

Abstract
Chromosomal DNA is a dynamic structure that can adopt a variety of non-canonical (i.e. non-B) conformations. In this regard, at least ten different forms of non-B DNA conformations have been identified, and many of them have been found to be mutagenic, and associated with human disease development. Despite the importance of non-B DNA structures in genetic instability and DNA metabolic processes, mechanisms remain largely undefined. The purpose of this review is to summarize current methodologies that are used to address questions in the field of non-B DNA structure-induced genetic instability. Advantages and disadvantages of each method will be discussed. A focused effort to further elucidate the mechanisms of non-B DNA-induced genetic instability will lead to a better understanding of how these structure-forming sequences contribute to the development of human disease.
Keywords: DNA Structure, Genetic instability, Methods, Reporter gene, Plasmid, Bacteria, Yeast, Mammal, Chromosome
DNA is not a uniform molecule and can adopt more than 10 types of non-B DNA conformations (DNA structures that are different from the classic Watson and Crick B-DNA helix). Although most regions of genomic DNA are in the right-handed double helix B-form, alternative DNA structures can exist transiently in the genome. The factors that influence secondary DNA structure formation include the nucleotide sequence, DNA metabolic activities, and the intracellular environment (1). Sequencing of the genomic DNA from various species (including human) has revealed an abundance of repetitive sequences, which have the capacity to adopt a variety of non-canonical DNA structures (2-4). For example, long simple repeat sequences can form slippage structures initiated by misalignment of the two strands, which can then result in looped-out repeat units (5); if the nucleotides in the loop regions are self-complementary and can form intra-strand Watson and Crick base pairs, such as in CNG (“N” can be any one of the 4 nucleotides), then a hairpin structure containing mismatches in the stem can form (6). Because purine bases can exist in a syn conformation, alternating purine/pyrimidine sequences, such as GT and GC repeats, are readily converted into a left-handed Z-DNA conformation, which is named because of the zig-zag shape of the phosphate backbone (7, 8). Polypurine-polypyrimidine regions with mirror repeat symmetry support the formation of H-DNA, where one of the single strands from half of the tract winds back and pairs with the purine-rich strand in the duplex through the major groove to form a three-stranded helix, with the complement of the third strand remaining unpaired (9, 10). Certain guanine-rich regions of the genome can form G4 DNA, which contains four guanines aligned to each other via Hoogsteen hydrogen bonding (11). Some specific sequences have the potential to form more than one non-canonical DNA structure, e.g., CG repeats can form both Z-DNA and hairpin/cruciform structures (12), and GAA triplet repeat can form H-DNA, or sticky DNA (a structure formed by two interacting triplex-like structures), in addition to the slippage structure due to its simple repeat feature (13, 14).
Recent studies have revealed that many types of non-B DNA structures play important roles in recombination, DNA replication/transcription, and particularly, genetic instability, and have been related with more than 20 human diseases (1, 15). Therefore, it is important to develop reliable experimental systems to study the roles of non-B DNA structures in human disease development and the mechanisms involved. In this review, we summarize a number of methods used to study non-B DNA structures in vitro and in vivo, with a focus on strategies to determine non-B DNA-induced genetic instability in different model systems, particularly on those methods established and used in this laboratory over the past several years.
1. Detecting non-B DNA structures in vitro
Non-B DNA structures differ from canonical B-form DNA in many aspects, and some of these differences can be used to detect the presence of non-B DNA structures within a background of canonical B-DNA. As discussed above, most non-B DNA structures contain single-stranded regions that can serve as substrates for single-strand specific chemicals and enzymes. For example, thymines in unpaired regions of the DNA are hyper-reactive to osmium tetroxide and potassium permangenate (16-21); cytosines in single-stranded DNA are sensitive to hydroxylamine (22); adenines and cytosines in single-stranded regions can be modified by bromoacetaldehyde and chloroacetaldehyde. The reaction of chloroacetaldehyde with DNA is less sensitive and slower than the reaction of bromoacetaldehyde with DNA. However, chloroacetaldehyde is more stable in solution than bromoacetaldehyde (23). Haloacetaldehyde reacts with adenines in single-stranded regions (24), and carbodiimide reacts with unpaired thymines and guanines (25). Dimethylsulfate methylates guanines in single-stranded DNA, but it is not able to modify guanines in duplexes or folded structures (e.g., tetraplex DNA) (26, 27). Diethyl pyrocarbonate reacts preferentially with purines at B-Z junctions (28, 29) or unpaired purines in H-DNA structures (30). Chemical probing is usually more sensitive to small distortions within regions of non-B DNA than are enzymatic reactions, and the reactions can be performed under a wider range of conditions (such as pH, temperature, and salt concentrations; see below). However, the modifications per se are not generally directly detectable and require a second step such as piperidine cleavage of the modified base to convert the modification into a break, followed by end-labeling or primer extension to visualize the break. Single-strand specific enzymes such as P1 and S1 nucleases directly cleave single-stranded DNA and can be used to detect non-B DNA structures in one step, but this type of reaction usually requires more stringent conditions. For example, the activity of S1 nuclease is very specific to single-stranded DNA and has been widely used to detect non-B DNA structures including cruciforms and hairpins (31, 32), H-DNA (27, 33, 34), Z-DNA (35, 36), and tetraplex DNA (37), but it works under acidic conditions (pH 4-5) and requires Zn2+ for activity. P1 nuclease is less sensitive than S1 nuclease, but it works under neutral conditions (38). Mung bean nuclease cleaves the single-stranded regions at the tips of hairpin structures, at the junctions of the stem of a cruciform structure (39, 40), and at H-DNA structures (41). T4 endonuclease VII selectively cleaves cruciform structures (42, 43).
Most types of non-B DNA structures require negative supercoiling to provide energy for transition, and a specific number of negative supercoil turns are relaxed once the non-B DNA structure is formed. This topological alteration has been used to detect the presence non-B DNA structures (44, 45) by two-dimensional gel electrophoresis. Briefly, this protocol includes: 1) treatment of DNA with topoisomerase I and increasing concentrations of ethidium bromide (EtBr) to generate various families of topoisomers, which are then combined (~1 μg of each topoisomer); 2) separate the topoisomer mixture on the 1st dimension gel in TBE buffer; 3) cut the DNA strip from the gel after the 1st dimension electrophoresis and turn it 90° and place it across the top of the gel for the 2nd dimension, which contains chloroquine; 4) run the gel in TBE buffer containing chloroquine at the same voltage as used in the 1st dimension electrophoresis; and finally 5) visualize the migration of the topoisomers after soaking the gel in EtBr solution. Because topoisomers migrate according to the number of supercoil turns, the 1st dimension generates a ladder of increasingly negative supercoiled species, with the closed circular (no supercoiled turns) molecules migrating the slowest, at the top of the gel. Chloroquine adds a certain number of positive supercoils to all species, and reverts non-B DNA structure back to the canonical B-DNA form when the reduced negative supercoiling is not enough to support the non-B DNA formation. Topoisomers migrate in the 2nd dimension according to the net sum of supercoil turns present prior to exposure to chloroquine + the effect of chloroquine on supercoil turns + any additional negative turns acquired from the non-B to B-DNA transition. Without a B-DNA to non-B DNA transition, the overall topoisomer distribution adopts a smooth arc-like shape in the 2nd dimension electrophoresis, with the slowest species in the 2nd dimension corresponding to the topoisomer with a net sum of supercoil turns equal to zero. Spots located above the smooth arc-shaped path indicate the presence of non-B DNA conformations at those levels of negative supercoiling (15, 46) since the number of supercoils lost during the structural transition caused these topoisomers to run slower in the 1st dimension.
Structure-, rather than sequence-specific antibodies are powerful tools for detecting non-B DNA structures. Using poly[d(Tm5dC).poly[d(GdA)] to immunize mice, a monoclonal antibody was developed to detect H-DNA. The antibody bound specifically to triplex DNA, particularly to those triplex structures formed at GA repeats, with no detectable binding to single-stranded DNA or to duplex DNA (47). H-DNA structures formed in plasmids or in mammalian chromosomes have been detected in assays using this H-DNA antibody (48-50). An anti-Z-DNA antibody was generated by immunizing rabbits with brominated poly[d(GdC).poly[d(G-dC)], and was used for detecting Z-DNA in chromosomes of Drosophila melanogaster by immunofluorescent staining (51) and Z-DNA formed at a GT(32) sequence in a negatively supercoiled plasmid in vitro (52). Monoclonal antibodies were also developed to recognize cruciform and T-shaped DNA structures and were able to protect the cruciforms against cleavage by mung bean nuclease or T7 endonuclease III (39).
Other methods have been employed for detecting non-B DNA structures. For example, structures that induce bending on DNA strands, such as cruciforms, H-DNA, and G-loops (large structures containing G4 DNA on the G-rich strand and a RNA/DNA hybrid on the other) have been visualized directly by electron microscopy (53-57). DNA modifying enzymes have also been used to detect the presence of non-B DNA structures. For example, the formation of Z-DNA protects that region of DNA from methylation modification and restriction digestion (58).
2. Methods to test expansion or contraction within simple repeat sequences
Expansion of trinucleotide repeats has been associated with more than 20 neurological disorders, and numerous studies have been performed to detect the expansion and contraction of repeat units in different model systems and in patient samples. The most commonly used method is direct measurement of the length of the repeat sequences by PCR amplification of DNA purified from tissues or cultured cells (59). If the repeats of different lengths are amplified at the same efficiency, then this method can provide a global view of non-B DNA-induced genetic instability of both expansion and contraction events. The limitation of the PCR-based repeat length measurement is its sensitivity; rare mutation events (<5%) will not be detected as a distinguishable band in the gel.
To detect lower frequency events occurring at simple repeats, selectable or screenable reporter systems have been developed based on length-dependent gain or loss of function of the reporter gene. Insertion of repeats within the reporter (e.g., AAAG(16) or CA(16) repeats in the green fluorescent protein (GFP) gene (60), or tk-neo gene (61)) in the coding region can disrupt the reading frame of the reporter gene, and some expansion or contraction events can restore the reading frame and reactivate reporter expression. Similarly, one can design a forward mutation-screening assay by inserting the repeats in-frame and then screen for frame-shift mutations that inactivate the reporter gene. For the greatest sensitivity, the reporter gene containing the repeats should be integrated as a single copy.
Although expansion or deletion within trinucleotide repeats (TNRs) does not change the reading frame, long triplet repeats can impact transcription or RNA splicing processes and inactivate reporter genes. For example, CAG triplet repeats were inserted at the single-copy chromosomally integrated hypoxanthine phosphoribosyl transferase (HPRT) mini-gene in its intron. The long CAG tract is spliced into the HPRT mRNA and inactivates the HPRT gene. Contraction to <39 CAG repeats reactivates the HPRT gene and permits cells to survive HPRT+ selection (62). Various sequences, located at different positions in reporter genes may have different length thresholds of inactivation or reactivation, and using a sequence length near the threshold should increase the sensitivity of the assay.
The threshold of TNRs for contraction or expansion has been investigated thoroughly in yeast, and this information has been applied to the design of reporter systems (63-65). For example, Lahue’s group developed a selectable yeast system to monitor TNR instability in a chromosomal context, with a detection level of ~2×10-8 per cell generation (66). In this system, a number of trinucleotide repeats were inserted between the TATA box and the URA3 gene, under the Schizosaccharomyces pombe adh1 promoter. The permissive distance between the TATA box and the transcription initiation site for transcription is between 55-125 bp in Saccharomyces cerevisiae (67), which is near the threshold of TNR expansions observed in human patients (~30-40 repeats, (68, 69)). To measure contractions, the distance from the TATA box to the URA3 gene in the initial construct, which was separated by TNR insertion, was longer than the permissive distance so that the transcription machinery was forced to use the upstream ATG start site, which generates an out-of-frame URA3 transcript. Contraction of TNRs brought the URA3 ORF near the TATA box, generating the correct transcript and a functional URA3 gene product. These cells can be selected for by growth on synthetic complete medium in the absence of uracil (64). Similarly, this system can be used to detect TNR expansions that separate the TATA box and the ATG start site over the permissive distance, inactivating the URA3 gene, rendering cells viable in the presence of medium containing 5-fluoroorotic acid (FOA) (70).
Recently, a vector derived from this yeast system was established for monitoring TNR instability in human cells (71). This vector contains the URA3 reporter to monitor for contraction and expansion events (as described above), an SV40 replication origin, and a large T antigen gene to insure replication in the human cells. The vector was incubated in human cells, collected and transformed into yeast for analysis. This in vitro system displayed high sensitivity (~10-6, limited by the background instability in yeast) in detecting contraction or expansion events. This reporter system has been used for studying several aspects of CTG/CAG repeat-induced expansion and contraction (e.g., length threshold, effect of TNR sequence purity, replication orientation), and the results suggested that this system can reflect the process of TNRs in human cells (68, 72-74).
3. Methods to detect non-B DNA-induced double-strand breaks, large-scale deletions, and rearrangement events
Expansion and contraction of repeat units are specific mutation types most common to simple repeat sequences. However, DNA strand breaks have been related with most types of non-B DNA structures (1). Many breakage hotspots that are associated with human disease (75) are mapped in or near sequences that have the potential to adopt non-B DNA conformations (76-81). For example, segments of the human c-MYC gene capable of adopting non-B structures (e.g., H-DNA) are found in the promoters P1, P2, and upstream of the P1 promoter (82), overlapping with the major breakage hotspots found in c-MYC-induced lymphomas and leukemias (83-87). Multiple Z-DNA motifs have been found near breakpoints in the c-MYC P1 promoter and the 3’ downstream region (88, 89), and the 5’ breakage hotspot in the BCL-2 gene is surrounded by sequences capable of adopting Z-DNA structures (80, 81). Breakpoints in lymphoid tumor-specific translocations also map to Z-DNA-forming sequences (90). Many chromosomal breakpoints associated with t(11;22) translocations in human cells are localized at the center of AT-rich palindromic sequences on 11q23 and 22q11 (91-93).
In experimental systems, these non-B DNA structures have been found to cause large-scale deletion and rearrangements, resulting from DNA double strand breaks (DSBs). Model Z-DNA-forming sequences (CG)14 inserted into a mutation reporter plasmid pUCNIM (described in detail below) induced a mutation frequency ~12-fold greater than the background control in mammalian COS-7 cells, and >95% of the (CG)14-induced mutants were large deletions and/or rearrangements resulting from DSBs located upstream, downstream, and within the (CG)14 repeats in mammalian cells (94). We have also discovered that a native H-DNA-forming sequences from the human c-MYC promoter, and model H-DNA-forming sequences are intrinsically mutagenic in mammalian cells and lead to DSBs (95). Palindromic sequences capable of adopting cruciform structures can induce DSBs (55, 96) and stimulate deletion, amplification, and recombination in bacteria (97), yeast (98, 99), and mammalian cells (100-102).
Although the model systems of bacteria, yeast, and mice share many important features of DNA metabolism with humans, it is important to note that the fate of non-B DNA structures in different species is not always the same. For example, in bacteria, the majority of the Z-DNA-forming CG repeat-induced mutations are small expansions and contractions of CG units within the repeat, likely generated by misalignment during replication (103, 104). However, we have shown that CG repeats in mammalian cells induce a very different kind of mutation event, i.e., DSBs surrounding the Z-DNA structure that can lead to large-scale deletions and rearrangements, and the occurrence of these mutations does not require replication (94). Interestingly, H-DNA-forming sequences, which cause DSBs and large-scale deletions in mammalian COS-7 cells, are stable in bacterial DH5α cells (95). Long CNG triplet repeats tend to cause expansions in human cells, but the majority of mutants are deletions in bacteria, yeast, and mice (104-107). Therefore, we have developed several reporter systems for studying non-B DNA-induced genetic instability in bacteria, yeast, transgenic mice, and cultured mammalian cells (see below).
Shuttle vector systems for studying non-B DNA-induced mutation in bacterial and mammalian cells
Shuttle vectors can replicate in both bacterial and mammalian cells, and are powerful tools for studying non-B DNA-induced genetic instability. Mutation reporter genes in the plasmids can be used to reflect the processing of non-B DNA sequences in bacteria or mammalian cells, where a considerable portion of transiently transfected plasmids are capable of being properly chromatinized (108), replicated, damaged, and repaired, thus mimicking the mammalian genomic DNA. The function of the reporter gene recovered from mammalian cells can be determined in an appropriate bacterial host cell. The advantages of this method are that it is facile, sensitive (capable of detecting mutation frequency as low as 10-5), and individual mutants can be characterized.
The supF mutation-reporter system
The supF mutation-reporter system is a forward mutation assay that allows detection of point mutations and a 6variety of deletion and rearrangement events. The pSP189 plasmid (Figure 1) is an SV40-based shuttle vector that can replicate in human cells due to the SV40 origin of replication and T antigen gene, and also in bacterial cells by virtue of the pBR327 origin. The supF gene encodes a suppressor tRNA that suppresses amber mutations in the β-galactosidase gene in an indicator bacterial strain, such as MBM7070, so that blue colonies can be seen resulting from wild-type supF genes and white colonies from mutant supF genes when grown on plates containing 5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-Gal) and isopropyl β-D-thiogalactoside (IPTG). The small size of the supF reporter gene (85-nucleotide mature tRNA, after processing) reduces the spontaneous mutation frequency. Non-B DNA-forming sequences are inserted at the XhoI restriction site 4-bp upstream of the mature tRNA sequence. The non-B DNA-containing plasmids are transfected into human cell lines, using appropriate transfection protocols according to the cell type, such as GenePorter transfection reagent (Gene Therapy Systems, Inc., San Diego, CA) or electroporation (Amaxa Nucleofector System, Cologne, Germany), and allowed to replicate for at least 48 hours prior to analysis. Plasmid DNA is then isolated from the cells (see below), analyzed by blue/white screening, and the mutation frequency of the supF gene calculated by dividing the number of white mutant colonies by the total number of colonies (white+blue). Mutants generated can be analyzed at the molecular level by DNA sequencing. Data obtained can be analyzed for statistical significance using Student’s T test.
Figure 1.
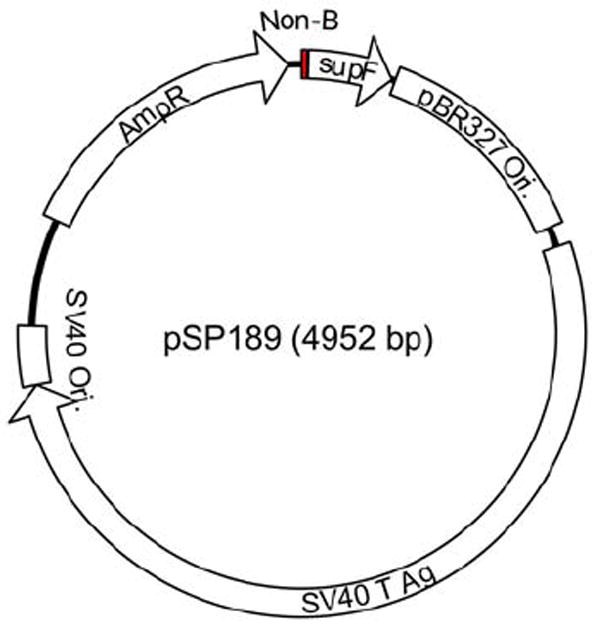
Schematic structure of the mutation-reporter shuttle plasmid, pSP189. Sequences capable of forming non-B DNA structures (shown as “Non-B” in the figure) can be inserted between the supF gene and the promoter located in the ampR gene.
The lacZ mutation-reporter system
We have also constructed a mutation-reporter shuttle vector, pUCNIM (Figure 2A), containing the lacZ gene expressing the amino-terminal fragment of the lacZ gene product (β-galactosidase), and the SV40 and the pBR327 origins of replication. Non-B DNA-forming sequences can be inserted at the multiple cloning site located within the lacZ gene (replacing codons 6-7 of the original lacZ gene). If the insertion is in-frame and does not introduce a stop codon, then sequences of interest can be as long as 80-100 bp and still maintain the function of the lacZ gene. The E. coli lac operon containing a CAP protein binding site, Plac promoter, and a lac repressor binding site provides a system of controllable transcription through the inserted non-B DNA-forming sequences in bacteria, and in mammalian cells that express the LacI protein (109). In addition, we have included a dexamethasone-inducible MMTV-LTR promoter restricted from pMAMneo in reporter plasmid pUCNIM. Upon adding dexamethasone at a final concentration of 10 μM, transcription through the non-B DNA-forming sequences and the lacZ gene was enhanced at least 10-fold over the levels of transcription in the absence of dexamethasone (Figure 2B). The controllable transcription system provides an opportunity to study the effect of transcription on non-B DNA metabolism (94). DNA structure-induced instability, including DSBs, illegitimate recombination, or frame-shift mutations due to small-scale deletion/insertion within the insert, which are not detectable via the supF system, result in dysfunction of the lacZ gene and can be detected by blue/white screening in indicator bacteria (e.g., DH5α) grown on plates containing X-Gal and IPTG. Since the supF and lacZ reporter systems are appropriate for detecting different types of mutations, using both systems enables one to screen a broad range of mutations in cell-based systems with high sensitivity.
Figure 2.
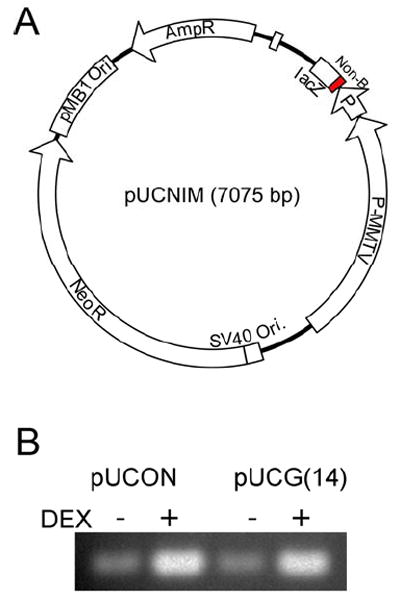
A. Schematic structure of the mutation reporter shuttle plasmid, pUCNIM. The non-B DNA-forming sequences (shown as “Non-B”) or the control sequence can be inserted between the lacZ gene and the promoter such that the function of the lacZ gene is maintained. B. RT-PCR analysis of dexamethasone-induced transcription through the non-B DNA-forming sequence (CG14) or control sequence in pUCG14 and pUCON plasmids. Primers were designed to bind sequences in the lacZ gene. RT-PCR products were subjected to agarose gel electrophoresis and visualized by ethidium bromide staining.
Plasmid recovery from mammalian cells
Plasmid DNA recovery from mammalian cells is usually performed according the methods described by Hirt (110) with slight modification. Briefly, approximately 0.2-2×107 mammalian cells are washed twice with PBS, and lysed by adding 1 ml Hirt’s lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM EDTA, 0.6% SDS, and 0.7 μg/ml of proteinase K) at 37°C for 30 minutes. The viscous cell lysate is then transferred into a 1.5 ml Eppendorf tube and mixed gently but thoroughly with 5% volume (0.25 ml) of 5 M NaCl, and incubated at 4°C overnight. The supernatant collected after centrifugation for 30 minutes at maximum speed on a microfuge at 4°C is purified by phenol/chloroform extraction and ethanol precipitation. Alternatively, plasmid DNA can be recovered through an alkaline lysis method, such as using a Qiagen Miniprep Kit (Qiagen Science, Maryland). In our experience, Hirt’s method can recover more nicked or linear plasmid DNA from repair/replication intermediates than the alkaline lysis method. Thus, it is appropriate for directly determining the damage and breakage events that occurred on the recovered plasmid DNA, while the alkaline lysis method recovers more supercoiled plasmids, and therefore is a better option for screening mutations. Purified plasmid DNA is digested with DpnI which cleaves methylated GATC sites as in dam+ bacterial strains, and has no activity on plasmids that have been replicated in mammalian cells, to ensure that all the plasmids transformed into indicator bacteria for mutation screening have been replicated in mammalian cells.
Determining the non-B DNA-induced genetic instability on a yeast chromosome
Investigation of non-B DNA-induced DSB formation in yeast has been performed on modified chromosomes (111) or on constructed yeast artificial chromosomes (YACs) (112). In both systems, one end of the chromosome was modified to contain only the selection marker (CAN1 and ADE2, or URA3) and no other essential genes. The non-B DNA-induced DSBs promote the complete loss of that end of the chromosome, including the selection marker. Thus, the chromosomal breakage event can be detected at a frequency as low as 10-10 (CAN1 and ADE2) or 10-8 (URA3).
We use the YAC system to monitor non-B DNA-induced breakage in a chromosomal context (Figure 3) as described by Freudenreich’s group (112). Non-B DNA-forming sequences are placed on the right arm of a YAC, upstream of a URA3 gene. Just proximal to the non-B DNA tract is a “backup telomere”, a 108-bp stretch of C4A4/T4G4 telomeric sequence from Oxytricha, used for telomere addition by yeast telomerase (113). The left arm of the YAC contains the LEU2 gene, for maintaining the YAC, a yeast origin of replication (ARS1), and a centromere (CEN4). Non-B DNA-induced breakage should result in loss of the right arm of the YAC, including the URA3 gene, giving rise to FOA resistance (FOAR). The exposed C4A4 sequence acts as a seed for addition of a yeast telomere for efficient YAC recovery. Thus, measurement of the rate of generation of FOAR cells can be used as a monitor of the rate of breakage.
Figure 3.
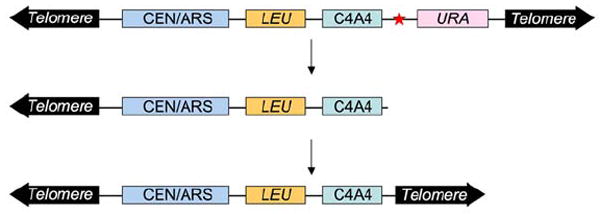
The YAC breakage assay. Cells containing YAC CF1 (not to scale), which has a non-B DNA-forming sequence (shown as a red star) and URA3 gene, are FOAS. If breakage occurs in the region around the non-B DNA insertion, the YAC will lose the URA3 gene, resulting in FOAR. The YAC can be rescued by addition of a yeast telomere onto the C4A4 seed sequence. Adapted from Callahan et al (112).
Since the non-B DNA-forming sequences are inserted ~770 bp away from the 3’-end of the URA3 gene, FOA selection would include only the complete loss of the right arm or large-scale deletion of the YAC. Small deletions, such as mutations from Z-DNA detected by using the lacZ system in bacteria (94), cannot be detected using this YAC system. Furthermore, while the human disease-related GAA/TCC and CAG/CTG repeats have been shown to cause chromosome loss in yeast (111, 112), expansions or recombination events arethe predominant form of genetic instability seen in human patients, suggesting a difference between yeast and human cells. However, it is possible that loss of one end of a chromosome is a costly and deadly event for human cells, and those cells might not survive long enough to be detected.
Determining the non-B DNA-induced genetic instability on a mammalian chromosome using a recoverable mutation-reporter construct
Although shuttle-vector reporter systems and yeast systems are sensitive and convenient, and can mimic the mammalian genomic DNA in many aspects, in some studies it is important to assess the effect of non-B DNA structures in a chromosomal context in mammalian cells. Chromosomally integrated selectable markers such as the hypoxanthine phosphoribosyl transferase (HPRT) have been used to determine the non-B DNA-induced instability in cultured mammalian cells (62), however, this strategy is not applicable in a living animal.
To develop an in vivo system to screen for genetic instability induced by non-B DNA-forming sequences, we constructed a recoverable shuttle-vector mutation reporter, p2RT (Figure 4). Recoverable plasmid-based transgenic mice or bacteriophage lambda shuttle vectors are established models for facile detection of mutations generated in living animals. We chose a plasmid-based reporter system because it allows for the detection of a variety of mutation types (e.g., translocations, deletions, DSBs), and unlike lambda vectors, the recovery of plasmid reporter DNA is not size-dependent, so mutants with deletions >50 bp can be recovered. This is important because these constitute the majority of mutants caused by non-B DNA, such as H-DNA and Z-DNA (94, 95). The plasmid, p2RT, contains a neomycin resistant (Neo) gene, which provides a selection marker for establishing a stably transected cell line, and only replicates in cells that express the SV40 large T antigen. To integrate this vector in the chromosome of SV40-transformed cell lines, the SV40 Ori should be removed prior to transfection. In both transgenic cell lines and mouse models, the vector containing the non-B DNA-forming sequences are randomly integrated into the mammalian genome (see detail below). To avoid possible position effects due to random integration of the reporter plasmid, two mutation reporter genes were included in the same vector; a lacZ reporter gene, same as in pUCNIM as described previously, and a supF reporter gene located ~2,000 bp away from the lacZ gene. Both reporter genes contain unique restriction sites between the reporter (supF tRNA or lacZ coding region) and the promoter (XhoI and NheI sites in the supF gene, and EcoRI, SacI, SalI, AccI, and SbfI sites in the lacZ gene). Non-B DNA-forming sequences to be studied can be inserted at one of two mutation reporter genes, and the other reporter gene can serve as an internal control for the background mutation frequencies (which might differ among different integration sites). The LacI binding site in the lacZ promoter (Plac) and an additional LacI binding site located ~600 bp away serve as tags during recovery of the plasmid DNA (as described below). The additional LacI site allows for recovery of deletion mutants that may have lost the Plac site. The cut & ligation site (C&L) containing SpeI-EcoRV-SpeI restriction sites can be used to recover the integrated plasmid DNA in single units. This recoverable lacZ-supF dual reporter system allows rapid analysis of a large number of mutants generated in cells and in mouse tissues.
Figure 4.
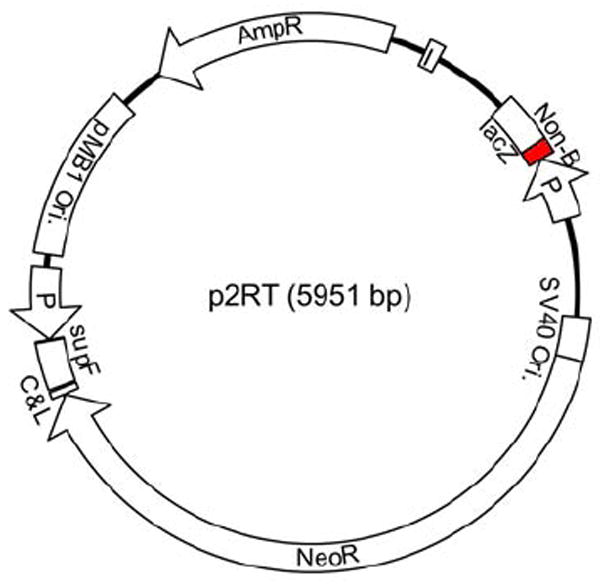
Schematic structure of the recoverable dual mutation-reporter construct p2RT. The non-B DNA forming sequences (shown as “Non-B”), or the control sequence can be inserted in either the supF gene or the lacZ gene (as shown in figure). The additional LacI binding site (shown as “I” in the figure), and an original one in Plac are used to pull down the plasmid DNA from the genomic DNA using the LacI protein coupled to magnetic beads. Plasmid DNA can be linearized by EcoRV digestion so that the vector fragment in the genome is flanked by SpeI sites on each side to facilitate isolation of the plasmid DNA from the genomic DNA, and religation for mutation screening.
Construction of mutation-reporter human cell lines
To develop mutation-reporter human cell lines to measure non-B structure-induced mutation frequencies in a chromosomal context, the recoverable dual mutation-reporter construct, p2RT containing non-B DNA sequences or control inserts, following linearization by EcoRV digestion at the cut & ligation site, were transfected into mammalian cells using appropriate transfection methods. Cells were selected in G418 (250-500 μg/ml) and maintained in medium containing 100 μg/ml G418. The copy numbers of incorporated reporter plasmids in individual clones were determined by Southern blot analysis or quantitative Real Time PCR (Wang et al., JNCI, 2008, in press).
Generation of transgenic mutation-reporter mice
The construct for generating transgenic mice was described in our recent publication (Wang et al., JNCI, 2008, in press). Briefly, dual-reporter plasmids containing the structure-forming or control sequences were linearized at the unique EcoRV site, and were microinjected into fertilized mouse oocytes to develop the transgenic mice. The integrated fragments in the genome are therefore flanked by two SpeI sites, which are used for plasmid DNA recovery from the genomic DNA. Transgenic founders were identified by PCR analysis of genomic DNA isolated from tail clips using primers specific to the plasmid sequence. Characterized strains with the maximum copy number of the reporter plasmid were further utilized for the mutation screening studies.
Detection of mutations in chromosomal DNA from transgenic human cells lines or tissues of transgenic mice
The cell lines or transgenic mice described above can be used to measure the non-B DNA-induced mutation frequencies in a chromosomal context. Genomic DNA is isolated from the cells or from the tissues. Purification of the mutation-reporter containing DNA has been described by Vijg and co-workers (114-117) as outlined below (Figure 5). Briefly, genomic DNA is digested with SpeI to release the mutation reporter plasmid DNA (containing SpeI sites on both ends). Plasmid DNA is then separated from the genomic DNA using the LacI-LacZ fusion protein coupled with magnetic beads, which can bind to either one of two LacI binding sequences in the plasmid, to pull down the DNA fragments containing the reporter gene. The recovered linear plasmid sequences are circularized using T4 DNA ligase, and electroporated into DH5α cells to screen for mutations in the lacZ gene, and into MBM7070 cell to screen for mutations in the supF gene. Non-B DNA-induced mutation frequencies on the reporter gene should be normalized to the background mutation frequency measured from the other reporter gene. Statistical analysis can be performed as described (116). Nucleotide sequences that have the capacity to adopt non-B DNA structures have been found to cause genetic instability in the form of deletions, expansions, and DSBs, which can be detected by appropriate methods described above. Further studies in this field are warranted and will lead to a better understanding of the mechanisms of human diseases and genetic evolution, and may provide new strategies to reduce genetic instability in human cells (118). To facilitate these studies, we list some experimental procedures that we have developed or modified in detail below. Other detailed protocols can be found in the references listed throughout the text.
Figure 5.
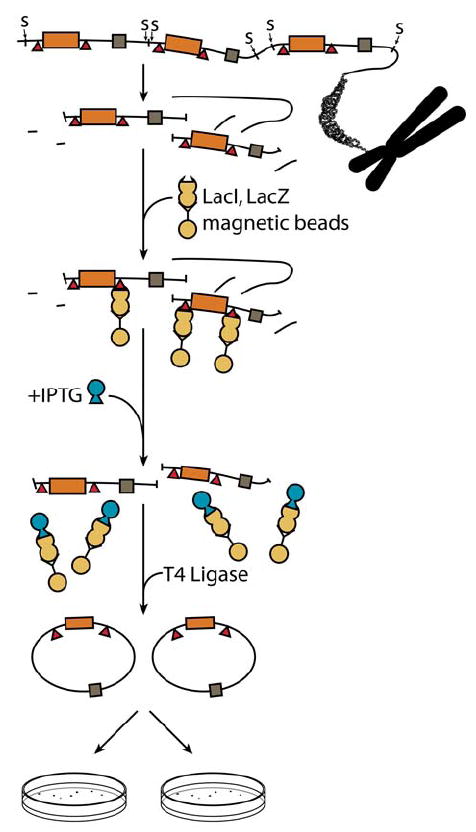
Schematic diagram of p2RT plasmid rescue from mammalian genomic DNA, and mutation analysis. The detailed procedure is described in the text. “S”: SpeI restriction sites flanking the integrated reporter sequence; LacI binding sites are shown as red triangles; a brown rectangle indicates the lacZ reporter gene, and a gray rectangle indicates the supF reporter gene.
p2RT reporter plasmid recovery from mammalian genomic DNA
This method was originally developed in Vijg’s group (114-117). Preparation of the LacI-LacZ fusion protein and its conjugation to magnetic beads coated with sheep anti-mouse IgG through the anti-β-galactosidase monoclonal antibody has been described in detail by Garcia et al (119). Below is a modified protocol for recovery of the p2RT plasmid from mammalian genomic DNA.
Extract genomic DNA from cultured cells or mouse tissue using DNeasy Blood & Tissue Kit (Qiagen) according to the manual.
Digest genomic DNA (10-50 μg) with SpeI (4μl, 40 U) in NEBuffer 2 (NEB, Ipswich, MA) + BSA in 200 μl reaction, for 4 hrs.
Purify the DNA by phenol/chloroform extraction and ethanol precipitation.
Dissolve DNA in 80 μl 1x binding buffer (10 mM Tris-HC1 pH 6.8, 1 mM EDTA, 10 mM MgCl2, 5% glycerol).
Pellet 60 μl of LacI-LacZ magnetic beads on the magnetic bed. Incubate the LacI-LacZ coated beads with DNA in 80 μl binding buffer for 1 h at 37°C while rotating.
For each sample, prepare a final IPTG-elution mixture by adding 75 μl IPTG-elution buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 125 mM NaC1), 5 μl IPTG solution at 25 mg/ml, and 20 μl 10 × NEBuffer 2 to 100 μl water.
After incubation, pellet the beads on a magnetic bed and remove the clear supernatant. Wash the beads 3 times with 250 μl 1x binding buffer (vortex gently and pellet the beads on the magnetic bed). After removal of the clear supernatant, add 200 μl of final IPTG-elution mixture prepared in step 5. Vortex gently and incubate for 30 min at 37°C while rotating.
Incubate at 65°C for 20 min and allow to cool to room temperature. Spin briefly to collect liquid, resuspend the beads by gentle vortexing, and pellet on magnetic bed. Transfer the supernatant to a clean Eppendorf tube and ethanol precipitate the DNA in the presence of 30 μl glycogen.
Resuspend the DNA in 10 μl buffer containing 1 μl 10x ligation buffer and 0.2 μl T4 DNA ligase (NEB). Incubate in 16°C overnight.
Heat sample to 65°C for 10 min to inactivate ligase.
Mix 2.5 μl of ligation product and 40 μl ElectroMAX™ DH5α-E™ cells (Invitrogen) or MBM7070 electrocompetent cells, and add the mixture into a chilled 0.1 cm cuvette and electroporate. We use the following conditions on a BioRad GenePulser® II electroporator for transformation: 1.7 kV, 200 Ω, 25 μF, and time constant 5 ms.
Immediately add 1 ml SOC medium and incubate 30-60 minutes with moderate shaking at 37°C. Plate the cells on LB agar plates containing 100 μg/ml ampicillin, 20 mg/ml X-Gal and 50 mg/ml IPTG.
Count the number of total (white+blue) colonies, and calculate the mutation frequencies of the supF and lacZ genes as the number of white colonies divided by the number of total colonies. Non-B DNA-induced mutation frequency should be normalized to the background mutation frequency determined on the other reporter gene.
Identification of DNA strand breaks caused by non-B DNA structures on recovered episomal vectors
Screened or selected mutants induced by non-B DNA structures are the final products of processing and repair. Initial characterization of the mutant plasmids can be performed by restriction digestion analysis and visualized on agarose gels, or by direct DNA sequencing, (e.g., large deletion areas and the existence of microhomologies at the junctions of the deletions would suggest an error-prone end-joining at a DSB (94, 95)) However, it may be useful to determine if strand breaks were in fact generated at the non-B DNA structures. To directly map strand breaks, we have developed a modified linker-mediated PCR (LM-PCR) assay to explore non-B DNA-induced DSB hotspots in plasmids recovered from mammalian cells (94, 95).
Preparation of a double-stranded linker: The linker sequence could be random and one of the oligonucleotides (LMCRP2, as shown below) will be used as a reverse primer, and should be able to work with the specific forward primer on the plasmid near the non-B DNA region (e.g., similar Tms, but not complementary to each other). For example, we used the oligonucleotides LMPCR1: 5’-CGTACATTCACAACGATAGCGACTGA-3’, and LMPCR2: 5’-GCTATCGTTGTGAATGTACG-3’. After annealing the two oligonucleotides to form the linker that has a blunt-end on one side and a 3’-overhang on the other, phosphorylate the 5’-ends to facilitate the ligation. Purify the double-stranded linker from an agarose gel, if necessary, by eluting the DNA from the gel by dialysis. Adjust the final concentration to 2.5 μM.
Plasmid DNA preparations: Plasmids should be purified from mammalian cells using Hirt’s method to recover the linear plasmid DNA, as described above (without DpnI digestion). Treat the plasmid DNA with 1-2 U DNA Polymerase I Klenow Fragment (NEB) with 33 μM dNTP in 20 μl total reaction at 25°C for 15 min to blunt the 3’-and 5-’ overhangs. Heat the mixture to 75°C for 30 min to inactive the enzyme and allow the reaction to cool to room temperature. Purify the DNA by phenol/chloroform/isoamyl alcohol (25:24:1) extraction and ethanol precipitation, and dissolve the DNA in 20-40 μl of dH2O.
Ligation: Mix 15 μl DNA Polymerase I Klenow Fragment treated plasmid DNA from step 2, 10 pmol phosphorylated linker, 3.5μl 10x T4 DNA ligase buffer and 0.5 μl T4 DNA ligase (NEB) in total volume of 35 μl by gentle vortexing and incubate the mixture at 16°C overnight. Heat the mixture to 75°C for 30 min to inactive the enzyme and allow it to cool to room temperature.
PCR: Mix 6 μl linker-conjugated plasmid DNA from step 3, 10 pmol LMPCR2 and 5 pmol specific primer in a standard PCR reaction mixture, and amplify the region between specific primer and the linker. The following conditions can be used for the PCR reaction; 95°C for 30 sec for denaturation, 54°C for 30 sec for annealing, and 72°C for 20 sec for extension, for 28 cycles. Separate the PCR products on a 1.5-2.0% agarose gel. The length of each specific band can be measured by image analyzing software such as the KODAK 1DTM Imaging Station.
Further analysis of the PCR products: each band can be recovered from an agarose gel, cloned into a PCR cloning vector and sequenced, as previously reported (120).
Acknowledgments
We thank Dr. Albino Bacolla for useful comments on the manuscript and Sarah Henninger for manuscript preparation. Support was provided by an NIH/NCI grant to K.M.V. (CA093729) and an NIH/NIEHS grant to K.M.V. (ES015707).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wells RD. Trends Biochem Sci. 2007;32:271–278. doi: 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, et al. Nature. 2001;409:860–921. [Google Scholar]
- 3.Schroth GP, Chou PJ, Ho PS. J Biol Chem. 1992;267:11846–11855. [PubMed] [Google Scholar]
- 4.Catasti P, Chen X, Mariappan SV, Bradbury EM, Gupta G. Genetica. 1999;106:15–36. doi: 10.1023/a:1003716509180. [DOI] [PubMed] [Google Scholar]
- 5.Djian P. Cell. 1998;94:155–160. doi: 10.1016/s0092-8674(00)81415-4. [DOI] [PubMed] [Google Scholar]
- 6.Sinden RR, Potaman VN, Oussatcheva EA, Pearson CE, Lyubchenko YL, Shlyakhtenko LS. J Biosci. 2002;27:53–65. doi: 10.1007/BF02703683. [DOI] [PubMed] [Google Scholar]
- 7.Malfoy B, Rousseau N, Vogt N, Viegas-Pequignot E, Dutrillaux B, Leng M. Nucleic Acids Res. 1986;14:3197–3214. doi: 10.1093/nar/14.8.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnston BH. Methods Enzymol. 1992;211:127–158. doi: 10.1016/0076-6879(92)11009-8. [DOI] [PubMed] [Google Scholar]
- 9.Mirkin SM, Frank-Kamenetskii MD. Annu Rev Biophys Biomol Struct. 1994;23:541–576. doi: 10.1146/annurev.bb.23.060194.002545. [DOI] [PubMed] [Google Scholar]
- 10.Htun H, Dahlberg JE. Science. 1989;243:1571–1576. doi: 10.1126/science.2648571. [DOI] [PubMed] [Google Scholar]
- 11.Sen D, Gilbert W. Nature. 1990;344:410–414. doi: 10.1038/344410a0. [DOI] [PubMed] [Google Scholar]
- 12.Wang AH, Hakoshima T, van der Marel G, van Boom JH, Rich A. Cell. 1984;37:321–331. doi: 10.1016/0092-8674(84)90328-3. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, Wells RD. Mol Cell. 1999;3:465–475. doi: 10.1016/s1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- 14.Potaman VN, Oussatcheva EA, Lyubchenko YL, Shlyakhtenko LS, Bidichandani SI, Ashizawa T, Sinden RR. Nucleic Acids Res. 2004;32:1224–1231. doi: 10.1093/nar/gkh274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinden RR. DNA structure and function. Academic Press; San Diego: 1994. [Google Scholar]
- 16.Lilley DM, Palecek E. EMBO J. 1984;3:1187–1192. doi: 10.1002/j.1460-2075.1984.tb01949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palecek E. Methods Enzymol. 1992;212:139–155. doi: 10.1016/0076-6879(92)12010-n. [DOI] [PubMed] [Google Scholar]
- 18.Palecek E. Methods Enzymol. 1992;212:305–318. doi: 10.1016/0076-6879(92)12019-m. [DOI] [PubMed] [Google Scholar]
- 19.Raghavan SC, Swanson PC, Wu X, Hsieh CL, Lieber MR. Nature. 2004;428:88–93. doi: 10.1038/nature02355. [DOI] [PubMed] [Google Scholar]
- 20.Rahmouni AR, Wells RD. Science. 1989;246:358–363. doi: 10.1126/science.2678475. [DOI] [PubMed] [Google Scholar]
- 21.Sasse-Dwight S, Gralla JD. J Biol Chem. 1989;264:8074–8081. [PubMed] [Google Scholar]
- 22.Ferl RJ, Paul AL. Plant Mol Biol. 1992;18:1181–1184. doi: 10.1007/BF00047722. [DOI] [PubMed] [Google Scholar]
- 23.McLean MJ, Larson JE, Wohlrab F, Wells RD. Nucleic Acids Res. 1987;15:6917–6935. doi: 10.1093/nar/15.17.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohwi-Shigematsu T, Kohwi Y. Methods Enzymol. 1992;212:155–180. doi: 10.1016/0076-6879(92)12011-e. [DOI] [PubMed] [Google Scholar]
- 25.Lebowitz J, Garon CG, Chen MC, Salzman NP. J Virol. 1976;18:205–210. doi: 10.1128/jvi.18.1.205-210.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yafe A, Etzioni S, Weisman-Shomer P, Fry M. Nucleic Acids Res. 2005;33:2887–2900. doi: 10.1093/nar/gki606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnston BH. Science. 1988;241:1800–1804. doi: 10.1126/science.2845572. [DOI] [PubMed] [Google Scholar]
- 28.Nejedly K, Klysik J, Palecek E. FEBS Lett. 1989;243:313–317. doi: 10.1016/0014-5793(89)80152-8. [DOI] [PubMed] [Google Scholar]
- 29.Guo Q, Lu M, Shahrestanifar M, Sheardy RD, Kallenbach NR. Biochemistry. 1991;30:11735–11741. doi: 10.1021/bi00115a001. [DOI] [PubMed] [Google Scholar]
- 30.Vojtiskova M, Mirkin S, Lyamichev V, Voloshin O, Frank-Kamenetskii M, Palecek E. FEBS Lett. 1988;234:295–299. doi: 10.1016/0014-5793(88)80102-9. [DOI] [PubMed] [Google Scholar]
- 31.Asakura Y, Kikuchi Y, Yanagida M. J Biochem. 1985;98:41–47. doi: 10.1093/oxfordjournals.jbchem.a135270. [DOI] [PubMed] [Google Scholar]
- 32.Lilley DM, Markham AF. EMBO J. 1983;2:527–533. doi: 10.1002/j.1460-2075.1983.tb01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson KL, Becker NA, Pahwa GS, Hollingsworth MA, Maher LJ., 3rd J Biol Chem. 1996;271:18061–18067. doi: 10.1074/jbc.271.30.18061. [DOI] [PubMed] [Google Scholar]
- 34.Pestov DG, Dayn A, Siyanova E, George DL, Mirkin SM. Nucleic Acids Res. 1991;19:6527–6532. doi: 10.1093/nar/19.23.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaho JA, Larson JE, McLean MJ, Wells RD. J Biol Chem. 1988;263:14446–14455. [PubMed] [Google Scholar]
- 36.McLean MJ, Wells RD. J Biol Chem. 1988;263:7370–7377. [PubMed] [Google Scholar]
- 37.Kumar P, Verma A, Maiti S, Gargallo R, Chowdhury S. Biochemistry. 2005;44:16426–16434. doi: 10.1021/bi051452x. [DOI] [PubMed] [Google Scholar]
- 38.Romier C, Dominguez R, Lahm A, Dahl O, Suck D. Proteins. 1998;32:414–424. [PubMed] [Google Scholar]
- 39.Frappier L, Price GB, Martin RG, Zannis-Hadjopoulos M. J Biol Chem. 1989;264:334–341. [PubMed] [Google Scholar]
- 40.Xodo LE, Manzini G, Quadrifoglio F, van der Marel G, van Boom J. Nucleic Acids Res. 1991;19:1505–1511. doi: 10.1093/nar/19.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bacolla A, Wu FY. Nucleic Acids Res. 1991;19:1639–1647. doi: 10.1093/nar/19.7.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lilley DM, Kemper B. Cell. 1984;36:413–422. doi: 10.1016/0092-8674(84)90234-4. [DOI] [PubMed] [Google Scholar]
- 43.Naylor LH, Lilley DM, van de Sande JH. EMBO J. 1986;5:2407–2413. doi: 10.1002/j.1460-2075.1986.tb04511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peck LJ, Wang JC. Proc Natl Acad Sci U S A. 1983;80:6206–6210. doi: 10.1073/pnas.80.20.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kladde MP, Kohwi Y, Kohwi-Shigematsu T, Gorski J. Proc Natl Acad Sci U S A. 1994;91:1898–1902. doi: 10.1073/pnas.91.5.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowater R, Aboul-Ela F, Lilley DM. Methods Enzymol. 1992;212:105–120. doi: 10.1016/0076-6879(92)12007-d. [DOI] [PubMed] [Google Scholar]
- 47.Agazie YM, Lee JS, Burkholder GD. J Biol Chem. 1994;269:7019–7023. [PubMed] [Google Scholar]
- 48.Raghavan SC, Chastain P, Lee JS, Hegde BG, Houston S, Langen R, Hsieh CL, Haworth IS, et al. J Biol Chem. 2005;280:22749–22760. doi: 10.1074/jbc.M502952200. [DOI] [PubMed] [Google Scholar]
- 49.Lee JS, Burkholder GD, Latimer LJ, Haug BL, Braun RP. Nucleic Acids Res. 1987;15:1047–1061. doi: 10.1093/nar/15.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JS, Latimer LJ, Haug BL, Pulleyblank DE, Skinner DM, Burkholder GD. Gene. 1989;82:191–199. doi: 10.1016/0378-1119(89)90044-9. [DOI] [PubMed] [Google Scholar]
- 51.Nordheim A, Pardue ML, Lafer EM, Moller A, Stollar BD, Rich A. Nature. 1981;294:417–422. doi: 10.1038/294417a0. [DOI] [PubMed] [Google Scholar]
- 52.Nordheim A, Rich A. Proc Natl Acad Sci U S A. 1983;80:1821–1825. doi: 10.1073/pnas.80.7.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mikheikin AL, Lushnikov AY, Lyubchenko YL. Biochemistry. 2006;45:12998–13006. doi: 10.1021/bi061002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shlyakhtenko LS, Potaman VN, Sinden RR, Lyubchenko YL. J Mol Biol. 1998;280:61–72. doi: 10.1006/jmbi.1998.1855. [DOI] [PubMed] [Google Scholar]
- 55.Kurahashi H, Inagaki H, Yamada K, Ohye T, Taniguchi M, Emanuel BS, Toda T. J Biol Chem. 2004;279:35377–35383. doi: 10.1074/jbc.M400354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vetcher AA, Napierala M, Iyer RR, Chastain PD, Griffith JD, Wells RD. J Biol Chem. 2002;277:39217–39227. doi: 10.1074/jbc.M205209200. [DOI] [PubMed] [Google Scholar]
- 57.Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lukomski S, Wells RD. Proc Natl Acad Sci U S A. 1994;91:9980–9984. doi: 10.1073/pnas.91.21.9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Slebos RJ, Oh DS, Umbach DM, Taylor JA. Cancer Res. 2002;62:6052–6060. [PubMed] [Google Scholar]
- 61.Yamada NA, Parker JM, Farber RA. Environ Mol Mutagen. 2003;42:75–84. doi: 10.1002/em.10179. [DOI] [PubMed] [Google Scholar]
- 62.Lin Y, Dion V, Wilson JH. Nat Struct Mol Biol. 2006;13:179–180. doi: 10.1038/nsmb1042. [DOI] [PubMed] [Google Scholar]
- 63.Miret JJ, Pessoa-Brandao L, Lahue RS. Mol Cell Biol. 1997;17:3382–3387. doi: 10.1128/mcb.17.6.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dixon MJ, Lahue RS. DNA Repair (Amst) 2002:1. 763–770. doi: 10.1016/s1568-7864(02)00095-2. [DOI] [PubMed] [Google Scholar]
- 65.Rolfsmeier ML, Dixon MJ, Pessoa-Brandao L, Pelletier R, Miret JJ, Lahue RS. Genetics. 2001;157:1569–1579. doi: 10.1093/genetics/157.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dixon MJ, Bhattacharyya S, Lahue RS. Methods Mol Biol. 2004;277:29–45. doi: 10.1385/1-59259-804-8:029. [DOI] [PubMed] [Google Scholar]
- 67.Furter-Graves EM, Hall BD. Mol Gen Genet. 1990;223:407–416. doi: 10.1007/BF00264447. [DOI] [PubMed] [Google Scholar]
- 68.Paulson HL, Fischbeck KH. Annu Rev Neurosci. 1996;19:79–107. doi: 10.1146/annurev.ne.19.030196.000455. [DOI] [PubMed] [Google Scholar]
- 69.Mirkin SM. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 70.Miret JJ, Pessoa-Brandao L, Lahue RS. Proc Natl Acad Sci U S A. 1998;95:12438–12443. doi: 10.1073/pnas.95.21.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pelletier R, Farrell BT, Miret JJ, Lahue RS. Nucleic Acids Res. 2005;33:5667–5676. doi: 10.1093/nar/gki880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leeflang EP, Zhang L, Tavare S, Hubert R, Srinidhi J, MacDonald ME, Myers RH, de Young M, et al. Hum Mol Genet. 1995;4:1519–1526. doi: 10.1093/hmg/4.9.1519. [DOI] [PubMed] [Google Scholar]
- 73.Farrell BT, Lahue RS. Nucleic Acids Res. 2006;34:4495–4505. doi: 10.1093/nar/gkl614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Claassen DA, Lahue RS. Hum Mol Genet. 2007;16:3088–3096. doi: 10.1093/hmg/ddm270. [DOI] [PubMed] [Google Scholar]
- 75.Popescu NC. Cancer Lett. 2003;192:1–17. doi: 10.1016/s0304-3835(02)00596-7. [DOI] [PubMed] [Google Scholar]
- 76.Echlin-Bell DR, Smith LL, Li L, Strissel PL, Strick R, Gupta V, Banerjee J, Larson R, et al. Hum Genet. 2003;113:80–91. doi: 10.1007/s00439-003-0936-2. [DOI] [PubMed] [Google Scholar]
- 77.Wiener F, Ohno S, Babonits M, Sumegi J, Wirschubsky Z, Klein G, Mushinski JF, Potter M. Proc Natl Acad Sci U S A. 1984;81:1159–1163. doi: 10.1073/pnas.81.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akasaka T, Akasaka H, Ueda C, Yonetani N, Maesako Y, Shimizu A, Yamabe H, Fukuhara S, et al. J Clin Oncol. 2000;18:510–518. doi: 10.1200/JCO.2000.18.3.510. [DOI] [PubMed] [Google Scholar]
- 79.Kovalchuk AL, Muller JR, Janz S. Oncogene. 1997;15:2369–2377. doi: 10.1038/sj.onc.1201409. [DOI] [PubMed] [Google Scholar]
- 80.Adachi M, Tsujimoto Y. Oncogene. 1990;5:1653–1657. [PubMed] [Google Scholar]
- 81.Seite P, Leroux D, Hillion J, Monteil M, Berger R, Mathieu-Mahul D, Larsen CJ. Genes Chromosomes Cancer. 1993;6:39–44. doi: 10.1002/gcc.2870060108. [DOI] [PubMed] [Google Scholar]
- 82.Michelotti GA, Michelotti EF, Pullner A, Duncan RC, Eick D, Levens D. Mol Cell Biol. 1996;16:2656–2669. doi: 10.1128/mcb.16.6.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Joos S, Haluska FG, Falk MH, Henglein B, Hameister H, Croce CM, Bornkamm GW. Cancer Res. 1992;52:6547–6552. [PubMed] [Google Scholar]
- 84.Haluska FG, Tsujimoto Y, Croce CM. Nucleic Acids Res. 1988;16:2077–2085. doi: 10.1093/nar/16.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saglio G, Grazia Borrello M, Guerrasio A, Sozzi G, Serra A, di Celle PF, Foa R, Ferrarini M, et al. Genes Chromosomes Cancer. 1993;8:1–7. doi: 10.1002/gcc.2870080102. [DOI] [PubMed] [Google Scholar]
- 86.Care A, Cianetti L, Giampaolo A, Sposi NM, Zappavigna V, Mavilio F, Alimena G, Amadori S, et al. EMBO J. 1986;5:905–911. doi: 10.1002/j.1460-2075.1986.tb04302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilda M, Busch K, Klose I, Keller T, Woessmann W, Kreuder J, Harbott J, Borkhardt A. Genes Chromosomes Cancer. 2004;41:178–182. doi: 10.1002/gcc.20063. [DOI] [PubMed] [Google Scholar]
- 88.Rimokh R, Rouault JP, Wahbi K, Gadoux M, Lafage M, Archimbaud E, Charrin C, Gentilhomme O, et al. Genes Chromosomes Cancer. 1991;3:24–36. doi: 10.1002/gcc.2870030106. [DOI] [PubMed] [Google Scholar]
- 89.Wolfl S, Wittig B, Rich A. Biochim Biophys Acta. 1995;1264:294–302. doi: 10.1016/0167-4781(95)00155-7. [DOI] [PubMed] [Google Scholar]
- 90.Boehm T, Mengle-Gaw L, Kees UR, Spurr N, Lavenir I, Forster A, Rabbitts TH. EMBO J. 1989;8:2621–2631. doi: 10.1002/j.1460-2075.1989.tb08402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kurahashi H, Emanuel BS. Hum Mol Genet. 2001;10:2605–2617. doi: 10.1093/hmg/10.23.2605. [DOI] [PubMed] [Google Scholar]
- 92.Edelmann L, Spiteri E, Koren K, Pulijaal V, Bialer MG, Shanske A, Goldberg R, Morrow BE. Am J Hum Genet. 2001;68:1–13. doi: 10.1086/316952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kurahashi H, Shaikh TH, Hu P, Roe BA, Emanuel BS, Budarf ML. Hum Mol Genet. 2000;9:1665–1670. doi: 10.1093/hmg/9.11.1665. [DOI] [PubMed] [Google Scholar]
- 94.Wang G, Christensen LA, Vasquez KM. Proc Natl Acad Sci U S A. 2006;103:2677–2682. doi: 10.1073/pnas.0511084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang G, Vasquez KM. Proc Natl Acad Sci U S A. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nasar F, Jankowski C, Nag DK. Mol Cell Biol. 2000;20:3449–3458. doi: 10.1128/mcb.20.10.3449-3458.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cromie GA, Millar CB, Schmidt KH, Leach DR. Genetics. 2000;154:513–522. doi: 10.1093/genetics/154.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gordenin DA, Lobachev KS, Degtyareva NP, Malkova AL, Perkins E, Resnick MA. Mol Cell Biol. 1993;13:5315–5322. doi: 10.1128/mcb.13.9.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nag DK, Kurst A. Genetics. 1997;146:835–847. doi: 10.1093/genetics/146.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Akgun E, Zahn J, Baumes S, Brown G, Liang F, Romanienko PJ, Lewis S, Jasin M. Mol Cell Biol. 1997;17:5559–5570. doi: 10.1128/mcb.17.9.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tanaka H, Tapscott SJ, Trask BJ, Yao MC. Proc Natl Acad Sci U S A. 2002;99:8772–8777. doi: 10.1073/pnas.132275999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou ZH, Akgun E, Jasin M. Proc Natl Acad Sci U S A. 2001;98:8326–8333. doi: 10.1073/pnas.151008498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Freund AM, Bichara M, Fuchs RP. Proc Natl Acad Sci U S A. 1989;86:7465–7469. doi: 10.1073/pnas.86.19.7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wells RD. J Biol Chem. 1996;271:2875–2878. doi: 10.1074/jbc.271.6.2875. [DOI] [PubMed] [Google Scholar]
- 105.Chastain PD, Sinden RR. J Mol Biol. 1998;275:405–411. doi: 10.1006/jmbi.1997.1502. [DOI] [PubMed] [Google Scholar]
- 106.Lahue RS, Slater DL. Front Biosci. 2003;8:s653–665. doi: 10.2741/1107. [DOI] [PubMed] [Google Scholar]
- 107.Bowater RP, Wells RD. Prog Nucleic Acid Res Mol Biol. 2001;66:159–202. doi: 10.1016/s0079-6603(00)66029-4. [DOI] [PubMed] [Google Scholar]
- 108.Jeong SW, Stein A. J Biol Chem. 1994;269:2197–2205. [PubMed] [Google Scholar]
- 109.Figge J, Wright C, Collins CJ, Roberts TM, Livingston DM. Cell. 1988;52:713–722. doi: 10.1016/0092-8674(88)90409-6. [DOI] [PubMed] [Google Scholar]
- 110.Hirt B. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 111.Kim HM, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, Lobachev KS. EMBO J. 2008;27:2896–2906. doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Callahan JL, Andrews KJ, Zakian VA, Freudenreich CH. Mol Cell Biol. 2003;23:7849–7860. doi: 10.1128/MCB.23.21.7849-7860.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pluta AF, Zakian VA. Nature. 1989;337:429–433. doi: 10.1038/337429a0. [DOI] [PubMed] [Google Scholar]
- 114.Gossen JA, de Leeuw WJ, Bakker AQ, Vijg J. Mutagenesis. 1993;8:243–247. doi: 10.1093/mutage/8.3.243. [DOI] [PubMed] [Google Scholar]
- 115.Gossen J, Vijg J. Trends Genet. 1993;9:27–31. doi: 10.1016/0168-9525(93)90069-T. [DOI] [PubMed] [Google Scholar]
- 116.Boerrigter ME, Vijg J. Environ Mol Mutagen. 1997;29:221–229. [PubMed] [Google Scholar]
- 117.Boerrigter ME, Dolle ME, Martus HJ, Gossen JA, Vijg J. Nature. 1995;377:657–659. doi: 10.1038/377657a0. [DOI] [PubMed] [Google Scholar]
- 118.Jain A, Wang G, Vasquez KM. Biochimie. 2008;90:1117–1130. doi: 10.1016/j.biochi.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Garcia AM, Busuttil RA, Rodriguez A, Cabrera C, Lundell M, Dolle ME, Vijg J. Methods Mol Biol. 2007;371:267–287. doi: 10.1007/978-1-59745-361-5_20. [DOI] [PubMed] [Google Scholar]
- 120.Wojciechowska M, Napierala M, Larson JE, Wells RD. J Biol Chem. 2006 doi: 10.1074/jbc.M603888200. [DOI] [PubMed] [Google Scholar]