

Abstract
The proinflammatory cytokines TNFα and Il-1β orchestrate the progression of CNS inflammation, which substantially contributes to neurodegeneration in many CNS pathologies. TNFα and Il-1β stimulate actin filament reorganization in non-neuronal cells often accompanied by the formation of reactive oxygen species (ROS). Actin filament dynamics is vital for cellular plasticity, mitochondrial function, and gene expression despite being highly susceptible to oxidative damage. We demonstrated that, in neuronal cells, TNFα and Il-1β stimulate a transient, redox-dependent reorganization of the actin cytoskeleton into lamellipodia under the regulation of Rac1 and a neuronal NADPH oxidase as the source of ROS. Persistent presence of intracellular ROS provoked oxidative damage (carbonylation) to actin coinciding with loss of lamellipodia and arrest of cellular plasticity. Inhibition of NADPH oxidase activity or Rac1 abolished the adverse effects of cytokines. These findings suggest that oxidative damage to the neuronal actin cytoskeleton could represent a key step in CNS neurodegeneration.
Keywords: Rac1, oxygen radicals, actin, lamellipodia, cytokines, NADPH oxidase
INTRODUCTION
Inflammation accompanied by oxidative stress is widespread in acute, chronic, and psychiatric CNS pathologies and plays a key role in the progression of neurodegenerative processes (Mrak and Griffin, 2005; Lucas et al., 2006). Persistent expression of the two proinflammatory cytokines tumor necrosis factor α (TNFα) and interleukin-1β (Il-1β) compromises plasticity and survival of neuronal cells (Rothwell and Luheshi, 2000; Allan and Rothwell, 2001). Disruption of proper actin filament dynamics and oxidative damage to actin in neuronal cells is prevalent in both acute CNS injury and chronic CNS pathologies and might represent a pivotal step in neurodegeneration (Dalle-Donne et al., 2001; Beal, 2002). Many cellular processes aside from motility are intimately linked to actin filament dynamics including gene expression, mitochondrial homeostasis, and vesicle traffic (Lin et al., 1997; Gourlay and Ayscough, 2005; Percipalle and Visa, 2006). Munch et al. recently demonstrated that proinflammatory mediators released from microglia attenuated neurite outgrowth and caused neurite retraction (Munch et al., 2003). Exposure of hippocampal neurons to TNFα in vitro attenuated neurite outgrowth and reduced branching of neurites through a RhoA-dependent mechanism (Neumann et al., 2002). TNFα and Il-1β caused significant rearrangement of actin filament architecture in non-neuronal cells mediated by small Rho GTPases (Wojciak-Stothard et al., 1998; Peppelenbosch et al., 1999; Puls et al., 1999; Hanna et al., 2001). Rho GTPases are vital in converting a plethora of extrinsic stimuli into a coordinated reorganization of actin filaments in non-neuronal cells as well as neurons (Nobes and Hall, 1995; Hall and Nobes, 2000b; Luo, 2002). Recent observations in non-neuronal cells demonstrated that Rac1 and ROS are implicated in actin filament reorganization and cell motility (Moldovan et al., 1999; Karnoub et al., 2001; Nimnual et al., 2003). Actin filament dynamics is highly sensitive to oxidative modification and distinct ROS differentially affect assembly, disassembly, and stability of actin filament (Krieger-Brauer and Kather, 1995; Sulciner et al., 1996; Milzani et al., 1997; Dalle-Donne et al., 2001; Lassing et al., 2007). Notably, proinflammatory cytokines, growth factors, and hormones induce ROS formation in many non-neuronal cell types often under the regulation of Rac1 (Sundaresan et al. 1996). Indeed, Rac1-dependent ROS intermediates are key to many physiological processes including cellular proliferation, gene expression, ion channel modulation, and actin reorganization (Droge, 2002). Only recently members of the NADPH oxidoreductase family have emerged as primary sources of physiological redox signaling and pathological oxidative stress. The classic NADPH oxidase of phagocytes represents the prototypic member of this family defined by the large membrane subunit gp91phox (new nomenclature NOX2) (Lambeth, 2002). NOX isoforms have been identified in various non-phagocytic cell types including hematopoietic stem cells, endothelial cells, epithelial cells, myocardial cells, muscle cells, hepatocytes, and neurons (Bedard and Krause, 2007). NOX activities are multi-subunit protein complexes composed of two membrane-bound subunits (gp91phox and p22phox), and at least three cytosolic subunits (p67phox, p47phox, and p40phox) or their homologues. Most NOX isoforms (NOX1-4) require Rac1 or Rac2 as a key regulator for functional assembly of cytosolic subunits. In this study, we demonstrated that TNFα and Il-1β stimulate a transient and redox-dependent reorganization of the actin cytoskeleton into lamellipodia in neuronal cells under the regulation of Rac1. Persistent increases in ROS, likely superoxide generated by a NOX2 activity, are necessary for actin filament reorganization into lamellipodia yet also results in carbonylation of actin, an irreversible and damaging oxidative modification, accompanied by a loss of lamellipodia formation. These findings suggest that inflammation-mediated disruption of actin filament reorganization in neuronal cells through oxidative damage could represent a key step in CNS neurodegeneration.
RESULTS
TNFα and Il-1β induce a transient reorganization of the neuronal actin cytoskeleton
Fibroblasts, endothelial cells, and smooth muscle cells all respond to the proinflammatory cytokines TNFα and IL-1β with a significant reorganization of the actin cytoskeleton into lamellipodia and filopodia (Wojciak-Stothard et al., 1998; Peppelenbosch et al., 1999; Hanna et al., 2001). Expanding on these findings, we examined the effects of TNFα and IL-1β on the actin cytoskeleton of SH-SY5Y human neuroblastoma cells since neuronal plasticity in the intact, developing, and regenerating CNS is almost universally linked to dynamics changes of the actin cytoskeleton (Luo, 2002).
Serum-starved SH-SY5Y cells displayed extensive lamellipodia formation and membrane ruffling accompanied by cell spreading upon acute exposure to TNFα or Il-1β (200 ng/ml each, 10 μl/500 μl medium) for 15 min, whereas exposure to an equal volume of PBS had not effect and SH-SY5Y cells retained a condensed, atrophic morphology (Fig. 1A–C). As a quantitative measure, we defined actively responding cells as cells exhibiting two or more distinct regions with lamellipodia and/or membrane ruffling. According to this criterion, we measured 80±4% (*p<0.0001, n=180) in response to 200 ng/ml TNFα and 83±4% (*p<0.0001, n=180) active cells in response to 200 ng/ml Il-1β, respectively, in stark contrast to 15±4% (n=180) active cells under control conditions. Both proinflammatory cytokines elicited a dose-dependent increase in the percentage of SH-SY5Y cells responding with lamellipodia formation and membrane ruffling (Table I). Remarkably, the reorganization of the neuronal actin cytokeleton ceased after prolonged exposure to either proinflammatory cytokine and SH-SY5Y cells reverted largely to their condensed, atrophic morphology typical for control conditions (Fig. 1D–G). After a 30 min presence of TNFα or Il-1β, the percentage of active SH-SY5Y cells was indistinguishable from control conditions (Table I). Taken together, these findings revealed that the proinflammatory cytokines TNFα and Il-1β elicited a dramatic yet transient reorganization of the neuronal actin cytokeleton into lamellipodia and membrane ruffles.
Figure 1. Proinflammatory cytokines stimulate a transient reorganization of the neuronal cytoskeleton.

Serum-starved SH-SY5Y human neuroblastoma cells grown on collagen were incubated with proinflammatory cytokines (200 ng/ml, 10 μl/500 μl medium) or PBS (10 μl/500 μl medium) either for 15 min (A–C) or 30 min (D–F) and immediately thereafter fixed with fixed paraformaldehyde fixation. Actin filaments were labeled with rhodamine phalloidin followed by confocal imaging. (A and D) Under control conditions (PBS), SH-SY5Y cells exhibited an atrophic, condensed morphology with virtually no lamellipodia or membrane ruffles, which persisted over time. (B) In contrast, exposure to 200 ng/ml TNFα for 15 min resulted in lamellipodia formation, membrane ruffling, and cell spreading (arrowheads). (C) Similarly, 200 ng/ml Il-1β for 15 min induced lamellipodia formation and membrane ruffling (arrowheads). (E) However after a 30 min in the presence of 200 ng/ml TNFα, SH-SY5Y cells resumed their atrophic, condensed morphology devoid of lamellipodia and membrane ruffles. The extent of lamellipodia formation and cell spreading was indistinguishable from controls. (F) SH-SY5Y cells also reverted to their original atrophic, condensed morphology after 30 min in the presence of or 200 ng/ml Il-1β. (Scale bar = 10 μm)
Table I.
TNFα and Il-1β elicit a transient formation of lamellipodia and membrane ruffles.
| Exposure | % active cells | |
|---|---|---|
| 15 min | 30 min | |
| PBS | 15±4 % | 12±4% |
| 20 ng/ml TNFα | 43±4 % * | 7±4 % |
| 50 ng/ml TNFα | 75±5 % * | 3±4 % |
| 200 ng/ml TNFα | 80±4 % * | 10±3 % |
| 20 ng/ml Il-1β | 48±3 % * | 8±4 % |
| 50 ng/ml Il-1β | 53±3 % * | 10±4 % |
| 200 ng/ml Il-1β | 83±3 % * | 7±4 % |
A total of at least 180 randomly chosen SH-SY5Y human neuroblastoma cells per condition were analyzed from at least three independent experiments. Cultures were scored for active cells according to our criterion; SH-SY5Y cells displaying at least two distinct regions with lamellipodia formation and/or membrane ruffling. Cytokines (200 ng/ml) and PBS were added to serum-free culture medium as volumes accounting for 2% or less of the total culture medium.
Increase in the percentage of cells responding with formation of lamellipodia and membrane ruffling that is significantly different from control (p<0.0001).
Cytokine-mediated reorganization of the neuronal actin cytoskeleton requires redox intermediates
Reactive oxygen species (ROS) have emerged as key signaling intermediates in many vital cellular processes including mitogenesis, gene expression, stress responses, ion channel function as well as adhesion and motility (Kourie, 1998; Finkel and Sullivan, 2001; Droge, 2002; Lambeth, 2004). Several reports establish a link between cytokines, ROS formation and actin filament reorganization in non-neuronal cells (Sulciner et al., 1996; Sundaresan et al., 1996; Bonizzi et al., 1999; Deshpande et al., 2000; Moldovan et al., 2000). Therefore, we explored whether redox intermediates are implicated in the transient reorganization of the actin cytoskeleton in SH-SY5Y cells in response to proinflammatory cytokines.
Both disruption of ROS formation with Diphenylene iodonium (DPI, an NADPH oxidoreductase inhibitor) or ROS scavenging with Manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP, a catalytic antioxidant and superoxide dismutase mimetic) abolished transient lamellipodia formation and membrane ruffling in response to TNFα or Il-1β (Fig. 2). As shown in Figure 2B, pretreatment of SH-SY5Y cells with 10 μM DPI for 30 min diminished the percentage of cells with lamellipodia and membrane ruffling upon acute addition of TNFα or Il-1β (200 ng/ml, 15 min) to control levels (13±4%, n=180) and 17±3%, n=180, respectively) compared to no DPI-pretreatment (65±5%, *p<0.001, n=180 and 57±4%, *p<0.001, n=180, respectively). DPI-treated SH-SY5Y cells exhibited the condensed morphology characteristic of control cells regardless of a presence of absence of cytokines (Fig. 2A). Basal levels of lamellipodia formation and membrane ruffling were not affected by DPI pretreatment. Similarly, MnTBAP-pretreatment of serum-starved SH-SY5Y cells (40 μM, 1 h) diminished lamellipodia formation and membrane ruffling upon acute addition of TNFα or Il-1β (15 min, 200 ng/ml) (Fig. 2A). As shown in Figure 2C, the extent of cytoskeletal reorganization in the presence of MnTBAP was substantially less (TNFα: 42±7%, **p<0.01, n = 180 and Il-1β: 40±7%, **p<0.01, n=180, respectively) compared to an absence of MnTBAP-pretreatment (TNFα: 85±7%, *p<0.01, n=180; and Il-1β: 80±7%, *p<0.01, n=180). Incubation of serum-starved SH-SY5Y cells with MnTBAP alone resulted in 17±7% active cells (n=180) identical to control conditions (28±7% active cells, n=180). Notably, SH-SY5Y cells in the presence of MnTBAP and to a lesser extend with DPI displayed considerable pseudopodia-like structures, which were not accounted for in our criterion. Taken together, a disruption of ROS formation (DPI) or scavenging of ROS (MnTBAP) abolished the transient reorganization of the neuronal actin cytoskeleton into lamellipodia in response to TNFα and Il-1β implying a role for ROS as signaling intermediates.
Figure 2. Proinflammatory cytokines mediate a redox-dependent reorganization of the neuronal actin cytoskeleton.

Serum-starved SH-SY5Y human neuroblastoma cells grown on collagen were incubated either with 10 μM DPI (an NADPH oxidoreductase inhibitor), or 40 μM MnTBAP (a catalytic superoxide dismutase mimetic) prior to acute cytokine exposure (200 ng/ml, 15 min). Following paraformaldehyde fixation, cultures were stained with rhodamine phalloidin and actin filament organization visualized by confocal microscopy. (A) Pretreatment with DPI (upper panel) or MnTBAP (lower panel) prevented transient rearrangement of the actin cyoskeleton of SH-SY5Y cells upon cytokine exposure. SH-SY5Y displayed a condensed morphology in the presence of DPI (upper panel, left). However, DPI pretreatment of SH-SY5Y cells negated lamellipodia formation, membrane ruffling, and cell spreading upon exposure to TNFα or Il-1β, respectively (upper panel, middle and right). SH-SY5Y cells also retained an atrophic morphology following preincubation with MnTBAP (lower panel, left). MnTBAP largely suppressed lamellipodia formation and membrane ruffling (arrowheads) upon addition of TNFα or Il-1β. Note, pseudopodia-like structures were observed in the presence of cytokines particularly in MnBAP-pretreated cultures. (Scale bar = 10 μm). (B) Quantitative analysis (see criterion in Experimental Methods) revealed that DPI-pretreatment suppressed lamellipodia formation and membrane ruffling upon acute exposure to TNFα or Il-1β (grey bars) compared to an absence of DPI (black bars, *p<0.001) to levels indistinguishable from controls (open bars). (C) Pretreatment with MnTBAP greatly reduced lamellipodia formation and membrane ruffling upon addition of cytokines (grey bars, **p<0.01 compared to control) as opposed to an absence of MnTBAP (black bars, *p<0.01). All values were normalized to controls and represent the mean of at least three independent experiments ± standard deviations n=180).
Rac1 mediates the redox-dependent reorganization of the neuronal actin cytoskeleton in response to TNFα and IL-1β
Member of the Rho family of small GTPases serve a pivotal function in the translation of extrinsic stimuli into a coordinated reorganization of the actin cytoskeleton with Rac1 activity tied to lamellipodia formation (Hall and Nobes, 2000a; Luo, 2000). Rac1 also serves as a key regulator of NADPH oxidase activities in non-phagocytic cell types stimulated by cytokines, hormones, and mitogenes (Freeman et al., 1996; Sulciner et al., 1996; Sundaresan et al., 1996). To assess whether Rac1 is implicated in the redox-dependent formation of lamellipodia in SH-SY5Y cells in response to cytokines, we transiently expressed dominant negative Rac1 (N17Rac1) or constitutively active Rac1 (V12Rac1) in serum-starved SH-SY5Y cells both Rac1 mutants carrying an N-terminal FLAG tag (> 70% transfection efficiency). N17Rac1-expressing SH-SY5Y cells exhibited a condensed, atrophic morphology with virtually no lamellipodia or membrane ruffling irrespective of a presence of TNFα or Il-1β (Fig. 3A). N17Rac1 was predominantly localized to the cell periphery. We focused our quantitative on FLAG-immunoreactive SH-SY5Y cells As shown in Figure 3B, N17Rac1 expression suppressed lamellipodia formation and membrane ruffling in SH-SY5Y cells upon acute exposure (200 ng/ml, 15 min) to either proinflammatory cytokine (TNFα: 40±7%, n=180 and Il-1β: 19±7%, n=180) compared to non-transfected SH-SY5Y cells (TNFα: 90±7% active cells, *p<0.01, n=180 and Il-1β: 70±7% active cells, *p<0.01, n=180, respectively). In the absence of cytokines, N17Rac1-transfected SH-SY5Y cultures contained 27±7% (n=180) active cells identical to 23±7% (n=180) active cells under control condition (non-transfected, PBS). FLAG immunoreactivity was readily detectable by immunocytochemistry as well as Western blotting (1 d postinfection) and colocalized with anti-Rac1 immunoreactivity in contrast to non-transfected cells (Fig. 3A and C).
Figure 3. Rac1 mediates cytokine-induced reorganization of the neuronal actin cytoskeleton.

N17Rac1 or V12Rac1 containing a N-terminal FLAG tag were expressed (CMV promoter) in serum-starved SH-SY5Y human neuroblastoma cells (collagen) using lipofectamine transfection (>70% transfection). (A) One day post-transfection, N17Rac1-expressing SH-SY5Y cells or non-transfected cells were exposed to 200 ng/ml TNFα or 200 ng/ml Il-1β for 15 min. After paraformaldehyde fixation, cultures were stained to reveal actin filament organization (upper panel, rhodamine phalloidin), and N17Rac1 expression (middle panel, anti-FLAG fluorescein), and visualized by confocal microscopy. Images were overlaid to extract colocalization of actin filaments and N17Rac1 (lower panel, yellow). Expression of N17Rac1 in SH-SY5Y cells strongly suppressed actin reorganization into lamellipodia or membrane ruffles upon acute exposure to either proinflammatory cytokines with cell morphologies indistinguishable from controls. N17Rac1-expressing SH-SY5Y cells exhibited a highly condensed, atrophic morphology in the absence of cytokines with N17Rac1 predominantly localized to the cell periphery (middle panel). (Scale bar = 10 μm). (B) Quantitative analysis was performed on FLAG-immunoreactive cells scoring for positive responding cells according to our criterion (see Experimental Methods). Expression of N17Rac1 abolished lamellipodia formation and membrane ruffling in cultures exposed to TNFα or Il-1β (grey bars) to basal levels (open bars) compared to non-transfected cultures (black bars, *p<0.01). All values were normalized to controls and data represent the mean of at least three independent experiments ± standard deviations (n=180). (C) One day post-transfection, SH-SY5Y cultures were solubilized and equal amounts of soluble protein subjected to SDS gel electrophoresis followed by westernblotting onto PVDF membranes. Immunoreactivity against FLAG was present in cultures infected with FLAG- V12Rac1 or FLAG- N17Rac1 (*) but not in non-transfected cultures. Anti-Rac1 immunoreactivity co-localized with FLAG immunoreactivity.
Since depletion of Rac1 activity (N17Rac1) as well as ROS scavenging or disruption of ROS formation abolished cytokine-mediated reorganization of the neuronal actin cytoskeleton, we tested whether Rac1 is implicated in the generation of ROS intermediates. As shown in Figure 4, expression of V12Rac increased lamellipodia formation and membrane ruffling accompanied by cell spreading in accordance with previous findings in many cell types. Yet, this function of Rac1 was exclusively dependent on redox intermediates. Whereas V12Rac-transfect SH-SY5Y cultures contained 93±7% active cells (*p<0.01, n=180), non-transfected SH-SY5Y cultures exhibited only 17±7% active cells (n=180). Inhibiting ROS formation in V12Rac-expressing SH-SY5Y cells with DPI (10 μM, 30 min) completely abolished lamellipodia formation and membrane ruffling (27±7% active cells, n=180). DPI-treated SH-SY5Y cells expressing V12Rac1 displayed an atrophic morphology typical for non-transfected SH-SY5Y cells yet in stark contrast to V12Rac1-expressing SH-SY5Y cells in the absence of DPI. Moreover, scavenging ROS intermediates with MnTBAP (40 μM, 1 h) in V12Rac1-expressing SH-SY5Y cells also negated lamellipodia formation and membrane ruffling (37±7% active cells, n=180) with cell morphologies characteristic for non-transfected cells. Taken together, these finding strongly suggest that Rac1 mediates the generation of ROS thus constituting a redox signaling pathway involved in the reorganization of the neuronal cytoskeleton in response to cytokines.
Figure 4. Rac1-dependent actin filament reorganization requires redox intermediates.

SH-SY5Y human neuroblastoma cells grown on collagen were transfected (lipofectamine) with FLAG-V12Rac1. One day post-transfection, V12Rac1-expressing SH-SY5Y cultures and non-transfected cultures were treated with 10 μM DPI (30 min) or 40 μM MnTBAP (1 h) and then fixed with paraformaldehyde. Cultures were then stained for actin filaments (rhodamine phalloidin, upper panel) FLAG-V12Rac1 (anti FLAG/fluorescein, middle panel). Representative confocal images of SH-SY5Y cells were acquired and image overlaid revealed colocalization of actin filaments and V12Rac1 expression (lower panel, yellow). Expression of V12Rac1 stimulated dramatic lamellipodia formation and spreading of SH-SY5Y cells. In contrast, both incubation with DPI or MnTBAP negated lamellipodia formation and cell spreading with cell morphologies similar to non-transfected cells. V12Rac1 was predominantly localized to the cell periphery. (Scale bar = 10 μm)
TNFα stimulates a sustained formation of ROS in neuronal cells
TNFα and Il-1β among other physiological stimuli, provoke the generation of ROS in several non-neuronal cell types others than phagocytes including fibroblast, endothelial cells, and hepatocytes (Sundaresan et al., 1996; Goossens et al., 1999; Chandel et al., 2001; Meier, 2001). We analyzed ROS formation in serum-starved SH-SY5Y human neuroblastoma cells utilizing the fluorescence indicators 2′,7′-dihydrodichlorofluorescein (H2DCF) and dihydroethidum (DHE), which both increase in fluorescence upon oxidation. Whereas H2DCF is oxidized to dichlorofluorescein (DCF) by H2O2, DHE is oxidized to ethidium (Eth) exclusively by superoxide. We found that TNFα exposure (100 ng/ml, 30 min) stimulated a sustained ROS formation in SH-SY5Y cells sensitive to both ROS scavenging and inhibition of NOX activity (Fig. 5A and B). Quantitative analysis demonstrated that TNFα caused a significant increase in relative DCF-fluorescence intensity (1.34±0.17, *p<0.05, n=5) in SH-SY5Y cells 7.5 minutes after addition that persisted for up to 60 min compared to control (0.95±0.08, n=3) indicative of ROS formation (Fig. 5A). TNFα-induced ROS formation was negated by a presence of 5 mM N-acetyl-L-cysteine (NAC; 0.86±0.04, n=3), or 5000 U/ml exogenous catalase (CAT; 1.14±0.10, n=3) but by exogenous superoxide dismutase (200 U/ml SOD; 1.20±0.08, n=3) (Fig. 5A). As expected, a presence of 10 μM DPI (0.97±0.06, n=4) blocked TNFα-mediated ROS formation (Fig. 5B). To further implicate a NOX activity in the cytokine-mediated formation of ROS, we targeted the lipid messenger arachidonic acid (AA), a potent stimulator of NOX activity. AA is generate by cytosolic phospholipase A2 (cPLA2), which is inhibited by arachidonyl trifluoromethylketone (ATK). Pretreatment of SH-SY5Y cells with 10 μM ATK completely prevented ROS formation (0.92±0.03, n=3). To verify the production of superoxide, the primary type of ROS generated by NOX activities, we included ethidium in our study, a superoxide-specific fluorescence indicator. TNFα (100 ng/ml, 15 min) also increased relative Eth-fluorescence intensity (1.53±0.09, *p<0.01, n=3) in SH-SY5Y cells indicative of superoxide production compared to control (1.00±0.1, n=5), which was abolished by a presence of 5 mM NAC (1.06±0.09, n=3), 10 μM DPI (1.05±0.08, n=3), or 10 μM ATK (0.90±0.06, n=3), respectively (Fig. 5C). Finally in SH-SY5Y cells loaded with both oxidation sensitive fluorescence indicators, TNFα primarily caused an increase in relative Eth-fluorescence intensity (1.47±0.07, *p<0.01, n=3) but not in relative DCF-fluorescence intensity (1.06±0.21, n=3) compared to their respective controls (Eth: 1.00±0.13, n=6 and DCF: 1.00±0.09, n=6) (Fig. 5D). As expected, TNFα-induced increases in relative Eth-fluorescence intensity (double loaded cells) were negated with 5 mM NAC (Eth: 1.00±0.08, n=3 and DCF: 0.63±0.09, n=3), 10 μM DPI (Eth: 0.92±0.03, n=3 and DCF: 0.95±0.27, n=3), or 10 μM ATK (Eth: 1.03±0.20, n=3 and DCF: 0.95±0.07, n=3). Taken together, these findings demonstrated that TNFα stimulated a sustained formation of ROS in SH-SY5Y human neuroblastoma cells. The sensitivity of ROS formation to DPI (NADPH oxidoreductase inhibitor) and ATK (cPLA2 inhibitor) together with the preferential oxidation of DHE even in the presence of H2DCF strongly implied a superoxide-generating mechanism; presumably a neuronal NOX activity
Figure 5. Proinflammatory cytokines elicit superoxide formation in SH-SY5Y neuroblastoma cells.
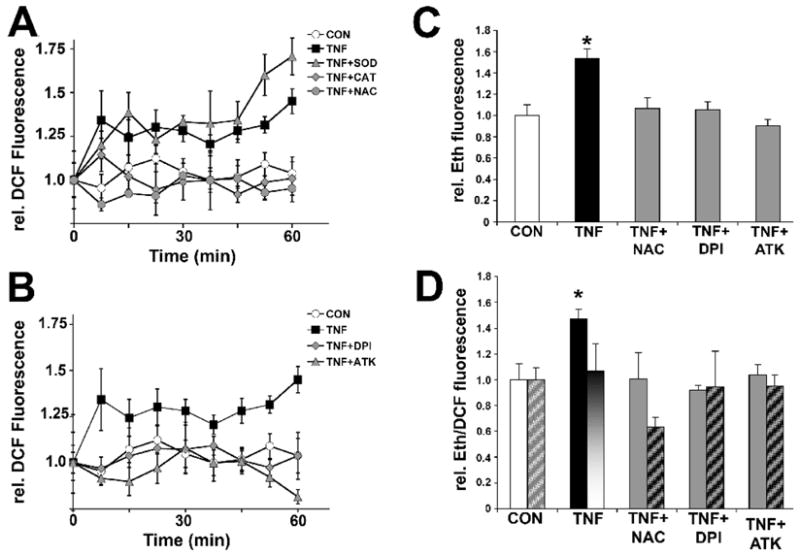
SH-SY5Y human neuroblastoma cells (1×105 cells) were loaded for 1h either with 50 μM 2′,7′-dihydrodichlorofluorescein diacetate (H2DCFDA) (A and B), 50 μM Dihydroethidium (DHE) (C), or 160 μM DHE together with 10 μM H2DCFDA (D) in the presence or absence of pharmacological inhibitors or antioxidant enzymes. Whereas H2DCFDA is preferentially oxidized to DCF by peroxide, DHE is exclusively oxidized to ethidium (Eth) by superoxide. Cultures were exposed to 100 ng/ml TNFα (15 min unless indicated otherwise) and maximum Eth or DCF-fluorescence intensity was quantified in whole cell lysates with all measurements normalized to the average maximum fluorescence intensity under control conditions (relative Eth or DCF-fluorescence values, respectively). (A) TNFα stimulated ROS formation in SH-SY5Y cells (filled squares) within 7.5 min upon exposure lasting up to 60 min indicated by significant increases in relative DCF fluorescence intensities (*p<0.05, n=4 for each time point) compared to control (open circles). TNFα-stimulated ROS formation was blocked when preincubating cultures either with 5 mM NAC (grey circles) or 5,000 U/ml catalase (grey diamonds). In contrast, 200 U/ml superoxide dismutase (grey triangles) was ineffective. (B) Pretreatment of SH-SY5Y cells with 10 μM DPI (grey diamonds), a NOX inhibitor, or 10 μM ATK (grey triangles), a cPLA2 inhibitor, both completely abolished TNFα-stimulated ROS formation. Arachidonic acid generated by cPLA2 is a potent activator of NOX activity. Traces for TNFα only (filled squares) and control (open circles) were repeated as shown in A. (C) TNFα-exposure of SH-SY5Y cells loaded with DHE resulted in a significant increase in relative maximum Eth-fluorescence (filled bar), which was negated by a presence of 5 mM NAC, 10 μM DPI, or 10 μM ATK (grey bars, respectively) compared to control (open bar). (D) TNFα stimulated a rise in relative maximum Eth-fluorescence (filled bars) even in the presence of H2DCF (hatched bars) indicative of a superoxide-generating source. 5 mM NAC, 10 μM DPI, or 10 μM ATK all negated TNFα-induced superoxide formation. All values were normalized to control and data represent the mean of at least three independent experiments ± standard deviations (*p<0.05).
SH-SY5Y human neuroblastoma cells express a functional NOX activity
The classic NADPH oxidase in phagocytes is a multi-subunit complex composed of two membrane-bound subunits (gp91phox or NOX2, and p22phox), and at least three cytosolic subunits (p67phox, p47phox, and p40phox). Only recently, NOX2 homologues were identified in many non-phagocyte cell types including SH-SY5Y cells (Nikolova et al., 2005; Bedard and Krause, 2007). Using human subunit-specific antibodies, we found immunoreactivity against all subunits of the NOX2 complex in whole cell lysates of human SH-SY5Y neuroblastoma cells with apparent molecular weights of 65 kDa for NOX2, 20 kDa for p22phox, 65 kDa for p67phox, 45 kDa for p47phox, and 40 kDa for p40phox (Fig 6A). Double bands were consistently detected for both p67phox and p47phox. In phagocytes, phorbol 12-myristate 10-acetate (PMA) and arachidonic acid (AA) potently stimulate NOX2 activity reflected by a translocation of cytosolic subunits such as p67phox to the plasma membrane. In analogy, treatment of SH-SY5Y cells with 200 nM PMA and 50 μM AA caused a time-dependent translocation of the cytosolic subunit p67phox to the plasma membrane (Fig. 6B). More importantly, TNFα (100 ng/ml, 15 min) also induced a significant translocation of p67phox to the plasma membrane (1.48 ± 0.09, *p<0.05, n=3) in SH-SY5Y cells compared to control (1.01 ± 0.02, n=3), which was completely negated by the cPLA2 inhibitor ATK (10 μM; 1.13 ± 0.03, n=3) (Fig. 6C). Similarly, phosphorylation of the cytosolic subunit p40phox (1.65 ± 0.21, *p<0.05, n=3) significantly increased compared to control (1.02 ± 0.08, n=3) upon TNFα stimulation (100 ng/ml, 15 min) (Fig. 6D). Taken together, these findings provided evidence that human SH-SY5Y neuroblastoma cells and E7 cortical neurons both express a functional NOX2 complex.
Figure 6. SH-SY5Y human neuroblastoma cells express a functional NADPH oxidase activity.

(A) Whole cell lysates of human SH-SY5Y neuroblastoma cells revealed immunoreactivity against both membrane-bound subunits (gp91phox, p22phox) and the cytosolic subunits (p67phox, p47phox, and p40phox) of the NOX2 complex using anti-human subunit specific antibodies (loading-control: actin). Double bands were routinely detected for both p67phox and p47phox. (B) Treatment of SH-SY5Y cells over a 60 min time course with 200 nM PMA and 50 μM AA stimulated translocation of p67phox into plasma membrane (arrowhead). Equal amounts of total plasma membrane protein were separated by SDS-gel electrophoresis followed by western blotting against p67phox. (C) SH-SY5Y cells were incubated with 100 ng/ml TNFα for 15 min. Plasma membrane proteins were biotinylated with Sulfo-NHS-biotin and affinity-purified on streptavidin-agarose. Equal amounts of membrane protein were separated by SDS-gel electrophoresis followed by western blotting, and immunoreactivity against p67phox was quantified (chemiluminescence). TNFα elicited a significant translocation of p67phox to plasma membranes (black bar, TNFα) demonstrating NOX activity, which was blunted by 10 μM ATK (grey bar, TNF+ATK) to levels similar of control (open bar, CON). (D) SH-SY5Y cells were incubated with TNFα (100 ng/ml, 15 min) and then lysed. Whole cell lysates were subjected to gel electrophoresis followed by western blotting and quantification of phospho-p40phox immunoreactivity (chemiluminescence). TNFα significantly increased levels of phospho-p40phox (black bar, TNFα) in SH-SY5Y cells compared to control (open bars, CON) indicative of NOX activation. All values were normalized to control and data represent the mean of three independent experiments ± standard deviations (*p<0.05)
TNFα elicits oxidative damage to the neuronal actin cytoskeleton
Actin both in its monomeric and filamentous state is highly susceptible to oxidative damage, which ultimately compromises proper dynamics of the actin cytoskeleton (Dalle-Donne et al., 2001; Beal, 2002). Oxidative damage of actin adversely affects many pivotal cellular processes including cellular plasticity, gene expression, mitochondrial viability, vesicle trafficking, and ion channel function (Kheradmand et al., 1998; Kourie, 1998; Moldovan et al., 2000; Lassing et al., 2007). In fact, actin oxidation is prevalent in neurodegeneration associated with many chronic CNS disorders and acute CNS trauma (Aksenov et al., 2001; Leski et al., 2001; Beal, 2002). We examined whether TNFα might cause oxidative damage to the neuronal actin cytoskeleton using carbonylation as a marker of oxidative stress (Dalle-Donne et al., 2003b). Exposure of purified rabbit actin to peroxide in vitro caused substantial carbonylation as revealed by Dinitrophenyl (DNP) hydrazone modification and detection of DNP-immunoreactivity by westernblotting (Fig. 7A). Interestingly, we detected considerable actin carbonylation in SH-SY5Y cells acutely exposed to TNFα or Il-1β (100 ng/ml, 1 h) analyzing whole cell lysates by 2D gel electrophoresis and westernblotting compared to our negative control (buffer) or positive control (100 μM H2O2) (Fig. 7B). It is noteworthy that a residual actin oxidation was detectable in cell extracts from control cultures. To quantify the extent of carbonylation, total cellular actin was immunoprecipitated from SH-SY5Y cell extracts, subjected to DNPH modification, and then analyzed by SDS gel electrophoresis followed by westernblotting and detection of anti-DNP immunoreactivity (chemiluminescence). Exposure of serum-starved SH-SY5Y cells to TNFα (100 ng/ml, 60 min) resulted in a dramatic increase of relative actin carbonylation (1.24 ± 0.02, *p<0.05, n=3) compared to control (0.99 ± 0.02, n=3), which was completely negated to control levels after pretreatment of SH-SY5Y cells with 10 μM DPI (1.09 ± 0.01, n=3) or 10 μM ATK (0.98 ± 0.01, n=3) (Fig. 7C). Neither pharmacological treatment altered actin carbonylation in the absence of TNFα and residual carbonylation was indistinguishable from control conditions (data not shown). These findings provide evidence that prolonged TNFα exposure inflicts irreversible oxidative damage (carbonylation) to the actin cytoskeleton by stimulating a NOX2-like activity in neuronal cells.
Figure 7. Proinflammatory cytokines inflict carbonylation of actin in neuronal cells.
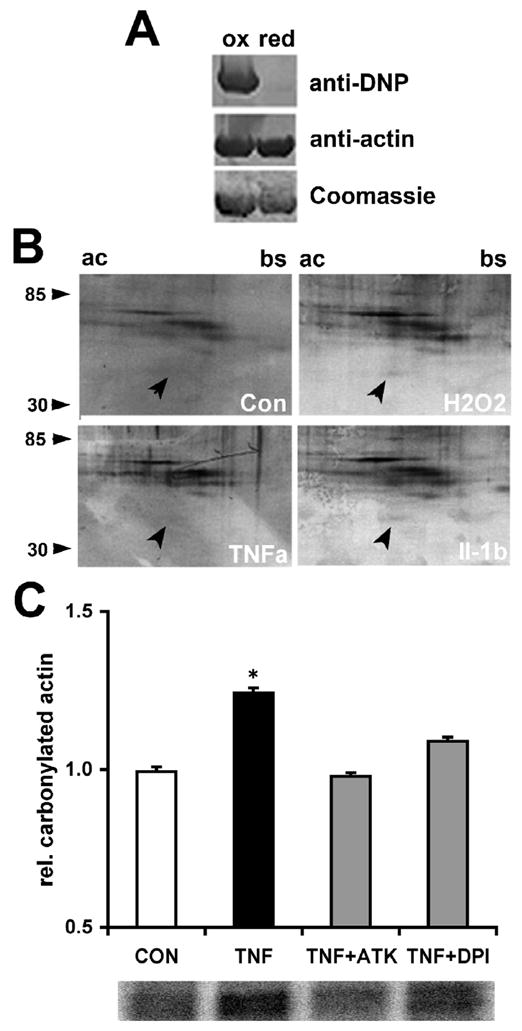
(A) Purified rabbit actin was exposed in vitro to 100 μM H2O2 (ox) or buffer (red) for 15 min. Actin was recovered by precipitation and carbonyl residues were modified with Dinitrophenyl (DNP) hydrazone. Equal amounts of oxidized or reduced actin (5 μg per lane) were subjected to 10% SDS gel electrophoresis followed by westernblotting and immunodetection of carbonyls (anti-DNP) or actin (anti-actin). Oxidation of actin with H2O2 caused substantial carbonylation. (B) SH-SY5Y cultures were exposed to TNFα (100 ng/ml, 1h), Il-1β (100 ng/ml, 1h), 100 μM H2O2 (30 min), or buffer. A soluble protein extract was obtained and carbonyls modified with DNP hydrazone. Protein samples (100 μg) were subjected to 2D gel electrophoresis using a pH gradient 3–10 for isoelectric focusing (ac=acidic, bs=basic) and 10% acrylamide gels for the second dimension (molecular weight range indicated). DNP immunoreactivity revealed carbonylated actin (arrowhead) in cultures exposed to TNFα, Il-1β, or H2O2 whereas only residual actin carbonylation was detectable in control cultures. (C) SH-SY5Y cells were incubated with 10 μM DPI or 10 μM ATK (60 min) prior to TNFα exposure (100 ng/ml, 60 min) followed by immunoprecipitation of total actin from whole cell lysates and derivatization of protein carbonyls with DNP hydrazone. Protein samples were subjected to SDS-gel electrophoresis followed by western blotting and quantification of DNP immunoreactivity (chemiluminescence). TNFα induced a significant increase in carbonylation of total actin (black bar) compared to control (white bar). Note, either DPI or ATK diminished actin carbonylation (grey bars) to control levels despite a presence of TNFα implying NOX as the major source of damaging ROS. All values were normalized to controls and represent the mean of three independent experiments ± standard deviations (*p<0.05).
DISCUSSION
The proinflammatory cytokines TNFα and Il-1β exert pleiotropic effects in the developing, adult, and injured CNS yet persistent, high levels of expression are neurotoxic due to oxidative stress and accumulation of toxic lipid second messengers (Mrak and Griffin, 2005; Lucas et al., 2006). Our findings revealed that TNFα and Il-1β provoked three principal responses in neuronal cells: (1) a transient, redox-dependent actin filament reorganization into lamellipodia and membrane ruffling under the regulation of Rac1, (2) a sustained ROS production through a neuronal NOX2 activity, and (3) irreversible, oxidative damage (carbonylation) to the neuronal actin cytoskeleton accompanied by loss of lamellipodia and membrane ruffling. Indeed, actin carbonylation is prevalent after acute CNS injuries, in many chronic CNS disorders, as well as in normal CNS aging (Beal, 2002; Floyd and Hensley, 2002; Dalle-Donne et al., 2003a). Conceivably, neurotoxicity of inflammation could arise in part from actin oxidation since disruption of actin filament dynamics would disable many vital cellular processes including plasticity, mitochondrial function, vesicle traffic, gene expression, and ion channel function (Lin et al., 1997; Kourie, 1998; Luo, 2002; Gourlay and Ayscough, 2005; Percipalle and Visa, 2006).
SH-SY5Y neuroblastoma cells grown on collagen displayed, after serum starvation, an atrophic, condensed morphology with virtually no lamellipodia, membrane ruffling, or pseudopodia. Upon acute exposure to cytokines, SH-SY5Y cells displayed a dramatic increase in cellular plasticity reflected by extensive lamellipodia formation and membrane ruffling followed by substantial cell spreading. However after 30 min, SH-SY5Y cells reverted back to their original morphology with mostly peripheral actin stress fibers. Interestingly, SH-SY5Y cells were no longer responsive to cytokine exposure even after a prolonged recovery phase (data not shown) suggesting a long lasting, irreversible effect on the actin cytoskeleton. Reorganization of actin filaments was demonstrated in several non-neuronal cell lines exposed to TNFα or Il-1β and consisted predominantly of lamellipodia formation followed by stress fiber formation (Wojciak-Stothard et al., 1998; Peppelenbosch et al., 1999). In regard to neuronal cells, TNFα reduces neurite branching, inhibits neurite outgrowth, and causes even neurite retraction in cultures of hippocampal neurons, reflecting a disruption of proper actin cytoskeleton reorganization (Neumann et al., 2002; Munch et al., 2003). It is well established that Rho GTPases (Cdc42, Rac1, and RhoA) are implicated in converting extrinsic stimuli into a coordinated reorganization of actin filament often in a hierarchical fashion (Nobes and Hall, 1995; Hall and Nobes, 2000b; Luo, 2000). Not surprisingly, depletion of Rac1 activity in SH-SY5Y cells by expressing N17Rac1 abolished lamellipodia formation and membrane ruffling in response to cytokines and SH-SY5Y cells retained their typical atrophic morphology in accordance with previous findings in non-neuronal cells. N17Rac1-expressing SH-SY5Y cells exhibited a more condensed morphology compared to control cells (qualitative observation). In fact, Rho GTPases do mediate cytoskeletal rearrangement induced by TNFα or Il-1β in various cellular systems (Peppelenbosch et al., 1999; Puls et al., 1999). In several non-neuronal cells, TNFα and Il-1β but also growth factors were shown to induce a Rac1-dependent ROS formation (Sulciner et al., 1996; Sundaresan et al., 1996; Bonizzi et al., 1999; Deshpande et al., 2000). And Rac1 and RhoA seem to have opposing roles in ROS generation in endothelial cells (Wojciak-Stothard et al., 2005). A connection was revealed among Rac1 activation, ROS formation, and TNF receptor associated factor (TRAF) recruitment (Chandel et al., 2001; Li et al., 2006). Based on reports by Moldovan et al. (1999), we demonstrated that (i) redox intermediates were required for cytokine-stimulated reorganization of actin filaments in SH-SY5Y cells, and (ii) Rac1 regulated the formation of redox intermediates. Sensory neurons responded to a NGF-withdrawal with a Rac1-dependent ROS formation (Suzukawa et al., 2000). We quantified cytokine-dependent ROS formation using the peroxide-sensitive fluorescence indicator H2DCF in conjunction with the superoxide-sensitive fluorescence indicator DHE (Tarpey and Fridovich, 2001). Acute exposure of SH-SY5Y cells to TNFα caused oxidation of both indicators as revealed by increases in DCF or Eth-fluorescence. The fact that DHE was preferentially oxidized in response to cytokines even in the presence of H2DCF suggested a superoxide-generating mechanism (Rothe and Valet, 1990). Several findings further support this conclusion. The 1e− transport inhibitor DPI, generally used as an NOX inhibitor, and the cPLA2 inhibitor ATK both abolished TNFα-mediated ROS formation. Addition of exogenous SOD did not quench maximum DCF fluorescence since dismutation of superoxide to peroxide is in favor of DCF oxidation. However, addition of exogenous catalase inhibited increases in DCF fluorescence suggesting that superoxide dismutation is naturally occurring. In addition, we demonstrated the presence of a functional NOX activity in SH-SY5Y cells based on immunological and biochemical evidence. Using human-specific antibodies, we found immunoreactivity against all subunits of NOX2 in human SH-SY5Y neuroblastoma cells in agreement with previous reports (Nikolova et al., 2005). We consistently detected p67phox immunoreactivity as double bands in SH-SY5Y cells whereas the same antibody revealed a single band when testing RAW 264 cell extracts (data not shown). The NOX2 activity in our assay system was both stimulus-dependent and functional as revealed both on the level of enzyme activity and protein subunit assembly. First, NOX disruption using DPI and ATK negated TNFα-stimulated ROS formation. Second, cytokine-stimulated ROS production was strictly Rac1-dependent but, in the absence of cytokines, enhanced by expressing constitutively active Rac1. Importantly, Rac1 is a key regulator of NOX isoforms and can substitute for Rac2 in phagocytes (Hordijk, 2006). Third, SH-SY5Y cells exposed to PMA and AA, two well-documented activators of NOX enzymes, induced the translocation of the cytosolic subunit p67phox to plasma membranes (Groemping and Rittinger, 2005). Fourth, a presence of ATK abolished plasma membrane translocation of p67phox induced by TNFα. Fifth, TNFα stimulated the phosphorylation of p40phox. These findings provide strong evidence for the presence of a neuronal NADPH oxidase as a pivotal source of oxidative stress in neurodegeneration (Lambeth, 2007). Several recent studies describe the expression of members of the multi subunit enzyme NADPH oxidase (NOX) in non-neuronal as well as neuronal cells (Lambeth, 2002; Lambeth, 2004; Bedard and Krause, 2007). The NOX2 isoform was found primary cortical neurons, hippocampal neurons, and sensory neurons (Noh and Koh, 2000; Tammariello et al., 2000; Tejada-Simon et al., 2005). We demonstrated that actin represents one target of oxidative damage resulting from a cytokine-stimulated ROS formation in neuronal cells. Both cytokines increased carbonyl residues in total actin of SH-SY5Y neuroblastoma cells, which was completely abolished by inhibiting NOX activity (DPI or ATK). Interestingly, we consistently detected residual oxidation in actin under control conditions even if performed in a strongly reducing environment. This finding might suggest that oxidative modification of actin could represent a physiological regulatory mechanism. It is noteworthy that ROS formation, actin carbonylation, and irreversible arrest of actin filament reorganization strongly coincided. SH-SY5Y cells displayed a redox-dependent formation of lamellipodia, highly motile subcellular structures, within 15 min upon cytokine addition. Irreversible arrest of actin filament reorganization occurred only after an exposure time of 30 min or longer. TNFα elicited ROS formation within 10 min after addition, which persisted for up to 60 min. Translocation of p69phox to plasma membranes, indicating NOX activation and thus ROS production, was found after 10 min following stimulation with PMA or TNFα. And lastly, actin carbonylation was readily detectable after incubation of SH-SY5Y cells with cytokines for over 30 min. Taken together, early increase in ROS produced by NOX2 in response to cytokines paralleled lamellipodia formation (dynamic actin filament structures) whereas sustained ROS generation (> 30 min) was accompanied by actin oxidation and arrest of motility. Presumably, limited exposure to cytokines could produce beneficial effects on neuronal motility and survival implying initially a physiological role for ROS intermediates whereas a prolonged presence of cytokines disrupts cellular plasticity via actin oxidation suggesting a pathological role for persistent high levels of ROS intermediates. Consequently, neurotoxicity of TNFα and Il-1β could be attributed, at least in part, to the oxidation of actin since (1) both cytokines elicit an irreversible reorganization of actin filaments, (2) both cytokines stimulate ROS formation, (3) actin exhibits a strong vulnerability to oxidative damage both in vivo and in vitro, (4) actin filament dynamics is essential role for many processes vital to cellular homeostasis, and (5) a presence of oxidized actin in CNS trauma and many chronic CNS disorders. A persistent inflammatory reaction is highly prevalent in most CNS pathologies whether acute, chronic, psychiatric, or even in normal aging with activated microglia and astrocytes releasing numerous neurotoxic mediators including cytokines and ROS (Mrak and Griffin, 2005; Lucas et al., 2006). Our findings provided evidence that the proinflammatory cytokines TNFα and Il-1β exert their neurotoxicity at least partially through a NOX-dependent, irreversible oxidative modification of actin.
EXPERIMENTAL METHODS
Reagents
Recombinant human tumor necrosis factor alpha (TNFα), interleukin-1β (Il-1β), a polyclonal rabbit anti-human p67phox antibody, a monoclonal mouse anti-human p40phox antibody, and a monoclonal mouse anti-FLAG antibody were purchased from Millipore (Temecula, CA). All other antibodies were obtained from Santa Cruz (Santa Cruz, CA). DMEM and Penicillin/Streptomycin were obtained from Mediatech (Herndon, VA). Glutamax, Hank’s Balanced Salt Solution (HBSS), trypsin/EDTA solution, and a polyclonal rabbit anti-DNP (dinitrophenyl) primary antibody were from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was received from Atlanta Biologicals (Atlanta, GA). Streptavidin-agarose beads, protease inhibitor cocktail (PIC), Pico Super Signal chemoluminescent kit, and a BCA protein assay kit were obtained from Pierce (Rockland, IL). Diphenylene iodonium chloride (DPI) and Manganese(III)tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP) were obtained from EMD (Gibbstown, NJ) and stock solutions (200x) of DPI or MnTBAP were prepared in DMSO or 10 mM NaOH, respectively, sterile filtered, and stored at −20°C protected from light. Rhodamine phalloidin was from Cytoskeleton Inc. (Denver, CO). Rattail collagen was from Roche Diagnostics (Indianapolis, IN). All other reagents were from Sigma (St. Louis, MO).
Cell Culture
SH-SY5Y human neuroblastoma cells were grown in 100 mm tissue culture dishes (Falcon) in medium composed of DMEM, 10% Fetal Bovine Serum (FBS), 1% Glutamax, 100 U/ml Penicillin and 100 U/ml Streptomycin (humidified atmosphere, 5% CO2, 37°C). For amplification, 80% confluent SH-SY5Y cultures were washed with HBSS incubated with 0.5 mg/ml trypsin/0.2mg/ml EDTA (5 min 37°C). Cells were rinsed off, collected by centrifugation (200×gmax), and plated at a dilution of 1:3 (100 mm dishes), 105 cells/well (96 well plates), or 2×105 per glass cover slip (confocal microscopy). Glass cover slips (22×22 mm2, #1) were mounted over a 1.5 cm diameter hole drilled into the bottom of 35 mm culture dishes. Glass cover slips were coated with 5 μg/cm2 collagen. As our acute exposure paradigm, SH-SY5Y cells were serum-starved overnight in DMEM, 1% Glutamax, 100 U/ml Penicillin and 100 U/ml Streptomycin (humidified atmosphere, 5% CO2, 37°C). Cytokines, pharmacological agents, and controls (PBS) were added to serum-free culture medium as volumes accounting for 2% or less of the total culture medium.
Plasmid Transfection
Constitutively active Rac1 (V12Rac1) or dominant negative Rac1 (N17Rac1) containing a N-terminal FLAG tag were cloned into the pShuttleCMV expression plasmid (Qbiogene, Carlsbad, CA) under a CMV promoter. SH-SY5Y cells grown on collagen-coated glass cover slips were transfected using Lipofectamine PLUS (Invitrogen, Carlsbad, CA). Briefly, plasmid DNA was digested with PmeI, treated with Calf Intestinal Phosphatase (Promega, Madison, WI), and subjected to 0.8% agarose electrophoresis. The linearized plasmid DNA was recovered and purified on glass milk (QBiogene, Carlsbad, CA). Purified, linearized plasmid DNA (0.5 μg) was resuspended in 20 μl of Opti-MEM serum-free media, mixed with 5 μl of PLUS reagent, and incubated at RT for 15 min. The transfection solution was combined with Lipofectamine (5 μl) in Opti-MEM (20 μl), incubated for 15 min at RT, and then added to SH-SY5Y cultures in 200 μl Opti-MEM. Transfection efficiencies were routinely greater than 70%. Following a 24 h expression period, cultures were treated with proinflammatory cytokines in the presence or absence of pharmacological inhibitors.
Confocal Microscopy
Human SH-SY5Y neuroblastoma cells were grown on collagen-coated glass cover slips (0.13 mm thick German glass) in serum-free medium for 24 h. Prior to acute addition of TNFα or Il-1β, serum-starved SH-SY5Y cultures were incubated with pharmacological inhibitors or transfected Rac1 mutants prior to acute exposure to 200 ng/ml TNFα or Il-1β for 15 min or 30 min unless indicated otherwise. SH-SY5Y cultures were fixed (RT, 30 min) with 4% paraformaldehyde in 10 mM MES, pH 6.1, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.5% Triton X-100, a cytoskeleton stabilizing buffer (Symons and Mitchison, 1991). After rinsing with 0.1% Triton X-100 in Tris-buffered saline (TX-TBS), cultures were incubated with rhodamine phalloidin in TX-TBS-BSA (1:10 dilution, 2% BSA, 20 min, RT). To reveal FLAG expression, SH-SY5Y cultures were incubated with a monoclonal mouse-anti-FLAG-antibody in TBS-TX-BSA (1:100, 1 h, RT) followed by incubation with a secondary FITC-conjugated goat-anti-mouse antibody (1:1000, 1 h, RT, light protected). Finally, cultures were rinsed with TX-TBS, and transferred into 60% glycerol/40% PBS. Images were acquired (40x, oil, Plan Fluor) with a Zeiss confocal microscope LSM 510 equipped with a He/Ne laser and an Argon laser using filter combinations for fluorescein and rhodamine fluorescence. Zeiss LSM Software was used for image acquisition and analysis. For each treatment condition, random fields of view were analyzed and 60 cells each from three independent sets of experiments (n=180) were scored for the presence of lamellipodia formation and/or membrane ruffling. As our criterion, SH-SY5Y cells exhibiting at least two distinct regions with extensive lamellipodia formation or membrane ruffling were considered active cells whereas pseupodia-like structures were not accounted for. Statistical significance between the means of control and treatments were calculated using Dunnett’s t-test. A Kruskal-Wallis test was employed to calculate significance between control distributions and treatment distributions.
Quantification of Reactive Oxygen Species Production
Increases in reactive oxygen species (ROS) were detected with the oxidation-sensitive fluorescence indicators 2′,7′-dihydrodichlorofluorescein diacetate (H2DCFDA) or Dihydroethidium (DHE). H2DCFDA is retained in the cytosol after deacetylation to dihydrodichlorofluorescein and increases in fluorescence upon oxidation by H2O2 to dichlorofluorescein (DCF). In contrast, DHE is oxidized to ethidium (Eth) preferentially by superoxide. SH-SY5Y cells (105 cells/well, 96-well plates, Falcon) were incubated for 1 h either with 50 μM H2DCFDA, 50 μM DHE, or a combination of both (160 μM DHE and 10 μM H2DCFDA) in the presence or absence of pharmacological inhibitors or ROS scavenging enzymes. Following a medium exchange, SH-SY5Y cells were exposed to 100 ng/ml TNFα, washed with PBS, and lysed (2 M Tris-Cl pH 8.0, 2% SDS, 10 mM Na3VO4). Maximum Eth or DCF-fluorescence intensity was quantified (100 μl cell lysate) using a Beckman Coulter Multimode DTX 880 microplate reader (Eth: 485 nm excitation filter, 610 nm emission filter and DCF: 495 nm excitation filter, 525 emission filter). All Eth or DCF-fluorescence intensity data were normalized to the average maximum Eth or DCF-fluorescence under control conditions (relative fluorescence values).
Plasma Membrane Translocation of p67phox
SH-SY5Y cells were treated with pharmacological inhibitors and then exposed either to a mixture of 200 nM PMA/50 μM arachidonic acid (AA) or 100 ng/ml TNFα. Plasma membrane fractions were enriched either by density centrifugation or biotinylation and streptavidin-affinity chromatography as described (Li and Shah, 2002). Regarding centrifugation, cell lysates were obtained by scraping SH-SY5Y cells into Buffer A (250 mM sucrose, 20 mM HEPES pH 7.4, 2 mM EDTA, 5 mM MgCl2, 1 mM dithiothreitol, 1 mM AEBSF, and 1% PIC), lysis by sonication, and collection of supernatants (1,200 × gmax, 5 min). Centrifugation (100,000 × gmax, 30 min, 4°C) of lysates separated membrane fractions (i.e. pellets) and cytosolic fractions (i.e. supernatants). Pellets were a resuspended in Buffer B (Buffer A containing 1% Triton X-100 and 0.01% saponin) and incubated on ice for 15 min. Following centrifugation (100,000 × gmax, 30 min, 4°C), membrane fractions were collected as supernatants. Regarding biotinylation, SH-SY5Y cultures (5×106 cells/well, 6-well plates) were washed with HBSS-CM (HBSS, 0.1 g/l CaCl2, 0.1 g/l MgCl2, pH 7.5), and incubated with 0.5 mg/ml Sulfo-NHS-Biotin (prepared in 20 mM HEPES, HBSS-CM, pH 8.0) for 40 min on ice. Excess Sulfo-NHS-Biotin was neutralized (on ice, 15 min) with 50 mM glycine (prepared in HBSS-CM), and cells were scraped into HBSS-CM. After centrifugation (200 × gmax, 2 min), cells were resuspended in 500 μL Buffer C (20 mM HEPES pH 7.5, HBSS, 1% Triton X-100, 0.2 mg/ml saponin, 1% PIC), sonicated, and cell lysates incubated with streptavidin-agarose beads (25 μl, 2 h, 4°C). Beads collected (2,500 × gmax, 2 min), and resuspended in 150 μL of Buffer C. After heating beads (5 min, boiling water bath), membrane proteins (2,500 × gmax, 2 min) were collected in the supernatant and total protein content was determined using a BCA protein assay.
Derivatization of protein carbonyls
Protein samples were prepared either using purified rabbit muscle actin, total protein of SH-SY5Y cell extracts, or actin immunoprecipitated from SH-SY5Y cells. SH-SY5Y cultures were exposed to 100 ng/ml TNFα (1 h), 100 ng/ml Il-1β (1 h), 100 μM H2O2 (30 min) or buffer (1 h) in serum-free culture medium following a preincubation with 10 μM DPI or 10 μM ATK (1 h), and then washed with Tris/sucrose (10 mM Tris-Cl pH 7.0, 250 mM sucrose). For 1D SDS gel electrophoresis, SH-SY5Y cultures were extracted with 2 M Tris-Cl pH 8.0, 2% SDS, 1 mM Na3VO4 (SDS) whereas for 2D gel electrophoresis, cultures were extracted with 8 M urea, 2% CHAPS, 80 μM DTT, 0.5 % Ampholytes pH 3–10, 5% glycerol, 0.001% bromophenol blue. Cell extracts were centrifuged (3,000 × gmax, 4°C, 20 min) and soluble protein subjected to carbonyl derivatization. For immunoprecipiation of actin, cell lysates (100 μl) were incubated with 1 μg normal mouse serum (45 min, 25°C, agitation), and normal IgG removed with protein A/G agarose beads (20 μl). Cleared lysates (100 μl) were mixed with 1 μg monoclonal mouse anti-human β-actin primary antibody (2 h, 4°C, agitation) followed by overnight incubation with protein A/G agarose beads (20 μl, 4°C, agitation). Protein A/G beads were collected by centrifugation (200 × gmax, 2 min), washed three times in lysis buffer, and bound proteins were released by boiling for 5 min. Carbonyl residues in protein samples were derivatized with 2,4-dinitrophenylhydrazine (DNPH) as described (Levine et al., 1994). Briefly, protein samples were mixed with an equal volume of 12% SDS, and further incubated with two sample volumes of 20 mM DNPH in 2M HCl (30 minutes, 25°C). Protein was recovered by chloroform/methanol precipitation and washed with ethylacetate/ethanol (1:1) followed by acetone (Wessel and Flugge, 1984). Protein samples were resuspended in 1D or 2D gel sample buffer and protein concentration was determined using a BCA assay.
Gel Electrophoresis and Western Blotting
For SDS-polyacrylamide gel electrophoresis, equal amounts of total protein (30–50 μg total cellular protein, or 5 μg actin) were separated (125 volts, 50 watts, 75 mA). For 2D gel electrophoresis, 100 μg total protein per sample was loaded onto rehydrated IPG strips (Invitrogen, Calsbad, CA) and isoelectric focusing performed at RT (Castegna et al., 2002). After equilibrating IPG strips with 1D sample buffer, strips were placed on NUPAGE 4–12% gels and separated (125 V, 30 mM) using a MES running buffer (50 mM MES, 50 mM Tris base, 0.1% SDS, 1 mM EDTA, pH 7.3). For western blotting, proteins were transferred onto PVDF membranes (2.5 hours, 50 volts, 50 watts, 250 mA) and membranes were blocked with TBST-BSA (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 0.1% Tween-20, 5 mg/ml BSA) (Towbin et al., 1979). After overnight incubation with the respective primary antibody (1 μg/μl in TBST), membranes were washed (TBST) and incubated with the corresponding secondary antibody (0.2 μg/μl in TBST, 45 min). Immunoreactivity was detected by chemiluminescence and quantified using a GE Healthcare Typhoon Imager running ImageQuant software (United Kingdom) or by colorimetric detection.
Statistical Analysis
Analysis of variance (ANOVA) was used to determine statistically significant differences between treatments (p<0.05). At least three independent experiments were performed for each condition. Post hoc comparisons of specific treatments were performed using Scheffe’s test to determine statistical significance based on the calculated ANOVA data. A Dunette’s test was used for multiple comparisons between treatments and a single control. A Kruskal-Wallis test was employed to calculate significance between control distributions and treatment distributions (confocal analysis). Error bars represent standard deviations of the mean.
Acknowledgments
We are grateful for the experimental support received from Dr. M. Wright and R. Parsley regarding the biochemical analysis of the membrane translocation of p67phox. We would like to extend our thanks to Dr. Schulte, Dr. Taylor, Dr. O’Brien and Dan Kirschner for their review of the manuscript and many constructive discussions of the research. This study was supported by National Institutes of Health U54 grant NS41069 (SNRP: NINDS, NIMH, NCRR, NCMHD), United States Department of Agriculture grant 2005-34495-16519 (TBK), and Christopher Reeve Paralysis Foundation grant KA1-0004-2 (TBK).
The abbreviations used are
- AA
arachidonic acid
- AEBSF
4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride
- ATK
arachidonyl trifluoromethylketone
- CHAPS
3-[(3-cholamido propyl)dimethylammonio]-1-propanesulfonate
- CNS
central nervous system
- DCF
dichlorofluorescein
- DHE
dihyroethidium
- DMEM
Dulbecco’s modified Eagle medium
- DNP
dinitrophenyl
- DNPH
dinitrophenyl hydrazine
- DPI
diphenylene iodonium
- DTT
ditiothreitol
- Eth
ethidium
- FBS
fetal bovine serum
- HBSS
Hank’s Balanced Salt Solution
- H2DCF
2′,7′-dihydrodichlorofluorescein
- Interleukin 1β
Il-1β
- MES
2-(N-morpholino)ethanesulfonic acid
- MnTBPA
Manganese (III) tetrakis (4-benzoic acid) porphyrin chloride
- NAC
N-acetyl-L-cysteine
- NOX
NADPH oxidoreductase
- PIC
protease inhibitor cocktail
- PMA
phorbol 12-myristate 10-acetate
- ROS
reactive oxygen species
- TBS
Tris-buffered saline
- TNFα
tumor necrosis factor α
- TX
Triton-X-100
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aksenov M, Aksenov M, Butterfield D, Geddes J, Markesbery W. Protein oxidation in the brain in Alzheimer’s Disease. Neurosci. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Allan S, Rothwell N. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Beal MF. Oxidatively modified proteins in aging and disease. Free Radic Biol Med. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Piette J, Schoonbroodt S, Greimers R, Havard L, Merville MP, Bours V. Reactive oxygen intermediate-depdendent NF-kB activation by interleukin-1beta requires 5-lipoxygenase or NADPH oxidase activity. Mol Cell Biol. 1999;19:1950–1960. doi: 10.1128/mcb.19.3.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT, Arch RH. Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J Biol Chem. 2001;276:42728–42736. doi: 10.1074/jbc.M103074200. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med. 2003a;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Lusini L, Milzani A, Di Simplicio P, Colombo R. Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic Biol Med. 2001;31:1075–1083. doi: 10.1016/s0891-5849(01)00690-6. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003b;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- Deshpande S, Angkeow P, Huang J, Ozaki M, Irani K. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB. 2000;14:1705–1714. doi: 10.1096/fj.99-0910com. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Finkel T, Sullivan D. Reactive oxygen species in proliferative signaling. In: Gutkind JS, editor. Signaling networks and cell cycle control: the molecular basis of cancer and other diseases. Humana Press Inc: Totowa, NJ; 2001. pp. 365–377. [Google Scholar]
- Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- Freeman J, Abo A, Lambeth J. Rac “insert region” is a novel effector region that is implicated in the activation of NADPH oxidase, but not PAK65. J Biol Chem. 1996;271:19794–19801. doi: 10.1074/jbc.271.33.19794. [DOI] [PubMed] [Google Scholar]
- Goossens V, De Vos K, Vercammen D, Steemans M, Vancompernolle K, Fiers W, Vandenabeele P, Grooten J. Redox regulation of TNF signaling. Biofactors. 1999;10:145–156. doi: 10.1002/biof.5520100210. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol. 2005;6:583–589. doi: 10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A, Nobes C. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philosoph Trans Royal Soc London B. 2000a;355:965–970. doi: 10.1098/rstb.2000.0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A, Nobes CD. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philosoph Trans Royal Soc London Biol Sci. 2000b;355:965–970. doi: 10.1098/rstb.2000.0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna AN, Berthiaume LG, Kikuchi Y, Begg D, Bourgoin S, Brindley DN. Tumor necrosis factor-alpha induces stress fiber formation through ceramide production: role of sphingosine kinase. Mol Biol Cell. 2001;12:3618–3630. doi: 10.1091/mbc.12.11.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- Karnoub A, Der C, Campbell S. The insert region of rac1 is essential for membrane ruffling but not cellular transformation. Mol Cell Biol. 2001;21:2847–2857. doi: 10.1128/MCB.21.8.2847-2857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. Role of rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280:898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol. 1998;275:C1–24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Krieger-Brauer HI, Kather H. The stimulus-sensitive H2O2-generating system present in human fat-cell plasma membranes is multireceptor-linked and under antagonistic control by hormones and cytokines. Biochem J. 1995;307:543–548. doi: 10.1042/bj3070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol. 2002;9:11–17. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassing I, Schmitzberger F, Bjornstedt M, Holmgren A, Nordlund P, Schutt CE, Lindberg U. Molecular and Structural Basis for Redox Regulation of beta-Actin. J Mol Biol. 2007;370:331–348. doi: 10.1016/j.jmb.2007.04.056. [DOI] [PubMed] [Google Scholar]
- Leski ML, Bao F, Wu L, Qian H, Sun D, Liu D. Protein and DNA oxidation in spinal injury: neurofilaments--an oxidation target. Free Radic Biol Med. 2001;30:613–624. doi: 10.1016/s0891-5849(00)00500-1. [DOI] [PubMed] [Google Scholar]
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Meth Enzymol. 1994;233:346–357. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ, Warren KS, Wamboldt DD, Wang T, Lin JL. Tropomyosin isoforms in nonmuscle cells. Int Rev Cytol. 1997;170:1–38. doi: 10.1016/s0074-7696(08)61619-8. [DOI] [PubMed] [Google Scholar]
- Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147:S232–240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Meier B. Superoxide generation of phagocytes and nonphagocytic cells. Protoplasma. 2001;217:117–124. doi: 10.1007/BF01289420. [DOI] [PubMed] [Google Scholar]
- Milzani A, Dalledonne I, Vailati G, Colombo R. Paraquat induces actin assembly in depolymerizing conditions. FASEB. 1997;11:261–270. doi: 10.1096/fasebj.11.4.9068615. [DOI] [PubMed] [Google Scholar]
- Moldovan L, Irani K, Moldovan N, Finkel T, Goldschmidt-Clermont P. The actin cytoskeleton reorganization induced by rac1 requires the production of superoxide. Antiox Redox Signal. 1999;1:29–43. doi: 10.1089/ars.1999.1.1-29. [DOI] [PubMed] [Google Scholar]
- Moldovan L, Moldovan N, Sohn R, Pakrish S, Goldschmidt-Clermont P. Redox changes of cultured endothelial cells and actin dynamics. Circ Res. 2000;86:549–557. doi: 10.1161/01.res.86.5.549. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Munch G, Gasic-Milenkovic J, Dukic-Stefanovic S, Kuhla B, Heinrich K, Riederer P, Huttunen HJ, Founds H, Sajithlal G. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp Brain Res. 2003;150:1–8. doi: 10.1007/s00221-003-1389-5. [DOI] [PubMed] [Google Scholar]
- Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci. 2002;22:854–862. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova S, Lee YS, Lee YS, Kim JA. Rac1-NADPH oxidase-regulated generation of reactive oxygen species mediates glutamate-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic Res. 2005;39:1295–1304. doi: 10.1080/10715760500176866. [DOI] [PubMed] [Google Scholar]
- Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- Nobes C, Hall A. Rho, rac and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci. 2000;20:1–5. doi: 10.1523/JNEUROSCI.20-23-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppelenbosch M, Boone E, Jones GE, van Deventer SJ, Haegeman G, Fiers W, Grooten J, Ridley AJ. Multiple signal transduction pathways regulate TNF-induced actin reorganization in macrophages: inhibition of Cdc42-mediated filopodium formation by TNF. J Immunol. 1999;162:837–845. [PubMed] [Google Scholar]
- Percipalle P, Visa N. Molecular functions of nuclear actin in transcription. J Cell Biol. 2006;172:967–971. doi: 10.1083/jcb.200512083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls A, Eliopoulos A, Nobes C, Bridges T, Young L, Hall A. Activation of the small GTPase cdc42 by the inflammatory cytokines TNF and IL-1, and by the Eppstein-Barr virus transforming protein LMP1. J Cell Sci. 1999;112:2983–2992. doi: 10.1242/jcs.112.17.2983. [DOI] [PubMed] [Google Scholar]
- Rothe G, Valet G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2′,7′-dichlorofluorescin. J Leukoc Biol. 1990;47:440–448. [PubMed] [Google Scholar]
- Rothwell NJ, Luheshi GN. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 2000;23:618–625. doi: 10.1016/s0166-2236(00)01661-1. [DOI] [PubMed] [Google Scholar]
- Sulciner D, Irani K, Yu Z, Ferrans V, Goldschmidt-Clermont P, Finkel T. Rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kB activation. Mol Cell Biol. 1996;16:7115–7121. doi: 10.1128/mcb.16.12.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan M, Yu Z, Ferrans V, Sulciner D, Gutkind J, Iranis K, Goldschmidt-Clermonts P, Finkel T. Regulation of reactive-oxygen-species generation in fibroblasts by rac1. Biochem J. 1996;318:379–392. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzukawa K, Miura K, Mitsushita J, Resau J, Hirose K, Crystal R, Kamata T. Nerve growth factor-induced neuronal differentiation requires generation of rac1-regulated reactive oxygen species. Journal of Biological Chemistry. 2000;275:13175–13178. doi: 10.1074/jbc.275.18.13175. [DOI] [PubMed] [Google Scholar]
- Symons M, Mitchison T. Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammariello S, Quinn M, Estus S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci. 2000;20:1–5. doi: 10.1523/JNEUROSCI.20-01-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ Res. 2001;89:224–236. doi: 10.1161/hh1501.094365. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV, Serrano F, Villasana LE, Kanterewicz BI, Wu GY, Quinn MT, Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29:97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Entwistle A, Garg R, Ridley A. Regulation of TNF alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by rho, rac, and cdc42 in human endothelial cells. J Cell Physiol. 1998;176:150–165. doi: 10.1002/(SICI)1097-4652(199807)176:1<150::AID-JCP17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Tsang LY, Haworth SG. Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288:L749–760. doi: 10.1152/ajplung.00361.2004. [DOI] [PubMed] [Google Scholar]