBOL – NCBI gene symbol; AUT – normalized average Log2 intensity across autistics samples; CTRL - normalized average Log2 intensity across control samples; ALR – average log2 ratio between autistic and control samples (AUT-CTRL); Pair pVal, Group pVal, Rank pVal – p-values obtained in a t-test performed in pairwise, groupwise or rank-based fashion. Gene probes highlighted in yellow report transcript increases, blue highlights denote probesets reporting decreased mRNA levels in the AUT subjects.
Supplemental material 2. Knowledge-based classification of differentially expressed transcripts revealed multi-transcript disturbances in genes involved in cytokine regulation, release and signaling, or/and immune response (Figure 3). The classification of these genes was performed based on the following references.
Supplemental Material 3. Gene enrichments in the AUT samples within the altered BioCarta pathways. Differentially expressed pathways are highlighted in yellow. For parameter descriptions, see GSEA 2.0 manual @ http://www.broad.mit.edu/cancer/software/gsea/doc/GSEAUserGuideFrame.html
Acknowledgments
We would like to thank Dr. Dominique Arion for assistance in processing tissue, Dr. Pat Levitt for critical input on project design and Dr. Luca Battistini for helpful comments on the manuscript. We also want to thank the donor families, the Autism Tissue Program (Princeton, N.J.) and the ATP Director, Dr. Jane Pickett, for providing the tissue resources for this study. The current research was supported by K02 MH070786 and R01 MH079299 (KM), a NARSAD Young Investigator fellowship (PE) and MIUR PRIN 200605819 (AP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62:711–721. doi: 10.1016/j.biopsych.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion D, Sabatini M, Unger T, Pastor J, Alonso-Nanclares L, Ballesteros-Yanez I, Garcia Sola R, Munoz A, Mirnics K, DeFelipe J. Correlation of transcriptome profile with electrical activity in temporal lobe epilepsy. Neurobiol Dis. 2006;22:374–387. doi: 10.1016/j.nbd.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Wills S, Van de Water J. The immune response in autism: a new frontier for autism research. J Leukoc Biol. 2006;80:1–15. doi: 10.1189/jlb.1205707. [DOI] [PubMed] [Google Scholar]
- Baird G, Cass H, Slonims V. Diagnosis of autism. Bmj. 2003;327:488–493. doi: 10.1136/bmj.327.7413.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranzini SE, Bernard CC, Oksenberg JR. Modular transcriptional activity characterizes the initiation and progression of autoimmune encephalomyelitis. J Immunol. 2005;174:7412–7422. doi: 10.4049/jimmunol.174.11.7412. [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S, Ring HA, Wheelwright S, Bullmore ET, Brammer MJ, Simmons A, Williams SC. Social intelligence in the normal and autistic brain: an fMRI study. Eur J Neurosci. 1999;11:1891–1898. doi: 10.1046/j.1460-9568.1999.00621.x. [DOI] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. The neuropathology of the autism spectrum disorders: what have we learned? Novartis Found Symp. 2003;251:112–122. discussion 122–118, 281–197. [PubMed] [Google Scholar]
- Bigler ED, Mortensen S, Neeley ES, Ozonoff S, Krasny L, Johnson M, Lu J, Provencal SL, McMahon W, Lainhart JE. Superior temporal gyrus, language function, and autism. Dev Neuropsychol. 2007;31:217–238. doi: 10.1080/87565640701190841. [DOI] [PubMed] [Google Scholar]
- Campbell DB, D’Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, Persico AM. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol. 2007;62:243–250. doi: 10.1002/ana.21180. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, Persico AM, Levitt P. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Chen N, Lau LF. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001;276:10443–10452. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- de Ledesma AM, Desai AN, Bolivar VJ, Symula DJ, Flaherty L. Two new behavioral QTLs, Emo4 and Reb1, map to mouse Chromosome 1: Congenic strains and candidate gene identification studies. Mamm Genome. 2006;17:111–118. doi: 10.1007/s00335-005-0107-y. [DOI] [PubMed] [Google Scholar]
- DeLong GR, Bean SC, Brown FR., 3rd Acquired reversible autistic syndrome in acute encephalopathic illness in children. Arch Neurol. 1981;38:191–194. doi: 10.1001/archneur.1981.00510030085013. [DOI] [PubMed] [Google Scholar]
- Dutta S, Das S, Guhathakurta S, Sen B, Sinha S, Chatterjee A, Ghosh S, Ahmed S, Usha R. Glutamate Receptor 6 Gene (GluR6 or GRIK2) Polymorphisms in the Indian Population: A Genetic Association Study on Autism Spectrum Disorder. Cell Mol Neurobiol. 2007 doi: 10.1007/s10571-007-9193-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM, Halt AR, Realmuto GR. Dysregulation of Reelin and Bcl–2 proteins in autistic cerebellum. J Autism Dev Disord. 2001;31:529–535. doi: 10.1023/a:1013234708757. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. 2002a;52:805–810. doi: 10.1016/s0006-3223(02)01430-0. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Earle J, Kanodia R, Kist D, Emamian ES, Patterson PH, Shi L, Sidwell R. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol Neurobiol. 2002b;22:25–33. doi: 10.1023/A:1015337611258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg JP, Lit L, Baron CA, Hertz-Picciotto I, Walker W, Davis RA, Croen LA, Ozonoff S, Hansen R, Pessah IN, Sharp FR. Gene expression changes in children with autism. Genomics. 2007 doi: 10.1016/j.ygeno.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Jamain S, Betancur C, Quach H, Philippe A, Fellous M, Giros B, Gillberg C, Leboyer M, Bourgeron T. Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry. 2002;7:302–310. doi: 10.1038/sj.mp.4000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz B, Benbir G, Demirbilek V. Migration abnormality in the left cingulate gyrus presenting with autistic disorder. J Child Neurol. 2006;21:600–604. doi: 10.1177/08830738060210070601. [DOI] [PubMed] [Google Scholar]
- Libbey JE, Sweeten TL, McMahon WM, Fujinami RS. Autistic disorder and viral infections. J Neurovirol. 2005;11:1–10. doi: 10.1080/13550280590900553. [DOI] [PubMed] [Google Scholar]
- Lord C, Cook EH, Leventhal BL, Amaral DG. Autism spectrum disorders. Neuron. 2000;28:355–363. doi: 10.1016/s0896-6273(00)00115-x. [DOI] [PubMed] [Google Scholar]
- Mimmack ML, Brooking J, Bahn S. Quantitative polymerase chain reaction: validation of microarray results from postmortem brain studies. Biol Psychiatry. 2004;55:337–345. doi: 10.1016/j.biopsych.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Pevsner J. Progress in the use of microarray technology to study the neurobiology of disease. Nat Neurosci. 2004;7:434–439. doi: 10.1038/nn1230. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Levitt P, Lewis DA. Critical appraisal of DNA microarrays in psychiatric genomics. Biol Psychiatry. 2006;60:163–176. doi: 10.1016/j.biopsych.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Vargas DL, Zimmerman AW. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry. 2005;17:485–495. doi: 10.1080/02646830500381930. [DOI] [PubMed] [Google Scholar]
- Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Persico AM, D’Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, Wassink TH, Schneider C, Melmed R, Trillo S, Montecchi F, Palermo M, Pascucci T, Puglisi-Allegra S, Reichelt KL, Conciatori M, Marino R, Quattrocchi CC, Baldi A, Zelante L, Gasparini P, Keller F. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry. 2001;6:150–159. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001;57:1618–1628. doi: 10.1212/wnl.57.9.1618. [DOI] [PubMed] [Google Scholar]
- Sabatini MJ, Ebert P, Lewis DA, Levitt P, Cameron JL, Mirnics K. Amygdala gene expression correlates of social behavior in monkeys experiencing maternal separation. J Neurosci. 2007;27:3295–3304. doi: 10.1523/JNEUROSCI.4765-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry RRG, Martinez-Murillo F, Spencer F. A Model Based Background Adjustment for Oligonucleotide Expression Arrays. Journal of the American Statistical Association. 2004;99(909):909–917. [Google Scholar]
- Yamashita Y, Fujimoto C, Nakajima E, Isagai T, Matsuishi T. Possible association between congenital cytomegalovirus infection and autistic disorder. J Autism Dev Disord. 2003;33:455–459. doi: 10.1023/a:1025023131029. [DOI] [PubMed] [Google Scholar]
- Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol (Berl) 2007;113:559–568. doi: 10.1007/s00401-006-0176-3. [DOI] [PubMed] [Google Scholar]
- Zirlinger M, Anderson D. Molecular dissection of the amygdala and its relevance to autism. Genes Brain Behav. 2003;2:282–294. doi: 10.1034/j.1601-183x.2003.00039.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material 1. Differentially expressed transcripts between the AUT and CONT samples. Abbreviations: PROBE – Affymetrix probe identifier; SYMBOL – NCBI gene symbol; AUT – normalized average Log2 intensity across autistics samples; CTRL - normalized average Log2 intensity across control samples; ALR – average log2 ratio between autistic and control samples (AUT-CTRL); Pair pVal, Group pVal, Rank pVal – p-values obtained in a t-test performed in pairwise, groupwise or rank-based fashion. Gene probes highlighted in yellow report transcript increases, blue highlights denote probesets reporting decreased mRNA levels in the AUT subjects.
Supplemental material 2. Knowledge-based classification of differentially expressed transcripts revealed multi-transcript disturbances in genes involved in cytokine regulation, release and signaling, or/and immune response (Figure 3). The classification of these genes was performed based on the following references.
Supplemental Material 3. Gene enrichments in the AUT samples within the altered BioCarta pathways. Differentially expressed pathways are highlighted in yellow. For parameter descriptions, see GSEA 2.0 manual @ http://www.broad.mit.edu/cancer/software/gsea/doc/GSEAUserGuideFrame.html