

Summary
Reversible covalent methylation of lysine residues on histone proteins constitutes a principal molecular mechanism that links chromatin states to diverse biological outcomes. Recently, lysine methylation has been observed on non-histone proteins, suggesting broad cellular roles for the enzymes generating and removing methyl moieties. Here we report that the lysine methyltransferase enzyme SET8/PR-Set7 is a novel regulator of the tumor suppressor protein p53. We find that SET8 specifically monomethylates p53 at lysine 382 (p53K382me1). This methylation event robustly suppresses p53-mediated transcription activation of highly responsive target genes, but has little influence on weak targets. Further, depletion of SET8 augments the pro-apoptotic and checkpoint activation functions of p53, and accordingly, SET8 expression is downregulated upon DNA damage. Together, our study identifies a novel p53-modifying enzyme, a new regulatory post-translational modification of p53, and begins to dissect how methylation may contribute to a dynamic post-translational code that modulates distinct p53 functions.
Introduction
Methylation events at distinct lysine residues within histone proteins are linked to diverse functional outcomes (Jenuwein and Allis, 2001). For example, methylation at histone H3 at lysine 4 (H3K4me) is largely detected at euchromatin and is thought to generally lead to increased DNA accessibility, whereas methylation of histone H3 at lysine 9 (H3K9me) is most commonly associated with heterochromatin and inaccessible DNA (Bannister and Kouzarides, 2004). One mechanism by which lysine methylation aids in the establishment of distinct chromatin states is by mediating modular protein-protein interactions (Daniel et al., 2005). In this regard, the proteins that recognize a methylated lysine within a specific sequence context can define the functional outcome of a lysine methylation event. Further, histone lysines can be mono-, di- or trimethylated, with a unique activity frequently being coupled to the specific state and extent of methylation on the lysine residue. Thus, methylation of lysine residues on a target protein can increase the signaling potential of the modified protein and as such lead to diverse physiologic consequences.
p53 is a transcription regulator that plays a central role in tumor suppression by directing cellular responses to diverse stresses (Laptenko and Prives, 2006; Toledo and Wahl, 2006). The levels and activity of p53 are regulated by a complex network of post-translational modifications that primarily occur within two regions of the protein: an N-terminal region that is phosphorylated at multiple sites and a C-terminal region rich in basic residues (Appella and Anderson, 2001; Toledo and Wahl, 2006). Recent reports indicate that p53 is monomethylated at two different lysine residues within the regulatory C-terminal region (Chuikov et al., 2004; Huang et al., 2006). Akin to how H3K4me and H3K9me are linked to opposing states of chromatin, the two known sites of p53 methylation are coupled to activities that oppose one another. Specifically, SET7/9-mediated monomethylation of p53 at K372 (p53K372me1) activates p53, postulated in part to occur via stabilization of chromatin-associated p53, whereas Smyd2-mediated monomethylation of p53 at K370 (p53K370me1) represents a repressive mark, the generation of which is impeded by p53K372me1 (Chuikov et al., 2004; Huang et al., 2006). In addition to methylation at K370 and K372, the C-terminal region of human p53 harbors several K residues that are subject to modification by acetylation, ubiquitylation, sumoylation and neddylation (reviewed in Toledo and Wahl, 2006). Notably, endogenous p53 protein from two independent mouse models in which these lysines were targeted for mutation did not display an alteration in stability, and the phenotypes of cells derived from the mice were relatively mild (Feng et al., 2005; Krummel et al., 2005). This work argues that in sum, the post-translational modifications (PTMs) on the p53 C-terminal region fine-tune p53 activity. However, as substitution of lysines will prevent all forms of PTMs, including mono-, di- and trimethylation, mutant phenotypes may indicate the elimination of both positive and negative regulatory effects. Thus, identifying and characterizing the enzymes that catalyze p53 modifications is critical for developing a molecular understanding of how p53 PTMs are coordinated to regulate p53 functions.
SET7/9 and Smyd2 were both first reported to function as histone methyltransferases (HMTs), suggesting that other HMTs might have non-histone substrates (Brown et al., 2006; Nishioka et al., 2002a; Wang et al., 2001). SET8/PR-Set7 is an HMT that adds a single methyl moiety to histone H4 tails at lysine 20 (H4K20me1), preferentially to nucleosomal H4 (Fang et al., 2002; Nishioka et al., 2002b). Mutation of the SET8/PR-Set7 gene in Drosophila melanogaster leads to lethality during development (Nishioka et al., 2002b). H4K20me1 generation by SET8 has also been shown to be important for gene silencing and mitotic regulation (Fang et al., 2002; Julien and Herr, 2004; Rice et al., 2002). Here we demonstrate a novel activity for SET8 as a p53 methyltransferase. We find that SET8-mediated methylation of p53 at K382 represses highly responsive p53 target genes to attenuate p53 pro-apoptotic and cell-cycle arrest functions. We propose a model in which SET8-mediated p53 methylation tips the balance of p53 function away from cell elimination towards cell survival.
Results
In vitro identification of SET8 as a p53K382 monomethyltransferase
To screen known HMTs to determine if they might function as p53 methyltransferases, we expressed recombinant SET7/9, Suv39h1, hDOT1L, SET8/PR-Set7 and Suv4-20h1, and performed in vitro methylation assays using full-length GST-p53 and histones as substrates (Figure 1A). As expected, SET7 methylated p53 and histone H3, but not nucleosomes (Figure 1A) (Chuikov et al., 2004). The other enzymes showed methyltransferase activity on their cognate histone substrate, but only SET8 was found to possess methylation activity on p53 (Figure 1A). We performed ms/ms analysis of the methylated p53 protein and mapped K382 as a residue modified by SET8 (Supplementary Figure 1; data not shown). To confirm these results, we generated several single site mutants in the context of full-length p53, and performed in vitro methylation assays with SET8 and SET7. As shown in Figure 1B, substitution of K382 with arginine (p53K382R) abolishes SET8-mediated p53 methylation, but has no effect on SET7-mediated activity. Substitutions at other lysines of p53 had no impact on SET8 methylation (Figure 1B). Based on these data, we conclude that SET8 methylates p53 exclusively at lysine 382.
Figure 1. SET8 monomethylates p53 at K382 in vitro.
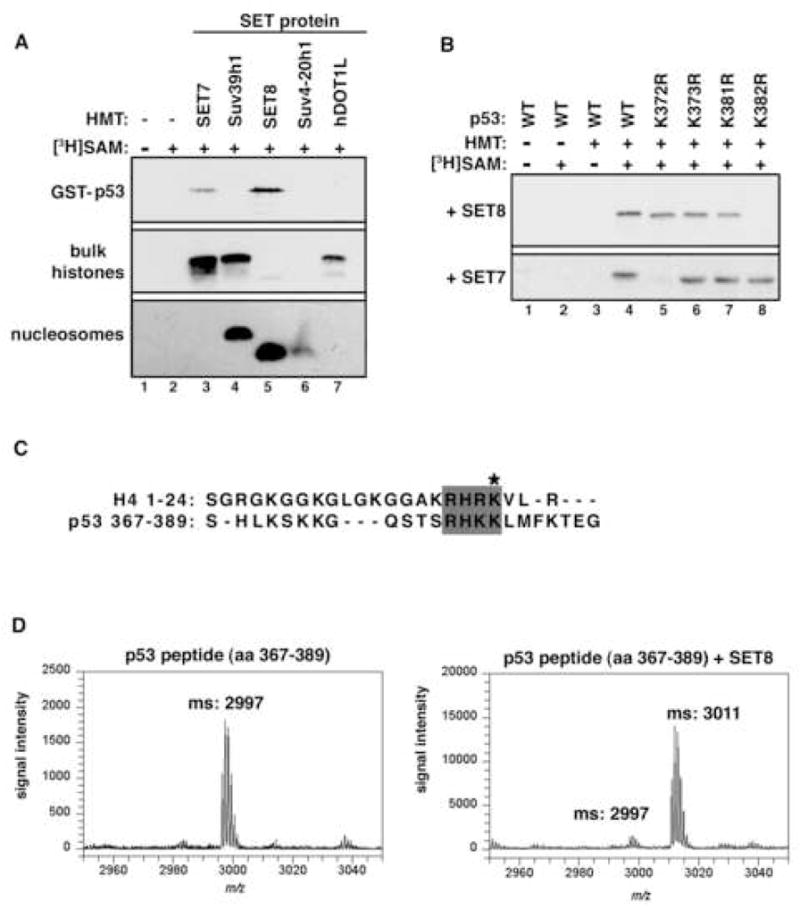
(A) Identification of SET8 as a novel p53 methyltransferase. Autoradiograms of methyltransferase assays with the indicated recombinant HMTs and substrates. (B) SET8 methylates p53 at K382. Autoradiograms of methyltransferase assays with SET8 or SET7 on wild-type p53 or the indicated p53 mutants. (C) Alignment of amino acid sequences of SET8 substrates histone H4 (aa 1–24) and p53 (aa 367–389). Asterisk indicates SET8 methylation sites on H4 and p53. (D) p53 is monomethylated by SET8 at K382. Mass spectrometry analysis of p53 peptide (aa 367–389) before (left panel) and after (right panel) SET8 methyltransferase reaction.
SET8 is a strict monomethyltransferase for H4K20 (Couture et al., 2005; Xiao et al., 2005). Alignment of the amino acid sequence surrounding the substrate site of SET8 in p53 (K382) and that of histone H4 (K20) reveals significant overlap (Figure 1C). We therefore reasoned that p53 is likewise monomethylated by SET8. To confirm the extent of methylation on p53K382 catalyzed by SET8, peptides bearing amino acids 367–389 of p53 were in vitro methylated by SET8 and analyzed by mass spectrometry. As shown in Figure 1D, SET8 induced a 14Da shift in the p53 peptide, indicating the addition of a single methyl group to K382. Under the conditions of our reaction, virtually all peptide was converted to the monomethyl species, with little to no dimethylation or trimethylation detected. Thus, in vitro, SET8 is a p53 monomethyltransferase.
Endogenous p53 protein is monomethylated at lysine 382
To confirm the existence of p53K382me1 in vivo, we performed a mass spectrometry analysis of endogenous p53 purified from HeLa nuclear extract (see Experimental Procedures). Two peaks representing unmethylated and monomethylated peptides containing p53K382 are shown in Figure 2A. The mass difference of 14Da indicates the addition of a single methyl moiety. We confirmed by ms/ms that the methyl moiety is specific to K382 and not to other residues present in the peptide fragment (data not shown). These data provide the first definitive confirmation of methylation being present on endogenous p53.
Figure 2. Characterization of a p53K382me1 modificaiton-specific antibody.
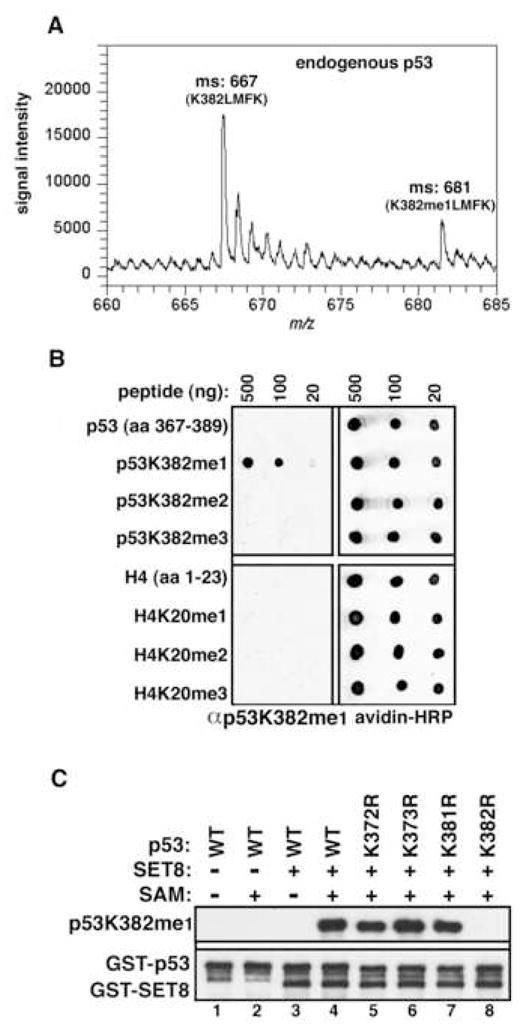
(A) Identification of endogenous p53 monomethylated at K382. Mass spectrometry analysis of endogenous p53 IPed from HeLa NE reveals trypsin-digested peptides containing lysine382 (382-KLMFK-386) present in peaks containing either unmethylated or monomethylated K382. (B) Specific recognition of p53K382me1 by the αp53K382me1 antibody. Dot blot analysis of the indicated biotinylated peptides (p53: top; H4: bottom) with αp53K382me1 antibody. Blots were probed with HRP-conjugated streptavidin to control for loading. (C) αp53K382me1 antibody recognizes p53 in vitro methylated at K382 by SET8. Immunoblot analysis of the indicated recombinant p53 protein or mutants ± methyltransferase assays with SET8 as indicated. Total p53 and SET8 were detected with GST antibody to show equal loading.
Characterization of an anti-p53K382me1 antibody
We next raised an antibody against the p53K382me1 epitope (hereafter referred to as αp53K382me1). This antibody specifically recognizes p53 peptides monomethylated at K382 and does not crossreact with unmodified p53, p53K382me2, p53K382me3 peptides or several additional p53 methylated residues (Figure 2B; data not shown). Notably, despite the sequence homology between the SET8 substrate sites of p53K382 and H4K20 (see Figure 1C), our antibody did not detect H4 peptides irrespective of methylation status at K20 (Figure 2B). We also tested the ability of αp53K382me1 to recognize p53 methylated at K382 in the context of the full-length p53 protein. As shown in Figure 2C, αp53K382me1 detects full-length SET8-methylated wild-type (wt) and various K>R mutant derivative p53 proteins (lane 4–7), but not unmethylated p53 (lane 1–3), or the p53K382R mutant (lane 8). Based on these results, we conclude that our αp53K382me1 antibody can be utilized to monitor SET8-dependent monomethylation of p53 at K382 for in vivo studies.
In vivo methylation of p53 at lysine 382
Next, to determine whether p53 is methylated by SET8 in vivo, U2OS cells were transfected with SET8 and either Flag-tagged wt-p53, p53K382R or p53K372R. As shown in Figure 3A, Western analysis detected p53K382me1 in whole cell extracts (WCE) and Flag-immunoprecipitates (IPs) from cells transfected with wt-p53 and the p53K372R mutant, but not with the p53K382R mutant. Moreover, SET8, but not a catalytically inactive mutant (SET8D338A), methylated endogenous p53 at lysine 382 in WCE and in αp53 IPs (Supplementary Figure 2; see Figure 3C; Couture et al., 2005). Thus, ectopic SET8 methylates endogenous p53 in vivo.
Figure 3. p53 is monomethylated at K382 in vivo by SET8.

(A) Ectopic SET8 specifically methylates p53 at K382 in vivo. Western blot analysis with αp53K382me1 antibody of Flag IPs or WCE (whole cell extracts) from U2OS cells expressing SET8 and the indicated Flag-tagged p53 derivatives. p53 and SET8 protein levels in WCE are shown. (B) Knockdown of endogenous SET8 decreases endogenous levels of p53K382me1 in U2OS cells. Western analysis of p53 levels present in αp53K382me1 IPs of U2OS cells treated with control or SET8 siRNAs. Total p53, SET8, H4K20me1 and tubulin present in the WCE are shown. (C) SET8 negatively regulates acetylation of p53 at K382. Western analysis with αp53K382me1, p53K382ac and p53 antibodies of p53 IPs from U2OS cells transfected with control vector or SET8, and treated for 2 hrs with 0.5 μg/ml NCS. SET8 and tubulin levels in the WCE are shown. Endogenous p53K382me1 is observed with longer exposure (Supplementary Figure 2C) (D) p53K382me1 levels decrease upon DNA damage. Western analyses with the indicated antibodies of αp53K382me1 and p53 (DO1) IPs from U2OS cells transfected with control or SET8 siRNA and treated with NCS for 2 hrs. Tubulin and total p53 levels present in WCE are shown to control for loading. (E) SET8 mRNA expression decreases in response to DNA damage. Real-time PCR analysis of SET8 and Smyd2 mRNA levels present in U2OS cells ± NCS treatment (0.5 μg/ml, 4 hrs). (F) SET8 protein levels decrease in response to DNA damage. Western analysis of SET8 in U2OS cells as in (E).
We next investigated whether SET8 is physiologically responsible for the monomethylation of p53K382. To this end, endogenous SET8 protein levels were knocked-down by RNA interference (RNAi) in the p53+ U2OS cell line, and levels of p53K382me1 determined (Figure 3B). The RNAi treatment did not alter p53 levels in whole cell extracts (WCE), but levels of p53 detected in αp53K382me1 IPs were specifically reduced by SET8 RNAi relative to control RNAi treatment, denoting a decrease of endogenous p53K382me1 levels in SET8 knock-down cells (Figure 3B). Three sequential transfections of SET8 RNAi are reported to result in a reduction of H4K20me1 levels (Botuyan et al., 2006); however, under our experimental conditions of a single round of SET8 RNAi treatment, a decrease in p53K382me1 levels is observed with no reduction in H4K20me1 levels detected (Figure 3B). The manifestation of p53K382me1 depletion without reduction of H4K20me1 upon acute SET8 knock-down is likely a consequence of (i) p53 protein levels being orders of magnitude lower than H4, (ii) the rapid kinetics of p53 protein turnover and (iii) the reported high stability of the H4K20me1 mark (Karachentsev et al., 2005). Taken together, we conclude that SET8 is required for maintenance of p53K382me1 levels in vivo.
DNA damage was reported to increase monomethylation at p53K372, suggesting the possibility that p53K382me1 might likewise be regulated (Chuikov et al., 2004). We therefore examined p53K382me1 levels in U2OS cells in response to the radiomimetic drug neocarzinostatin (NCS), in the presence of ectopic SET8 expression (Figure 3C) and upon knock-down of endogenous SET8 protein (Figure 3D). As expected, total p53 levels (and acetylation of p53 at K382 (p53K382ac)) increased in response to DNA damage induced by NCS treatment (Figure 3C and 3D). In contrast, levels of p53K382me1 decreased in response to DNA damage compared to control treatment (Figure 3C, compare lanes 3 and 4; Figure 3D, compare lanes 1 and 2). Consistent with these findings, SET8 mRNA and protein expression levels in U2OS cells are markedly repressed in response to NCS treatment, whereas Smyd2 expression does not change with DNA damage (Figure 3E and 3F). These data suggest the hypothesis that the generation of p53K382me1 by SET8 represses p53 functions, an activity that is itself curbed during the physiologic DNA damage response (see discussion). Finally, SET8 monomethylation at p53K382 impaired DNA damage-induced acetylation at the same residue (p53K382ac) (Figure 3C and 3D). The observations that p53K382me1 generation negatively correlates with DNA damage and that SET8 inhibits the formation of p53K382ac, a modification linked to DNA damage responses, raises the possibility that monomethylation at K382 inhibits p53-mediated DNA damage responses (Appella and Anderson, 2001; Toledo and Wahl, 2006).
SET8 suppresses p53-dependent transcription activation
To investigate the biological consequences for SET8-mediated methylation of p53, the ability of SET8 to regulate transcription of the p53 target genes p21 and PUMA was determined. For these experiments, we used H1299 cells, which lack endogenous p53 and can therefore be complemented with either wild-type or mutant p53, allowing for the selective investigation of the role of K382 (Knights et al., 2006; Lokshin et al., 2005). p53 reconstitution in H1299 cells resulted in marked induction of p21 mRNA (Figure 4A) and p21 protein (Figure 4B) levels relative to cells transfected with vector alone. The p53K382R mutant likewise triggered a strong induction of the p53 target genes, though the phenotype was slightly less pronounced than wild-type p53, indicating that K382 residue per se is not required for p53 transactivation activity (Figures 4A and 4B; Supplementary Figure 3A). Notably, co-expression of SET8 with p53, which did not affect p53 protein expression, largely abolished the induction of p21 mRNA and p21 protein elicited by wt-p53, yet did not impinge on the activity of the p53K382R mutant (Figures 4A and 4B). Equivalent results were obtained for PUMA, a second p53 target gene (Supplementary Figure 3). Additionally, we observed that in chromatin immunoprecipitation (ChIP) assays, SET8 co-expression reduced occupancy of p53 at the p21 and PUMA promoters (Figure 4C; Supplementary Figure 3B), but notably had no effect on H4K20me1 levels at these promoters (Figure 4D; Supplementary Figure 3C). p53K382me1 was not detected at the p21 and PUMA promoters, though global distribution of this species is grossly similar to that of total p53 (Supplementary Figure 4). Finally, the SET8 catalytic mutant SET8D338A, which fails to methylate p53 in vivo, likewise failed to inhibit p53 transactivation activity (Figure 4E; Supplementary Figure 3D). Taken together, these data argue for a model in which it is the direct addition of the methyl moiety to K382 by SET8, rather than alterations of H4K20me1 levels by SET8 or the failure to otherwise modify p53K382, which acts to inhibit p53 transcriptional activity.
Figure 4. SET8 methylation of p53 at K382 suppresses p53 transactivation activity.
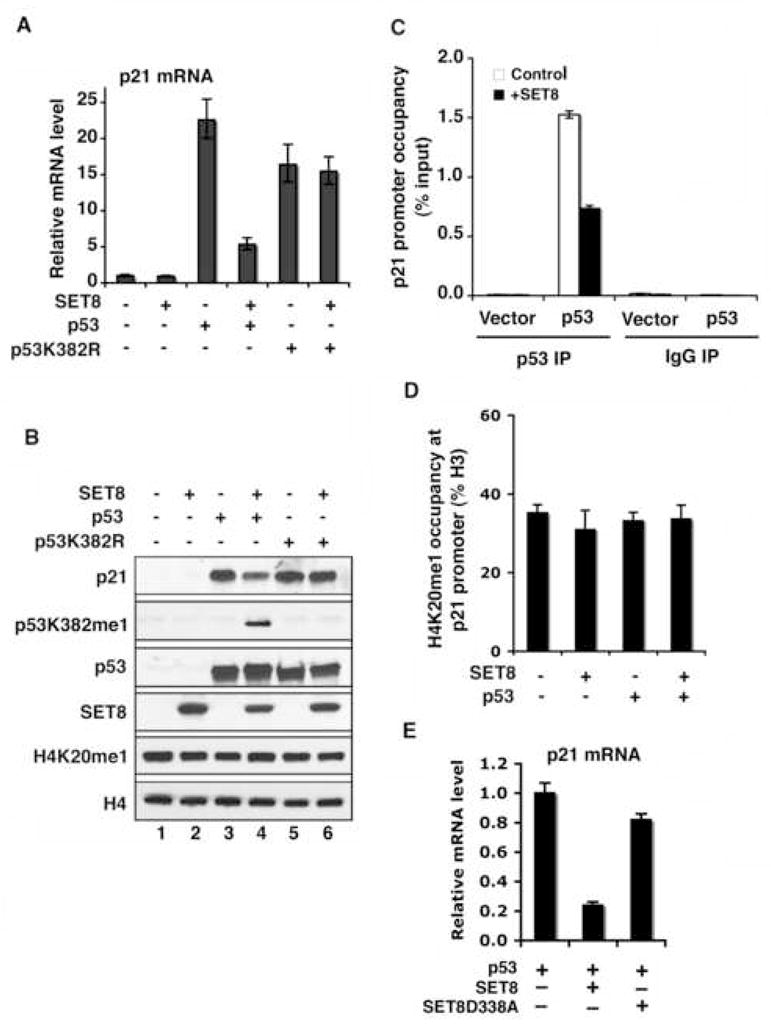
(A) and (B) SET8 inhibits induction of p21 transcription (A) and p21 protein expression (B) by wild-type p53, but does not affect the activity of the p53K382R mutant. (A) Real-time PCR analyses of p21 mRNA levels in H1299 cells transfected with control vector, p53 or p53K382R mutant, ± SET8. (B) Western analyses with the indicated antibodies of H1299 cells WCE as in (A). (C) SET8 expression attenuates occupancy of p53 at the p21 promoter. p53 occupancy at the p21 promoter in H1299 cells transfected with control vector or p53, ± SET8 was determined by ChIP analyses. DO-1 antibody was used for p53 ChIP and IgG was used as control. Occupancy values (ChIP/inputX100) were determined by real-time PCR. (D) SET8 expression does not alter H4K20me1 levels at the p21 promoter. Occupancy of H4K20me1 (H4K20me1 ChIP/H3 ChIPX100) at the p21 promoter was determined as in (C). Error bars in (A, C, D) indicate ± s.e.m. from three experiments. (E) SET8 catalytic mutant SETD338A fails to suppress p53 transactivation activity on target genes. Real-time PCR analyses of relative p21 mRNA levels in H1299 cells co-transfected with p53, SET8, SET8D338A or control vector as indicated.
To test this hypothesis in a more physiologic setting, endogenous SET8 protein levels were knocked-down in the p53+ U2OS cell line, and p53-dependent responses to NCS treatment were determined. Consistent with the overexpression data, knock-down of SET8 led to an increase of p21 mRNA levels and p21 protein expression induced by the genotoxic agent relative to the control siRNA treatment (Figure 5A and 5B). For these experiments, two independent siRNA pools were utilized to exclude off-target effects (Figure 5A and 5B). Like p21, the expression of PUMA was upregulated by SET8 RNAi treatment relative to control treatment (Supplementary Figure 5A).
Figure 5. SET8 RNAi augments p53 activity in response to DNA damage.
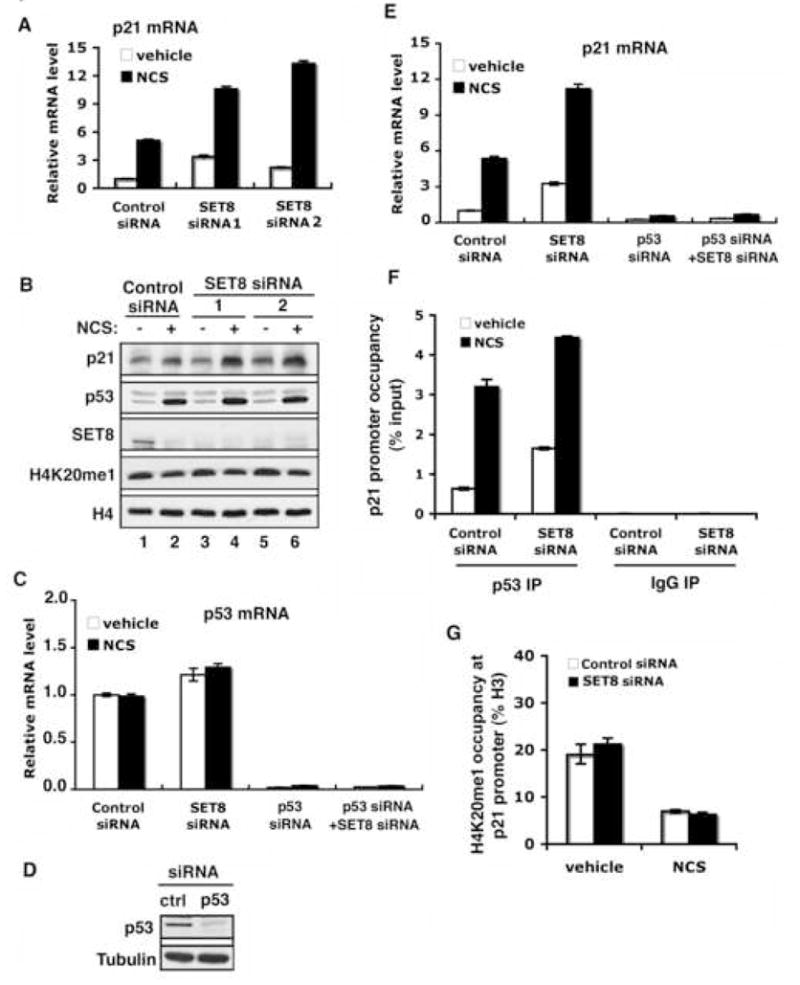
(A) and (B) Knock-down of SET8 augments expression of p21 mRNA (A) and p21 protein (B) in response to DNA damage. (A) Real-time PCR analyses of p21 mRNA in U2OS cells treated with 0.5 μg/ml NCS (4 hrs) and transfected with control or two different sets of SET8 siRNA. (B) Western analyses with the indicated proteins of WCE from U2OS cells as in (A). Both SET8 RNAi sets depleted endogenous SET8 protein levels without altering H4K20me1 levels. H4 levels are shown to control for equal loading. (C) Real-time PCR analysis of p53 mRNA in U2OS cells transfected with control or SET8 siRNA ± p53 siRNA under normal condition or NCS treatments. (D) Western analysis of p53 in U2OS transfected with control or p53 siRNA. Tubulin levels are shown to control for loading. (E) SET8 regulation of p21 expression is p53-dependent. Real-time PCR analysis of p21mRNA in U2OS cells as in (C). (F) Knockdown of endogenous SET8 augments p53 occupancy at the p21 promoter. ChIP assays as in (Figure 4C) in U2OS cells transfected with control or SET8 siRNA ± NCS treatment. (G) SET8 RNAi does not alter H4K20me1 levels at the p21 promoter. ChIP assays to determine H4K20me1 occupancy at the p21 promoter as in (Figure 4D) in U2OS cells as in (F). Error bars in (A, C, E, F, G) indicate ± s.e.m. from at least three experiments.
It is possible that the altered regulation of p21 and PUMA in response to DNA damage by SET8 knock-down is due to modulation of H4K20 methylation rather than p53 methylation. However, this possibility is highly unlikely for a number of reasons: (1) irrespective of the different treatments of our experiments, no SET8-dependent changes in H4K20me1 levels were observed globally (Figure 5B) or at specific promoters (Figure 5G; Supplementary Figure 5D); (2) the increase in p21 and PUMA induction upon DNA damage observed with SET8 knock-down is dependent on p53 protein, as co-knock-down of p53 (Figure 5C and 5D) eliminated any effects of SET8 (Figure 5E; Supplementary Figure 5B); (3) the SET8 knock-down mediated increase in gene expression of p21 and PUMA correlates with increased p53 occupancy at the cognate promoters, indicating SET8 directly alters p53 behavior (Figure 5F; Supplementary Figure 5C); (4) the regulation of highly responsive p53 target genes is specific, as expression of a constitutively active non-p53 regulated gene (actin) did not change upon SET8 knockdown, as might be expected were the phenotype due to global loss of H4K20me1 (Supplementary Figure 6); (5) knock-down of SET8 did not impact the expression of a number of weak p53 targets such as Bax and NOXA, and suppressed mRNA induction of the DNA repair factor GADD45, the inverse of what would be expected were the phenotype due to H4K20me1 depletion (Supplementary Figure 7; data not shown). Together, these observations strongly argue for direct methylation by SET8 at lysine 382 of p53 – and not lysine 20 of histone H4 – in the regulation of p53 responses.
SET8 depletion augments p53 cellular functions
We next addressed the role of SET8 in p53-dependent cell-cycle arrest and apoptosis. In the absence of DNA damage and consistent with a previous report (Julien and Herr, 2004), SET8 knock-down cells behave like control RNAi cells with respect to cell-cycle progression (Figure 6A). In contrast, upon DNA damage, SET8 knock-down renders cells more prone to apoptosis relative to control cells (9.9% Sub-G1 fraction in SET8 RNAi cells versus 3.1% for control cells; Figure 6A). Further, SET8 RNAi cells exhibit a higher fraction of cells in G1 than control cells (56.5% versus 38%, respectively; Figure 6A). These effects are strictly dependent on p53, as co-knock-down of p53 (see Figure 5D) eliminates the DNA damage-dependent phenotype of SET8 knock-down cells (Figure 6B). To further test whether the increased sensitivity of SET8 knock-down cells to DNA damage is mediated by p53, cell death induced by the DNA damage agent doxorubicin was also determined in the absence or presence of both SET8 and p53 knock-down. As shown in Figure 6C, whereas SET8 RNAi alone increases sensitivity of cells to DNA damage relative to control, this effect is abolished in a p53 knock-down background.
Figure 6. Monomethylation of p53 at K382 attenuates p53 biological function.

(A) SET8 knock-down renders cells more sensitive to cell death and cell-cycle arrest following DNA damage. Sub-G1 and cell-cycle distribution of U2OS cells ± SET8 siRNA and ± 24 hrs treatment with 1 μg/ml doxorubicin was determined by flow cytometry. (B) The increased sensitivity of SET8 knock-down cells to DNA damage is p53 dependent. Cell cycle distribution of SET8 knock-down cells as in (A) ± p53 knockdown. (C) Increased sensitivity of SET8 knock-down cells to DNA damage-induced cell death is p53-dependent. Cell death was determined in U2OS cells ± SET8 siRNA and ± p53 siRNA, in response to 2 μg/ml doxorubicin for 20 hrs. Error bars indicate ± s.e.m. from at least three experiments.
Discussion
Here we have shown that the HMT enzyme SET8/PR-Set7 specifically monomethylates p53 at lysine 382 in vitro and in vivo. We have also provided the first mass spectrometry evidence that endogenous p53 is methylated (see Figure 2A). Ectopic expression of SET8 suppresses p53 transactivation activity and knock-down of endogenous SET8 by RNAi augments a number of p53 activities, including induction of highly responsive target genes, and increased apoptosis and cell-cycle arrest (see model, Figure 7). Numerous experiments indicate that these phenotypes are a consequence of SET8 methylation of p53, and not an indirect effect of H4K20 methylation by SET8. For example, in H1299 cells, SET8 represses ectopic p53, but fails to do so if p53 harbors a mutation at K382 (p53K382R) (Figure 4A and 4B). If SET8 repression of p53 target genes occurred via H4K20 methylation rather than p53K382 methylation, then the transactivation activity of wild-type and mutant p53K382R proteins should both be equally affected by SET8. As we do observe a requirement for K382 to be intact, we conclude that availability of this residue for methylation by SET8 is needed for SET8 to repress p53. Further evidence for a direct regulation of p53 by SET8 is that all SET8 RNAi phenotypes associated with DNA damage (e.g. increased p21 induction and cell death) are absolutely dependent on the presence of p53 protein. Thus, SET8, like a number of other p53-regulatory enzymes, such as TIP60, SIRT1, SET7/9 and HDAC1, utilizes both p53 and histones as substrates – with distinct functions specific to the different substrates (Sykes et al., 2006; Tang et al., 2006) (Chuikov et al., 2004; Luo et al., 2001; Luo et al., 2000; Vaquero et al., 2004; Vaziri et al., 2001; Wang et al., 2001).
Figure 7. Model for SET8 regulation of p53.
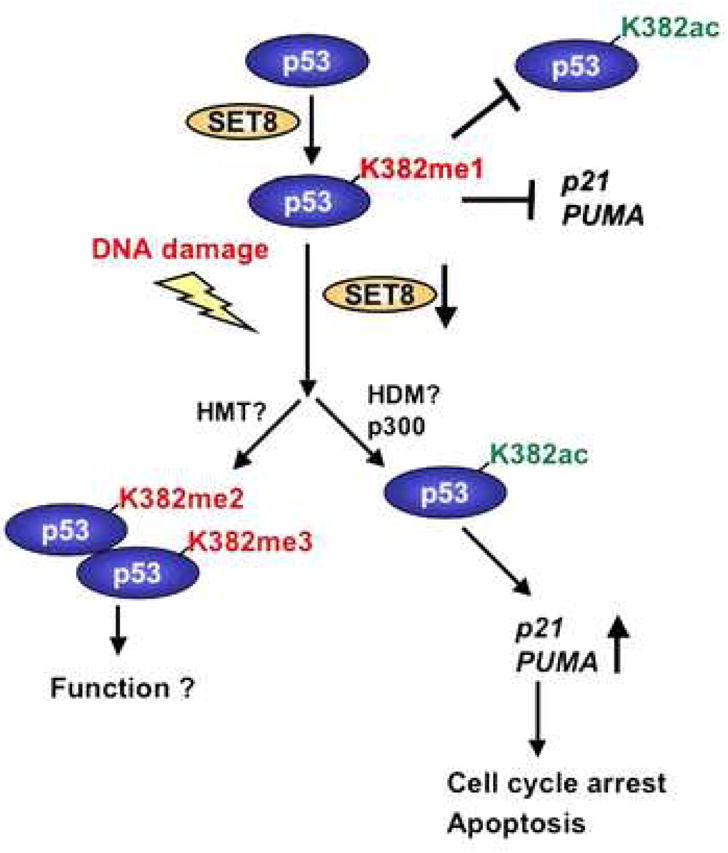
Under normal conditions, a population of p53 is monomethylated at K382 by SET8, which might render p53 inert in part by preventing acetylation at K382. Upon DNA damage, the inhibitory effect of p53K382me1 might be reversed by a combination of SET8 downregulation being coupled to increased p53 stability, and potentially via methylation of p53K382me1 to p53K382me2/3 by an as yet unknown histone methyltransferase (HMT) and/or demethylation by an as yet unknown demethylase (HDM).
We observe that p53K382me1 levels decrease with DNA damage. This observation, in conjunction with our functional characterization, argues that SET8 is a negative regulator of p53 activity. However, we do observe a SET8-dependent up-regulation of the DNA repair gene GADD45, indicating that p53K382 methylation might have more complex regulatory roles than simply contributing to on/off functions. In this regard, under normal conditions, monomethylation at K382 might render p53 largely inert but predisposed to respond to specific cellular stresses (Figure 7). Alternatively, this methylation event might dampen p53 as a mechanism for measured responses to mild insults, allowing for p53-dependent repair of DNA without the induction of cell death.
At the molecular level, how might K382 monomethylation be coupled to regulation of p53 functions? We postulate that the biological consequences of p53K382me1, alone, or in the context of additional modifications, might be dictated by distinct protein binding partners; different proteins can therefore define and channel p53 towards divergent activities. In this context, the MBT domain is a protein module found commonly on nuclear proteins and that has been shown to bind monomethyllysines (Kim et al., 2006), raising the possibility that a modular interaction between an MBT domain-containing protein and p53K382me1 acts to repress p53. There are several other methyllysine-binding domains that preferentially recognize higher methyl states than monomethyllysine (Daniel et al., 2005; Ruthenburg et al., 2007; Zhang, 2006). This raises the question of whether SET8 monomethylation of p53 at K382 leaves p53 poised to be further methylated in response to a specific signal, and that the higher methyl forms are linked to additional p53 functions (Figure 7). We have not detected methyltransferase activity on p53K382 by Suv4-20h1 and Suv4-20h2, two well-characterized H4K20 HMTs (X. Shi and O. Gozani, unpublished observations) (Schotta et al., 2004). In addition to these two enzymes, there are other putative H4K20 HMTs, such as ASH1 and NSD1, as well as numerous uncharacterized SET domain containing proteins, and perhaps one of these functions as a di- or trimethyltransferase at p53K382 (Beisel et al., 2002; Rayasam et al., 2003). In summary, this study reveals novel functions for the SET8 HMT in regulating the non-histone protein p53, and supports the hypothesis that protein lysine methylation likely modulates diverse cellular processes (Zhang and Dent, 2005).
Experimental Procedures
Plasmids, Antibodies and Peptides
SET8 cDNA was cloned into pcDNA3.1. p53 cDNA was cloned into pCAG-Flag and pGEX6p. SET8 and p53 mutants were generated by site-direct mutagenesis (Stratagene). Primer sequences are available upon request. αp53K382me1 antibody was generated in rabbits immunized with the monomethylated peptide: 377-TSRHKK(me)LMFKT-387. Other antibodies used in this study are: HRP-p53 (R&D systems); p53 (DO1) and p21 (EA10) (Calbiochem); p53K382ac (Cell Signaling); SET8, H4K20me1 and H3 (Abcam); p53(FL-393) and GST (E5) (Santa Cruz); Flag (M2) and tubulin (Sigma). p53 and histone peptides bearing different modifications were synthesized at the W.M. Keck Facility at Yale.
Cell Culture and Transfections
U2OS, H1299 and 293T were maintained in DMEM medium supplemented with 10% fetal bovine serum. Cells were transfected with plasmids or siRNA duplexes by TransIT-LT1 (Mirus) or DharmaFECT (Dharmacon), respectively, according to the manufacturers’ protocols.
Immunoprecipitation and Western Blots
Endogenous p53 or ectopically expressed Flag-tagged p53 were IPed with agarose conjugated p53 or Flag antibodies from whole cell extracts in cell lysis buffer (50 mM TrisHCl pH7.4, 250 mM NaCl, 0.5% Triton X100, 10% glycerol, 1 mM DTT, 1mM PMSF and protease inhibitors). After incubation at 4°C for overnight, the beads were washed 3X with the same buffer, and boiled in 2xLaemmli buffers. The IPed p53 was resolved on SDS-PAGE gel and detected by antibodies against state-specific p53 antibodies or HRP-αp53 to avoid crossreactivity with IgG heavy chain.
In Vitro methyltransferase assay
Methyltransferase assays were performed as previously described (Shi et al., 2006). Briefly, 2 μg of GST-p53, bulk histones, nucleosomes, or 1 μg of p53 or H4 peptides were incubated with 1μg of recombinant HMT and 0.1 mM S-adenosyl-methionine (SAM, Sigma), or 2 μCi 3H-SAM Amersham) in reaction buffer containing 50mM Tris-HCl pH 8.0, 10% glycerol, 20mM KCl, 5 mM MgCl2, 1 mM DTT and 1 mM PMSF, at 30°C for 30 min to 2hrs. The reaction mixtures were then subject to electrophoresis on SDS-PAGE, followed by either radiography or Westerns. The reactions with peptides were subject to mass spectrometry analysis.
Protein Purification and Mass Spectrometry
Hela nuclear extracts were prepared as previously described (Gozani et al., 1994). To IP endogenous p53 proteins, ~10 mg of nuclear extracts were incubated with 50 μl of DO1-conjugate agarose in buffer containing 20 mM Tris-Cl pH 8.0, 150 mM NaCl, 0.01% SDS, 1% Trition X 100, 1 mM EDTA and protease inhibitors with gentle rotation at 4°C for overnight. The beads were washed 2X with the same buffer, 2X with high salt buffer (20 mM Tris-Cl pH 8.0, 500 mM NaCl, 0.1% SDS, 1% Trition X 100, 2 mM EDTA), once with LiCl buffer (20 mM Tris, pH 8.0, 500 mM LiCl, 1% NP40, 1% deoxycholate, 1 mM EDTA), and once with TE buffer (10 mM TrisHCl 8.0, 1 mM EDTA). The p53 protein bound to the beads was subject to SDS-PAGE, excised from the gels, and incubated with trypsin overnight at 37 C. Pooled supernatants containing extracted peptides were dried and resuspended in 30% acetonitrile and 0.1% TFA prior to mass spectrometry analysis. Samples were analyzed on a reflectron time-of-flight mass spectrometer, MALDI-TOF instrument (Ultraflex, Bruker Daltonics, Billerica, MA), equipped with a 337 nm nitrogen laser and delayed ion extraction capability (delay times: 30–50 ns). Ion structure information was obtained by post source decay (PSD), using the mass gate feature, to select specific m/z window for fragmentation. The mass gate resolution was 1% of the precursor mass. Data was recorded in both positive and negative ion modes at 20kV acceleration, and mass analysis of ions determined using a dual micro-channel plate detector. Detector output was collected with a 1 GHz digitizer and displayed directly on a Windows NT based computer. Ten positive ion reflectron TOF mass spectra of 1000 laser shots were accumulated and externally calibrated with commercial peptide mix (Bruker Daltonics, Billerica, MA). For analysis of in vitro methylated synthetic peptides, the synthetic peptides untreated and treated with SET8 were equilibrated with 0.1% trifluoroacetic acid (TFA), and 50% acetonitrile with 0.1% TFA, and applied to the MALDI target plate with equal volumes of the matrix α-cyano-4-hydroxycinnamic acid (CHCA) (Sigma).
siRNA-Mediated Knockdown of SET8 and p53
The knockdown of SET8 was performed by transfection of U2OS cells for 48 hrs with two sets of Dharmacon on-target plus siRNA duplex targeting human SET8 (5′-AGUCAAAGAUCUAUUCCUAUU-3′/5′-GCAACUAGAGAGACAAAUCUU-3′) or (5′-GGAAACCAUUAGCCGGAAUUU-3′/5′-GUACGGAGCGCCAUGAAGUUU-3′) respectively, or with on-target plus SMARTpool SET8 siRNA, by using DharmaFECT according to the manufacturer’s protocol. p53 knockdown was carried out with Dharmacon on-target plus p53 siRNAs (5′-GAAAUUUGCGUGUGGAGUAUU-3′/GUGCAGCUGUGGGUUGAUUUU-3′). On-target plus siControl siRNA (5′-UGGUUUACAUGUCGACUAA-3′, Dharmacon) or on-target plus SMARTpool siControl siRNA were used as controls.
RT-PCR, Real-time PCR and ChIP Assays
RT-PCR and real-time PCR and ChIP assays were performed as previously described (Shi et al., 2006). Cells were treated with NCS (0.5 μg/ml, Sigma) for 2 to 4 hrs. mRNA was prepared using RNeasy plus kit (Qiagen) and reverse-transcribed using First Strand Synthesis kit (Invitrogen). Quantitative Real-time RT-PCR was performed on the ABI PRISM 7700 Sequence Detection System using Taqman Gene Expression Assay primer/probe sets (Applied Biosystems). Gene expressions were calculated following normalization to GAPDH levels by the comparative Ct (Cycle threshold) method. Primer sequences used for ChIP analyses are: p21 promoter: 5′-GTGGCTCTGATTGGCTTT-CTG-3′/5′-CTGAAAACAGGCAGCCCAAG-3′. PUMA promoter: 5′-GCGAGACTGTGGCCTTGTGT-3′/5′-CGTTCCAGGGTCCACAAAGT-3′. Other primer sequences are available upon request.
Cell Cycle and Cell Death Assays
For flow cytometry, U2OS cells were collected and fixed with 70% ethanol at 4°C for 4 hrs to overnight. Cells were then washed with PBS for 3 times and stained with 10 μg/ml propidium iodide and 100 μg/ml RNase. Cell death assays were performed as previously described (Shi et al., 2006; Vaziri et al., 2001). Briefly, following treatment, cells were stained with trypan blue and the fraction of cells uptaking the dye was determined utilizing a hemocytometer.
Supplementary Material
Acknowledgments
We thank Y. Zhang for GST-SET7 construct, T. Jenuwein for Suv4-20h1 and Suv4-20h2 GST constructs and M. Bedford for SET8, G9a, Suv39h1, hDOT1L GST constructs and E. Michishita for Flag-p53 construct. This work was supported in part by grants from the NIH to O.G and by the Intramural Research Program of the NIH (E.A., H.Y.). O.G. is a recipient of a Burroughs Wellcome Career Award in Biomedical Sciences and a Kimmel Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Histone methylation: recognizing the methyl mark. Methods Enzymol. 2004;376:269–288. doi: 10.1016/S0076-6879(03)76018-2. [DOI] [PubMed] [Google Scholar]
- Beisel C, Imhof A, Greene J, Kremmer E, Sauer F. Histone methylation by the Drosophila epigenetic transcriptional regulator Ash1. Nature. 2002;419:857–862. doi: 10.1038/nature01126. [DOI] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MA, Sims RJ, 3rd, Gottlieb PD, Tucker PW. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer. 2006;5:26. doi: 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 2005;19:1455–1465. doi: 10.1101/gad.1318405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Pray-Grant MG, Grant PA. Effector proteins for methylated histones: an expanding family. Cell Cycle. 2005;4:919–926. doi: 10.4161/cc.4.7.1824. [DOI] [PubMed] [Google Scholar]
- Fang J, Feng Q, Ketel CS, Wang H, Cao R, Xia L, Erdjument-Bromage H, Tempst P, Simon JA, Zhang Y. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr Biol. 2002;12:1086–1099. doi: 10.1016/s0960-9822(02)00924-7. [DOI] [PubMed] [Google Scholar]
- Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–5395. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Patton JG, Reed R. A novel set of spliceosome-associated proteins and the essential splicing factor PSF bind stably to pre-mRNA prior to catalytic step II of the splicing reaction. Embo J. 1994;13:3356–3367. doi: 10.1002/j.1460-2075.1994.tb06638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Julien E, Herr W. A switch in mitotic histone H4 lysine 20 methylation status is linked to M phase defects upon loss of HCF-1. Mol Cell. 2004;14:713–725. doi: 10.1016/j.molcel.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Karachentsev D, Sarma K, Reinberg D, Steward R. PR-Set7-dependent methylation of histone H4 Lys 20 functions in repression of gene expression and is essential for mitosis. Genes Dev. 2005;19:431–435. doi: 10.1101/gad.1263005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006 doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, Quong AA, Zhang X, Beerman T, Pestell RG, Avantaggiati ML. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci U S A. 2005;102:10188–10193. doi: 10.1073/pnas.0503068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 2006;13:951–961. doi: 10.1038/sj.cdd.4401916. [DOI] [PubMed] [Google Scholar]
- Lokshin M, Tanaka T, Prives C. Transcriptional regulation by p53 and p73. Cold Spring Harb Symp Quant Biol. 2005;70:121–128. doi: 10.1101/sqb.2005.70.046. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002a;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell. 2002b;9:1201–1213. doi: 10.1016/s1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- Rayasam GV, Wendling O, Angrand PO, Mark M, Niederreither K, Song L, Lerouge T, Hager GL, Chambon P, Losson R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. Embo J. 2003;22:3153–3163. doi: 10.1093/emboj/cdg288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JC, Nishioka K, Sarma K, Steward R, Reinberg D, Allis CD. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 2002;16:2225–2230. doi: 10.1101/gad.1014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Wang H, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P, Zhang Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8:1207–1217. doi: 10.1016/s1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, Martin SR, Sarma K, Reinberg D, Gamblin SJ, Wilson JR. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 2005;19:1444–1454. doi: 10.1101/gad.1315905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Dent SY. Histone modifying enzymes and cancer: going beyond histones. J Cell Biochem. 2005;96:1137–1148. doi: 10.1002/jcb.20615. [DOI] [PubMed] [Google Scholar]
- Zhang Y. It takes a PHD to interpret histone methylation. Nat Struct Mol Biol. 2006;13:572–574. doi: 10.1038/nsmb0706-572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.