

Abstract
The higher prevalence and risk for Alzheimer’s disease in women relative to men has been partially attributed to the precipitous decline in gonadal hormone levels that occur in women following the menopause. While considerable attention has focused on the consequence of estrogen loss, and thus, estrogen’s neuroprotective potential, it is important to recognize that the menopause results in a precipitous decline in progesterone levels as well. In fact, progesterone is neuroprotective, although the precise mechanisms involved remain unclear. Based on our previous observation that progesterone elicits the phosphorylation of ERK and Akt, key effectors of the neuroprotective MAPK and PI3-K pathways, respectively, we determined if activation of either of these pathways was necessary for progesterone-induced protection. Using organotypic explants (slice culture) of the cerebral cortex, we found that progesterone protected against glutamate-induced toxicity. Further, these protective effects were inhibited by either the MEK1/2 inhibitor, UO126, or the PI-3K inhibitor, LY294002, supporting the requirement of both the MAPK and PI-3K pathways in progesterone-induced protection. In addition, at a concentration and duration of treatment consistent with our neuroprotection data, progesterone also increased the expression of Brain-Derived Neurotrophic Factor (BDNF), at the level of both protein and mRNA. This induction of BDNF may be relevant to the protective effects of progesterone since inhibition of Trk signaling, using K252a, inhibited the protective effects of progesterone. Collectively, these data suggest that progesterone is protective via multiple and potentially related mechanisms.
Keywords: Progesterone, ERK, Akt, BDNF, Neuroprotection
INTRODUCTION
The menopause is characterized by a precipitous decline in the levels of circulating estrogen and progesterone. As the average lifespan of women has increased to approximately 80 years of age (Arias and Smith 2003), a more substantial portion of a woman’s life is spent in a hormone-deprived state. Since post-menopausal women have a two to three fold higher prevalence of Alzheimer’s disease (AD) than men (Andersen et al. 1999; Gao et al. 1998; Henderson 1997; Sherwin 1999), it is possible that these hormone deficits may play a significant role in enhancing the risk for the disease.
Numerous studies have indeed reported behavioral, neurochemical and molecular deficits following ovariectomy and found that estradiol, the biologically most potent and prevalent estrogen, can at least partially normalize these deficits (Bishop et al. 1995; Dubal et al. 1998; Fan et al. 2003; Gibbs 1998; Hoffman et al. 2003; Jezierski and Sohrabji 2000; Luine et al. 1975; Nordell et al. 2003; Panickar et al. 1997; Shi et al. 1998; Shi et al. 2000; Simpkins et al. 2004; Simpkins et al. 2005; Singh et al. 1994; Singh et al. 1995; Wen et al. 2004; Wise 2002). For example, ovariectomy results in a decrease in Brain-Derived Neurotrophic Factor (BDNF) levels (Singh et al. 1995) while estradiol has been shown in multiple models to elicit an increase in the expression of BDNF (Gibbs 1999; Gonzalez et al. 2004; Jezierski and Sohrabji 2000; Singh et al. 1995; Sohrabji et al. 1995; Solum and Handa 2002). Interestingly, our previous data suggested that estradiol only partially normalized the ovariectomy-induced deficit in BDNF expression in the cerebral cortex (Singh et al. 1995), leading us to suggest that the incomplete restoration of BDNF levels could have been due to the lack of added progesterone, the other major ovarian hormone that also diminished following ovariectomy.
Progesterone has, in fact, been shown to exert neuroprotective effects. For example, progesterone pre-treatment protects hippocampal neurons from FeSO4-, amyloid β- (Goodman et al. 1996) as well as glutamate-induced cell death (Nilsen and Brinton 2002; Nilsen and Diaz Brinton 2003). In addition, progesterone reduces MPTP-induced toxicity (Callier et al. 2001), a neurotoxin used in models of Parkinson’s disease. Further, secondary neuronal loss following cortical contusion injury and resulting cognitive impairment was significantly reduced in rats that received progesterone treatment relative to untreated controls (Asbury et al. 1998; Roof et al. 1994). And though evidence continues to increase with respect to the ability of progesterone to protect against a variety of insults, our understanding of the precise mechanisms underlying the neuroprotective effects of progesterone remain incomplete.
The purpose of the present study was to determine whether progesterone is protective against glutamate toxicity, a commonly used insult to simulate the excitotoxicity and oxidative stress that occurs with age. Utilizing slice cultures (organotypic explants) of the cerebral cortex, we assessed if progesterone’s ability to elicit the activation of ERK and Akt in cortical explants (Singh 2001), is required for progesterone’s protective effects. Additionally, since our earlier work suggested that estrogen only partially normalizes the ovariectomy-induced deficit in BDNF mRNA in the cerebral cortex, we hypothesized that the lack of progesterone contributed to the incomplete restoration of BDNF levels, and as such, predicted that progesterone may also increase the expression of BDNF. Our results demonstrate that progesterone is protective against glutamate-induced cytotoxicity in the organotypic explants of the cerebral cortex and that this protection was mediated by the ERK/MAPK and PI-3K/Akt pathways. Further, our data also support the induction of BDNF by progesterone as another important mediator of progesterone’s protective effects.
MATERIALS AND METHODS
Tissue Culture
Organotypic explants were derived from ~360 μm thick hemicoronal slices of postnatal day 3 (P3) frontal and cingulate cerebral cortex (day of birth = P1), obtained from pups born of C57B1/6J mice and maintained as roller tube cultures (Gähwiler 1981) on rat tail collagen-coated/poly-L-lysine pre-coated glass cover slips, as previously described (Singh et al. 2000). The cultures were maintained in steroid-deficient and phenol red-free maintenance medium (25% heat inactivated horse serum (Sigma, St. Louis, MO), 22.5% Hank’s Balanced Salt Solution (Mediatech, Inc., Herndon, VA), 50% minimum essential medium Eagle MEM (Sigma), 5.25 mg/ml of D(+)-glucose (Sigma), 2 m M L-glutamine (MediaTech, Inc.), 50 μg/mL of L-ascorbic acid (Sigma) supplemented with 2 nM 17-β estradiol (Steraloids, Newport, RI). Three hemicoronal slices were placed on each coverslip per Leighton tube. Antibiotics were not used.
Treatment of Cultures
The cultures were maintained in vitro for 6 days prior to treatment. On the 6th day in vitro, a 24 h “hormone washout” was performed, consisting of replacing the maintenance medium with media that did not contain exogenously added 17-β estradiol. On the 7th day in vitro, cultures were spiked with the appropriate treatment (hormone, inhibitor, and/or insult - see below) for the specified duration. Controls were also hormone-deprived and sham (vehicle)-treated to account for any consequences of procedural manipulation of the explants.
Progesterone (4-pregnen-3, 20-dione, Sigma) was dissolved in DMSO to a stock concentration of 100 μM and applied to the culture at a final concentration of 100 nM. Vehicle controls were performed in parallel such that control cultures were exposed to 0.1% DMSO. The concentrations of L-glutamate used in the studies described were based on concentration response curves performed in cortical explants (data not shown) identifying 5 - 15 mM as optimal concentrations for the purposes of reliably inducing a significant increase in LDH release. In evaluating the requirement of the ERK/MAPK or PI3-K pathways in progesterone’s ability to protect cultures from L-glutamate, the pharmacological inhibitors, 10 μM UO126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenyalthio] butadiene, Cell Signaling, Danvers, MA), or 15 μM LY294002 (2-[4-morpholinyl]-8-phenyl-4H-1-benzopyran-4-one, Cell Signaling), were used, respectively.
Assessment of Cell Viability
Progesterone’s protective effects were based on its ability to reduce L-glutamate–induced lactate dehydrogenase (LDH) release into the culture media. Progesterone was applied twenty-four hours prior to treatment with L-glutamic acid (Sigma) for 6 hr. This duration of glutamate treatment was chosen to ensure minimal degradation of the LDH in the conditioned media [t1/2 of LDH in solution has been reported to be ~9 hrs (CytoTox-ONE Homogeneous Membrane Integrity Assay Technical Bulletin #TB306, Promega, Madison, WI)]. A fluorometric assay (CytoTox-One Homogenous Membrane Integrity Assay Kit, Promega) was used for the measurement of lactate dehydrogenase (LDH) released from damaged or dying cells. The assay itself is based on the ability of LDH to promote the formation of a fluorescent product, resorufin, from the substrate, resazurin. Briefly, 100 μL of conditioned media was aliquoted into a black 96-well plate. The media was allowed to equilibrate to ambient temperature (~15 min), after which the CytoTox-One reagent was added to each well and incubated for 10 min at room temperature. Following termination of the enzymatic reaction, resulting fluorescence was measured [560 nm (excitation)/590 nm (emission)] using a Viktor3 ELISA plate reader (Perkin Elmer, Boston, MA). Relative fluorescent units (RFU) were normalized to the amount of protein in the explant culture (assessed using BIORAD DC protein assay kit) from which the conditioned media was derived. These values were subsequently normalized and expressed as a percentage of vehicle-treated control.
BDNF Enzyme-linked Immunosorbant Assay
An enzyme-linked immunosorbant assay was used to detect and quantify total cellular BDNF levels (Promega). A 96-well Nunc MaxiSorp surface polystyrene flat bottom immuno-plate plate was pre-coated with an anti-BDNF monoclonal antibody [diluted 1:1000 in coating buffer (25 mM sodium bicarbonate and 25 mM sodium carbonate, pH 9.7)]. After rinsing off unbound antibody with TBS-T buffer (20 mM Tris-HCl (pH 7.6), 150 mM NaCl and 0.05% (v/v) Tween-20), and blocking the plate to minimize non-specific binding, 100 μg of sample lysate (in a final volume of 100 μL) or volume of appropriate BDNF standard, ranging in concentration from 7.8 – 500 pg/ml, was added. After 5 washes with TBS-T, the captured BDNF was then incubated with the polyclonal anti-human BDNF antibody. The amount of specifically bound polyclonal antibody was then detected through the use of the anti-IgY-horseradish peroxidase (HRP) tertiary antibody, which when exposed to the chromogenic substrate (TMB reagent, Promega), changes color in proportion to the amount of BDNF present in the sample. The color intensity was quantified by measuring the absorbance at 450 nm using a Viktor3 ELISA plate reader (Perkin Elmer). Only values that were within the linear range of the standard curve were considered valid. BDNF levels were normalized to protein, and reported as a percentage of vehicle control.
Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
RNA isolation and cDNA synthesis
Total RNA was extracted from explant cultures and DNase-treated using the RNeasy Lipid Kit (Qiagen) according to manufacture’s instructions. RNA concentrations of extracted RNA were calculated from the absorbance at 260 nm. The quality of RNA was assessed by absorption at 260 nm and 280 nm (A260/A280 ratios of 1.9 to 2.10 were considered acceptable) and by electrophoresis through agarose gels and staining with ethidium bromide. The 18S and 28S rRNA bands were visualized under UV light. Total RNA (1.5 μg) was reverse transcribed into cDNA in a total volume of 50 μL using the High Capacity DNA Archive Kit (Applied Biosystems) according to manufacture’s instructions.
Primers and Probes for Quantitative Real-Time Reverse Transcription PCR
PCR primers and probes for the target gene, BDNF, and the endogenous control, 18S rRNA, were purchased as Assays-On-Demand (Applied Biosystems). The assays were supplied as 20X mix of PCR primers (900 nM) and TaqMan probes (200 nM). The BDNF assay (Mm00432069_m1) contained FAM (6-carboxy-fluorescein phosphoramidite) dye label at the 5′ end of the probes and minor groove binder and nonfluorescent quencher at the 3′ end of the probes. The 18S rRNA assays (4319413E) contained VIC-labeled probes. The assays are optimized for use on ABI prism Sequence Detection System using the default machine settings.
Quantitative Real-Time Reverse Transcription-PCR
The reaction mixture containing water, 2X qPCR™ Master Mix (Eurogentec), and 20X Assay-On-Demand for BDNF was prepared. A separate reaction mixture was prepared for the endogenous control, 18S rRNA. The reaction mixture was aliquoted in a 96-well plate and cDNA (11 ng of RNA converted to cDNA) was added to give a final volume of 30 μL. Each sample was analyzed in triplicate. Two nontemplate controls (RNase-free water) were included on each plate for reaction mixture containing BDNF and 18S rRNA assays. Amplification and detection were performed using the ABI 7300 Sequence Detection System (Applied Biosystems) with the following profile: 2 min hold at 50°C (UNG activation), 10 min hold at 95°C, followed by 40 cycles of 15 s at 95°C (denaturation) and 1 min at 60°C (annealing and extension). Sequence Detection Software 1.3 (Applied Biosystems) was used for data analysis.
The comparative CT method (2−ΔΔCT) was used to calculate the relative changes in target gene expression. In the Comparative CT Method, the amount of target, normalized to an endogenous control (18S) and relative to a calibrator (untreated control), is given by the 2−ΔΔCT equation. Quantity is expressed relative to a calibrator sample that is used as the basis for comparative results. Therefore, the calibrator was the baseline (vehicle-treated control) sample and all other treatment groups were expressed as an n-fold (or %) difference relative to the calibrator (Livak and Schmittgen 2001). The average and standard deviation of 2−ΔΔCT was calculated for the values from four independent experiments, and the relative amount of target gene expression for each sample was plotted in bar graphs using GraphPad Prism 4 software.
Statistical Analysis
Data from at least three independent experiments were subjected to ANOVA, followed by a single degree of freedom F-test within the main effect of the ANOVA (analysis performed using Systat–Systat, Inc., San Jose, CA). For statistical consideration of the BDNF mRNA and protein data, a single sample t-test was employed to compare the effects of progesterone against the vehicle-treated control. The data are presented as bar graphs depicting the means ± SEM, and were created using GraphPad Prism 4 software (San Diego, CA).
RESULTS
Progesterone protects against glutamate – induced cytotoxicity
In order to determine if progesterone (P4) protected against an excitotoxic/oxidative insult, we pre-treated organotypic explants of the cerebral cortex with progesterone for 24 hours prior to treatment with L-glutamate (10 - 15 mM, 6 hr). P4 (100 nM) significantly reduced the amount of glutamate-induced LDH release (Figure 1B) while a lower concentration of P4 (10 nM) did not result in a statistically significant reduction in glutamate-induced LDH release (Figure 1A). The concentration of glutamate chosen for these experiments was based on concentration-response curves in which we found that concentrations between 5 and 15 mM resulted in significant and reproducible increases in LDH release (data not shown). Further, our data suggest that the duration of glutamate treatment used in our studies (6 hr) induces cytotoxicity primarily through an NMDA receptor-sensitive mechanism, as MK801, the NMDA-receptor antagonist, prevented the glutamate-induced LDH release (Figure 1C).
Figure 1. Progesterone (P4) protects against glutamate-induced LDH release.

A. Cerebral cortical explants were pre-treated with either 10 nM or 100 nM P4 for 24 hr prior to the administration of L-glutamate (Glut, 5 mM for 6 hr). While 100 nM P4 was effective at reducing glutamate-induced LDH release, 10 nM P4 was not. LDH release was normalized to vehicle-treated control, whose value (set at 100%) is represented by the solid horizontal line. Data are presented as mean ± SEM. (**: p < 0.05 relative to vehicle-treated control)
B. Cerebral cortical explants were pre-treated with P4 (100 nM) for 24 hr prior to the administration of L-glutamate (Glut, 10 mM or 15 mM for 6 hr). P4 prevented glutamate-induced LDH release as these values were not statistically different from vehicle treated control. LDH release is expressed as a percentage of that seen in the vehicle-treated control (set at 100%), with the latter represented by the solid horizontal line. The graph shown represents data from 3 independent experiments. Data are presented as mean ± SEM. Following an Analysis of Variance (ANOVA), individual comparisons were made using a single degree of freedom F-test within the main effect of the ANOVA [F(6,30) = 6.545, p < 0.0001; *: p ≤ 0.001 versus vehicle control; #: p < 0.05 versus P4 + Glut (10mM) and P4 + Glut (15 mM)].
C. Cerebral cortical explants were pre-treated with the NMDA receptor antagonist, MK801 (10 or 20 μM), for 10 min. prior to the administration of L-glutamate (Glut, 10 mM or 15 mM for 6 hr). MK801 prevented glutamate – induced LDH release. LDH release was normalized to vehicle-treated control, whose value (set at 100%) is represented by the solid horizontal line. Data are presented as mean ± SEM. (*: p<0.05 versus vehicle-treated control).
Progesterone-induced cytoprotection is dependent on MEK and PI3-kinase activity
Our laboratory has previously shown that progesterone elicits the phosphorylation of ERK and Akt, key effectors of the neuroprotection-associated ERK/MAPK and PI3-K pathways, respectively, in explants of the cerebral cortex (Singh 2001). In order to determine if the activation of such signaling pathways was relevant to the protection afforded by progesterone treatment, we determined if inhibitors of these pathways would alter progesterone’s ability to exert its protective effects. Pre-treatment of the cortical cultures with the MEK inhibitor (UO126, 10μM) significantly attenuated progesterone’s ability to protect against glutamate-induced LDH release (Figure 2A). Similarly, pre-treatment of the cortical cultures with the PI3-K inhibitor also attenuated progesterone’s ability to protect against glutamate-induced LDH release (Figure 2B). Neither the vehicle (0.1% dimethylsulfoxide), UO126 or LY294002 alone were cytotoxic after 30 hrs of treatment (from time of initial application to the time of insult; data not shown).
Figure 2. Progesterone-induced protection is dependent on MEK1/2 kinase activity as well as PI3-K activity.
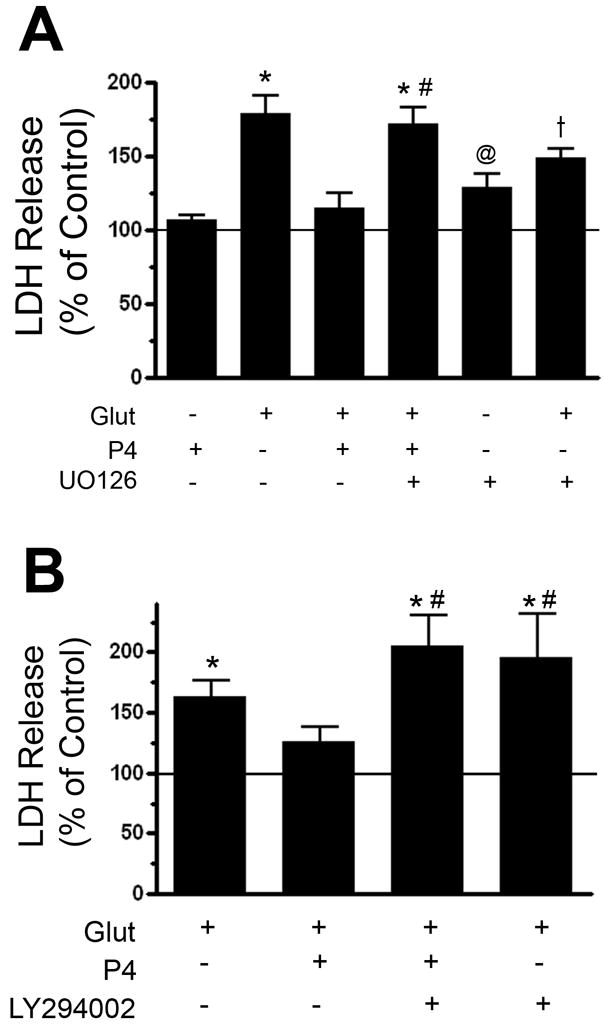
A. Cerebral cortical explants were pre-treated with the MEK1/2 inhibitor, UO126 (10 μM, 30 min), prior to the administration of 100 nM progesterone (P4, 24 hr) and subsequently treated with glutamate (Glut, 10 mM, 6 hr). UO126 reduced the ability of P4 to protect against glutamate-induced LDH release. LDH release was normalized to vehicle-treated control, whose value (set at 100%) is represented by the solid horizontal line. Following an Analysis of Variance (ANOVA), individual comparisons were made using a single degree of freedom F-test within the main effect of the ANOVA (F(6,44) = 14.226, p < 0.0001; symbols denoting individual group differences: *, p<0.0001 relative to vehicle-treated control; †: p < 0.001 relative to vehicle treated control; @: p < 0.05 relative to vehicle-treated control; #: p<0.0001 relative to P4 + Glut group).
B. Cerebral cortical explants were pre-treated with the PI3-kinase inhibitor, LY294002 (15 μM, 30min), prior to the administration of 100 nM progesterone (P4, 24 hr) and subsequently treated with glutamate (Glut, 10 mM, 6 hr). LY294002 reduced the ability of P4 to protect against glutamate-induced LDH release. LDH release, the marker of cytotoxicity, was normalized to vehicle-treated control, whose value (set at 100%) is represented by the solid horizontal line. Following an Analysis of Variance (ANOVA), individual comparisons were made using a single degree of freedom F-test within the main effect of the ANOVA (F(4,20) = 8.528, p < 0.001; symbols denoting individual group differences: *, p<0.01 relative to vehicle-treated control; #: p<0.01 relative to P4 + Glut group).
Progesterone elicits an increase in both BDNF protein and mRNA
We had previously shown that ovariectomy resulted in a substantial decrease in the expression of BDNF mRNA in both the hippocampus and cerebral cortex (Singh et al. 1995). Interestingly, however, estradiol replacement was able to only partially restore BDNF levels in the cerebral cortex (Singh et al. 1995), leading us to hypothesize that this incomplete normalization of BDNF mRNA may have been due to the omission of progesterone in the replacement paradigm. As such, we evaluated the effect of progesterone treatment, at a concentration and duration of treatment consistent with the cytoprotection studies above, on the expression of BDNF mRNA and protein in explants of the cerebral cortex. Using real time RT-PCR, our results showed that progesterone induces an approximately 75% increase in BDNF mRNA expression (Figure 3A). This effect size was nearly identical to that seen for BDNF protein levels (Figure 3B).
Figure 3. Progesterone elicits an increase in BDNF mRNA expression.
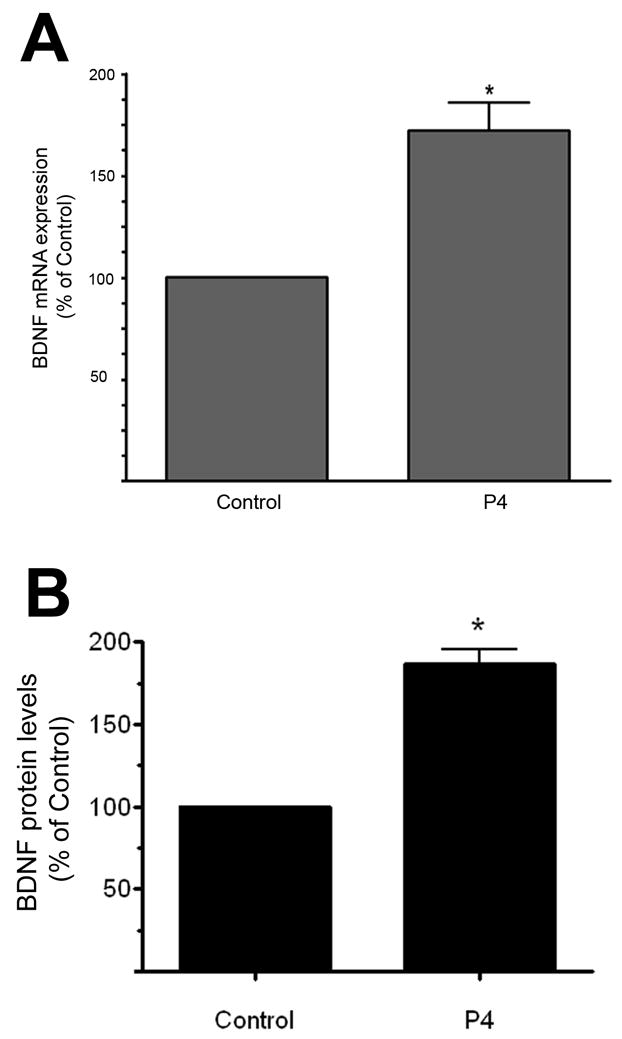
A. BDNF mRNA was assessed in cerebral cortical explants treated with progesterone (P4, 100 nM) for 18 hr. Using real time RT-PCR, we found that P4 induced an increase in BDNF mRNA. Statistical significance was determined using a single sample t-test (*: p ≤ 0.05). Data are presented as mean ± SEM.
B. BDNF protein levels were assessed in cerebral cortical explants treated with progesterone (P4, 100 nM) for 18 hr. Using an enzyme-linked immunosorbent assay (ELISA), we found that P4 induced an increase in BDNF expression. Statistical significance was determined using a single sample t-test (*: p ≤ 0.05). Data are presented as mean ± SEM.
The protective effects of progesterone are inhibited by K252a
In order to determine if the increase in BDNF elicited by progesterone was relevant to its protective effects, we used the pharmacological inhibitor of Trk receptor activation, K252a (6 nM). This concentration of K252a was chosen so as to maximize inhibition of Trk family of receptor activation (IC50 = 3 nM), while minimally influencing such alternative and potentially neuroprotection-relevant kinases as PKA (Ki = 18 nM) or PKC (Ki = 25 nM). Co-application of K252a at the time of progesterone treatment effectively prevented progesterone-induced protection (Figure 4) suggesting that the increase in BDNF expression was, at least in part, an important mediator of progesterone’s protective effects. K252a treatment by itself was without effect (data not shown).
Figure 4. K252a inhibits Progesterone–induced protection against glutamate toxicity.
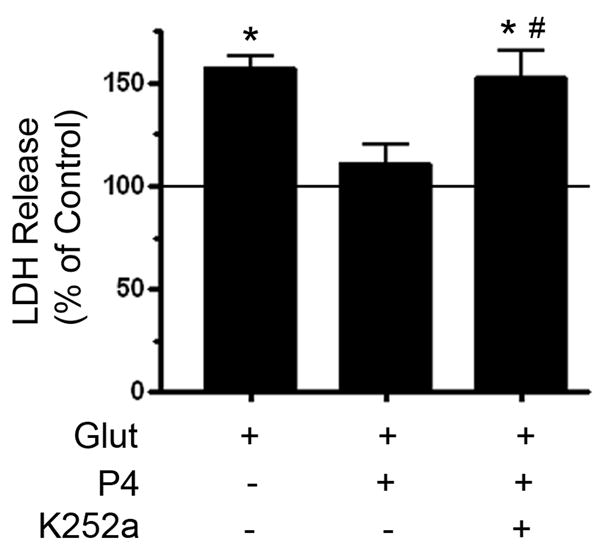
Cerebral cortical explants were pre-treated with the Trk receptor inhibitor, K252a (6 nM) together with the administration of 100 nM progesterone (P4, 24 hr), and subsequently treated with glutamate (Glut, 10 mM, 6 hr). While P4 reduced glutamate-induced LDH release, K252a inhibited P4’s protective effects. K252a by itself was without effect (data not shown, p = 0.439). LDH release was normalized to vehicle-treated control, whose value (set at 100%) is represented by the solid horizontal line. Following an Analysis of Variance (ANOVA), individual comparisons were made using a single degree of freedom F-test within the main effect of the ANOVA (F(4,12) = 7.487, p < 0.01; symbols denoting individual group differences: *, p<0.01 relative to vehicle-treated control; #: p<0.05 relative to P4 + Glut group).
DISCUSSION
Numerous studies have been published that support the neuroprotective effects of estradiol, the biologically most potent and prevalent estrogen. However, only recently has the role of the other major ovarian hormone, progesterone, received attention with respect to its neuroprotective effects. Though the molecular, biochemical and behavioral deficits that have been reported to result following ovariectomy, and the increased risk for AD seen following the menopause, have been heavily attributed to the loss of circulating estrogen, it is important to recognize that ovariectomy, like the menopause, results in a precipitous decline of not only circulating estrogen, but also in levels of progesterone. Thus, the decline in progesterone may have contributed to the deficits observed. As such, progesterone treatment may potentially have beneficial effects of its own.
We and others have previously reported that progesterone elicits the phosphorylation of extracellular-signal regulated kinase (ERK), a signaling protein within the Ras/Raf/MAP Kinase (MAPK) pathway (Nilsen and Brinton 2002; Singh 2001). In addition, we have also shown that progesterone elicits the phosphorylation of Akt, a key effector of the phosphoinositide (PI)-3 kinase pathway in cerebral cortical explants (Singh 2001). Both of these pathways have been implicated in mechanisms of neuroprotection (Datta et al. 1997; Desdouits-Magnen et al. 1998; D’Mello et al. 1997; Dudek et al. 1997; Klesse et al. 1999; Mills et al. 1997; Singer et al. 1999; Singer et al. 1996; Yan and Greene 1998). In the present study, we demonstrated that progesterone protected organotypic explants of the cerebral cortex against glutamate-induced cytotoxicity. Moreover, we found that this protection was mediated by the ERK/MAPK and PI3-K signaling pathways since pharmacological inhibitors of either of these pathways effectively inhibited progesterone’s protective effects.
The model in which we chose to assess the protective effects of progesterone (organotypic explants) has the unique advantage or retaining the cytoarchitecture of the brain, at least within the confines of the slice, and thus, preserves many of the cell-cell interactions that exist in vivo. This heterogeneous nature of the explant system does, therefore, reflect the complex nature of the intact brain may reliably predict the effect of progesterone in vivo and as such, complements previous published work that demonstrates the ability of progesterone to protect in primary dissociated cultures of the brain (Nilsen and Brinton 2002). Glutamate-induced cell damage/toxicity was chosen to mimic the excitotoxicity and oxidative stress that accompanies age-associated neuronal dysfunction and age-associated disorders such as Alzheimer’s Disease (Greenamyre et al. 1985; Siesjo 1981). Glutamate contributes to the cellular loss through both the induction of excitotoxicity (by dramatically increasing intracellular Ca2+ levels), and oxidative stress (Coyle and Puttfarcken 1993; Simonian and Coyle 1996). Glutamate-induced excitotoxicity occurs primarily through the NMDA receptor, while oxidative stress can occur as a result of perturbation in mitochondrial membrane potential (which could also be attributed to NMDA-induced Ca2+ overload), decreased ATP levels, and the generation of reactive oxygen species (Michaelis 1998). The latter is mediated, in part, by the inhibition of the glutamate/cysteine antiporter (Murphy et al. 1989), resulting in a depletion of cellular cysteine levels, the synthetic precursor to glutathione, and thus, causing the decline in the levels of this endogenous anti-oxidant. The co-existence of both neurons and glia in the explant model, however, did require a higher concentration of glutamate to elicit cell damage/death. While primary dissociated cells typically require mid-micromolar concentrations of glutamate to promote cytotoxicity, concentration response curves for glutamate-induced LDH release in cerebral cortical explants revealed that only low millimolar concentrations of glutamate were effective (data not shown). With regards to the mechanism of glutamate-induced toxicity in this tissue culture system, our data strongly implicate the NMDA receptor as the primary mediator (Figure 1C). As such, progesterone’s protective effects may be mediated through a regulation of the NMDA receptor.
To further explore the mechanism by which progesterone protects against glutamate induced LDH release, we addressed whether progesterone may increase the cellular levels of BDNF, a member of the neurotrophin family of growth factors known to have neuroprotective effects. Estrogen has been shown to regulate the levels of neurotrophins in a variety of experimental models, including the ovariectomized rodent. However, there is a paucity of information on the effect of progesterone alone (i.e., without concomitant estrogen treatment) in this regard. Further, given our previous data that showed an incomplete restoration of BDNF mRNA in the cerebral cortex of estrogen-treated, ovariectomized animals, we proposed that progesterone may play an important role in upregulating BDNF levels. We found that progesterone does indeed elicit an increase in both BDNF protein and mRNA levels in the cerebral cortex. Although the effects of combined estradiol and progesterone treatment were not evaluated here, we believe, based on our work and that of others, that progesterone would not inhibit the neurotrophin-inducing effects of estradiol. This hypothesis is based, in part, on reports showing that BDNF levels increased in estradiol alone- as well as estradiol + progesterone-treated, ovariectomized rats (Gibbs 1999). Further, we have also found that progesterone does not inhibit the induction of ERK phosphorylation elicited by estradiol in explants of the cerebral cortex, supporting the fact that progesterone does not always functionally antagonize the trophic effects of estradiol (data not shown). However, whether the effects of estradiol and progesterone are additive or synergistic is still unclear. Future experiments aimed at addressing not only the effect of concomitant estrogen and progesterone treatment, but also the relative timing of estrogen and progesterone administration (i.e., estrogen treatment prior to progesterone treatment or vice-versa) should provide greater insight into the consequences of combined estradiol and progesterone treatment on neurotrophin expression.
In an effort to determine if the progesterone-induced increase in BDNF levels may be relevant to progesterone’s protective effects, we examined if the pharmacological inhibitor of Trk receptor signaling, K252a, could alter progesterone’s protective effects. We recognize that the effects of K252a are not specific to TrkB, and as such, we cannot rule out the role of other Trk receptors (and thus, other neurotrophins) as mediators of progesterone’s protective effects. Nonetheless, the data support the importance of neurotrophin signaling in progesterone-induced protection was mediated, potentially through the induction of an increase in BDNF synthesis seen here.
Collectively, these data expand our understanding of progesterone neurobiology and offer plausible mechanisms through which progesterone may exert its protective effects. Knowledge of these novel mechanisms of action will be instrumental towards developing novel neuroprotective strategies for treating brain injury or age-associated diseases of the brain, including Alzheimer’s disease.
Acknowledgments
We would like to thank Drs. Michael Forster (Dept. Pharmacology & Neuroscience), Sejong Bae and Shande Chen (Dept. of Biostatistics, UNTHSC) for their expert advice on statistical analysis. This work was supported in part by funds from the National Institutes of Health (AG22550, AG23330, AG26672), and a National Alliance for Research on Schizophrenia and Depression (NARSAD)-sponsored Young Investigator Award to MS.
Abbreviations
- BDNF
Brain-derived neurotrophic factor
- ERK
extracellular-signal regulated kinase
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- P4
progesterone
- PI3-K
phosphoinositide-3 kinase
- RT-PCR
reverse transcriptase-polymerase chain reaction
References
- Andersen K, Launer LJ, Dewey ME, Letenneur L, Ott A, Copeland JR, Dartigues JF, Kragh-Sorensen P, Baldereschi M, Brayne C, Lobo A, Martinez-Lage JM, Stijnen T, Hofman A. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group. Neurology. 1999;53(9):1992–1997. doi: 10.1212/wnl.53.9.1992. [DOI] [PubMed] [Google Scholar]
- Arias E, Smith BL. Deaths: preliminary data for 2001. Natl Vital Stat Rep. 2003;51(5):1–44. [PubMed] [Google Scholar]
- Asbury ET, Fritts ME, Horton JE, Isaac WL. Progesterone facilitates the acquisition of avoidance learning and protects against subcortical neuronal death following prefrontal cortex ablation in the rat. Behav Brain Res. 1998;97(1–2):99–106. doi: 10.1016/s0166-4328(98)00031-x. [DOI] [PubMed] [Google Scholar]
- Bishop J, Simpkins JW, Singh M. Estradiol enhances brain glucose uptake in ovariectomized rats. Brain Res Bull. 1995;36(3):315–320. doi: 10.1016/0361-9230(94)00208-i. [DOI] [PubMed] [Google Scholar]
- Callier S, Morissette M, Grandbois M, Pelaprat D, Di Paolo T. Neuroprotective properties of 17beta-estradiol, progesterone, and raloxifene in MPTP C57Bl/6 mice. Synapse. 2001;41(2):131–138. doi: 10.1002/syn.1067. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262(5134):689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Desdouits-Magnen J, Desdouits F, Takeda S, Syu LJ, Saltiel AR, Buxbaum JD, Czernik AJ, Nairn AC, Greengard P. Regulation of secretion of Alzheimer amyloid precursor protein by the mitogen-activated protein kinase cascade. J Neurochem. 1998;70(2):524–530. doi: 10.1046/j.1471-4159.1998.70020524.x. [DOI] [PubMed] [Google Scholar]
- D’Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17(5):1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18(11):1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275(5300):661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Fan T, Yang SH, Johnson E, Osteen B, Hayes R, Day AL, Simpkins JW. 17beta-Estradiol extends ischemic thresholds and exerts neuroprotective effects in cerebral subcortex against transient focal cerebral ischemia in rats. Brain Res. 2003;993(1–2):10–17. doi: 10.1016/j.brainres.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Gahwiler B. Organotypic monolayer cultures of nervous tissue. J Neurosci Methods. 1981;4(4):329–342. doi: 10.1016/0165-0270(81)90003-0. [DOI] [PubMed] [Google Scholar]
- Gao S, Hendrie HC, Hall KS, Hui S. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch Gen Psychiatry. 1998;55(9):809–815. doi: 10.1001/archpsyc.55.9.809. [DOI] [PubMed] [Google Scholar]
- Gibbs RB. Levels of trkA and BDNF mRNA, but not NGF mRNA, fluctuate across the estrous cycle and increase in response to acute hormone replacement. Brain Res. 1998;787(2):259–268. doi: 10.1016/s0006-8993(97)01511-4. [DOI] [PubMed] [Google Scholar]
- Gibbs RB. Treatment with estrogen and progesterone affects relative levels of brain-derived neurotrophic factor mRNA and protein in different regions of the adult rat brain. Brain Res. 1999;844(1–2):20–27. doi: 10.1016/s0006-8993(99)01880-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez SL, Labombarda F, Gonzalez Deniselle MC, Guennoun R, Schumacher M, De Nicola AF. Progesterone up-regulates neuronal brain-derived neurotrophic factor expression in the injured spinal cord. Neuroscience. 2004;125(3):605–614. doi: 10.1016/j.neuroscience.2004.02.024. [DOI] [PubMed] [Google Scholar]
- Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66(5):1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Penney JB, Young AB, D’Amato CJ, Hicks SP, Shoulson I. Alterations in L-glutamate binding in Alzheimer’s and Huntington’s diseases. Science. 1985;227(4693):1496–1499. doi: 10.1126/science.2858129. [DOI] [PubMed] [Google Scholar]
- Henderson VW. Estrogen, cognition, and a woman’s risk of Alzheimer’s disease. Am J Med. 1997;103(3A):11S–18S. doi: 10.1016/s0002-9343(97)00261-1. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Moore N, Fiskum G, Murphy AZ. Ovarian steroid modulation of seizure severity and hippocampal cell death after kainic acid treatment. Exp Neurol. 2003;182(1):124–134. doi: 10.1016/s0014-4886(03)00104-3. [DOI] [PubMed] [Google Scholar]
- Jezierski MK, Sohrabji F. Region-and peptide-specific regulation of the neurotrophins by estrogen. Brain Res Mol Brain Res. 2000;85(1–2):77–84. doi: 10.1016/s0169-328x(00)00244-8. [DOI] [PubMed] [Google Scholar]
- Klesse LJ, Meyers KA, Marshall CJ, Parada LF. Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene. 1999;18(12):2055–2068. doi: 10.1038/sj.onc.1202524. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luine VN, Khylchevskaya RI, McEwen BS. Effect of gonadal steroids on activities of monoamine oxidase and choline acetylase in rat brain. Brain Res. 1975;86(2):293–306. doi: 10.1016/0006-8993(75)90704-0. [DOI] [PubMed] [Google Scholar]
- Michaelis EK. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol. 1998;54(4):369–415. doi: 10.1016/s0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- Mills J, Laurent Charest D, Lam F, Beyreuther K, Ida N, Pelech SL, Reiner PB. Regulation of amyloid precursor protein catabolism involves the mitogen-activated protein kinase signal transduction pathway. J Neurosci. 1997;17(24):9415–9422. doi: 10.1523/JNEUROSCI.17-24-09415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2(6):1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology. 2002;143(1):205–212. doi: 10.1210/endo.143.1.8582. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Diaz Brinton R. Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc Natl Acad Sci U S A. 2003;100(5):2842–2847. doi: 10.1073/pnas.0438041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordell VL, Scarborough MM, Buchanan AK, Sohrabji F. Differential effects of estrogen in the injured forebrain of young adult and reproductive senescent animals. Neurobiol Aging. 2003;24(5):733–743. doi: 10.1016/s0197-4580(02)00193-8. [DOI] [PubMed] [Google Scholar]
- Panickar KS, Guan G, King MA, Rajakumar G, Simpkins JW. 17beta-estradiol attenuates CREB decline in the rat hippocampus following seizure. J Neurobiol. 1997;33(7):961–967. [PubMed] [Google Scholar]
- Roof RL, Duvdevani R, Braswell L, Stein DG. Progesterone facilitates cognitive recovery and reduces secondary neuronal loss caused by cortical contusion injury in male rats. Exp Neurol. 1994;129(1):64–69. doi: 10.1006/exnr.1994.1147. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. Can estrogen keep you smart? Evidence from clinical studies. J Psychiatry Neurosci. 1999;24(4):315–321. [PMC free article] [PubMed] [Google Scholar]
- Shi J, Panickar KS, Yang SH, Rabbani O, Day AL, Simpkins JW. Estrogen attenuates over-expression of beta-amyloid precursor protein messager RNA in an animal model of focal ischemia. Brain Res. 1998;810(1–2):87–92. doi: 10.1016/s0006-8993(98)00888-9. [DOI] [PubMed] [Google Scholar]
- Shi J, Yang SH, Stubley L, Day AL, Simpkins JW. Hypoperfusion induces overexpression of beta-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 2000;853(1):1–4. doi: 10.1016/s0006-8993(99)02113-7. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1(2):155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- Simonian NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 1996;36:83–106. doi: 10.1146/annurev.pa.36.040196.000503. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Yang SH, Liu R, Perez E, Cai ZY, Covey DF, Green PS. Estrogen-like compounds for ischemic neuroprotection. Stroke. 2004;35(11 Suppl 1):2648–2651. doi: 10.1161/01.STR.0000143734.59507.88. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Yang SH, Wen Y, Singh M. Estrogens, progestins, menopause and neurodegeneration: basic and clinical studies. Cell Mol Life Sci. 2005;62(3):271–280. doi: 10.1007/s00018-004-4382-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19(7):2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;212(1):13–16. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- Singh M. Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine. 2001;14(3):407–415. doi: 10.1385/ENDO:14:3:407. [DOI] [PubMed] [Google Scholar]
- Singh M, Meyer EM, Millard WJ, Simpkins JW. Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female Sprague-Dawley rats. Brain Res. 1994;644(2):305–312. doi: 10.1016/0006-8993(94)91694-2. [DOI] [PubMed] [Google Scholar]
- Singh M, Meyer EM, Simpkins JW. The effect of ovariectomy and estradiol replacement on brain-derived neurotrophic factor messenger ribonucleic acid expression in cortical and hippocampal brain regions of female Sprague-Dawley rats. Endocrinology. 1995;136(5):2320–2324. doi: 10.1210/endo.136.5.7720680. [DOI] [PubMed] [Google Scholar]
- Singh M, Setalo G, Jr, Guan X, Frail DE, Toran-Allerand CD. Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-alpha knock-out mice. J Neurosci. 2000;20(5):1694–1700. doi: 10.1523/JNEUROSCI.20-05-01694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrabji F, Miranda RC, Toran-Allerand CD. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92(24):11110–11114. doi: 10.1073/pnas.92.24.11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solum DT, Handa RJ. Estrogen regulates the development of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus. J Neurosci. 2002;22(7):2650–2659. doi: 10.1523/JNEUROSCI.22-07-02650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, Simpkins JW. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004;1008(2):147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Wise PM. Estrogens and neuroprotection. Trends Endocrinol Metab. 2002;13(6):229–230. doi: 10.1016/s1043-2760(02)00611-2. [DOI] [PubMed] [Google Scholar]
- Yan CYI, Greene LA. Prevention of PC12 cell death by N-acetylcysteine requires activation of the Ras pathway. J Neurosci. 1998;18(11):4042–4049. doi: 10.1523/JNEUROSCI.18-11-04042.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]