

Abstract
With the emergence of Staphylococcus aureus as a prominent pathogen in community and healthcare settings, there is a growing need for effective reporter tools to facilitate physiology and pathogenesis studies. Fluorescent proteins are ideal as reporters for their convenience in monitoring gene expression, performing host interaction studies, and monitoring biofilm growth. We have developed a suite of fluorescent reporter plasmids for labeling S. aureus cells. These plasmids encode either green fluorescent protein (GFP) or higher wavelength reporter variants for yellow (YFP) and red (mCherry) labeling. The reporters were placed under control of characterized promoters to enable constitutive or inducible expression. Additionally, plasmids were assembled with fluorescent reporters under control of the agr quorum-sensing and Sigma factor B promoters, and the fluorescent response with wildtype and relevant mutant strains was characterized. Interestingly, reporter expression displayed a strong dependence on ribosome binding site (RBS) sequence, with the superoxide dismutase RBS displaying the strongest expression kinetics of the sequences examined. To test the robustness of the reporter plasmids, cell imaging was performed with fluorescence microscopy and cell populations were separated using florescence activated cell sorting (FACS), demonstrating the possibilities of simultaneous monitoring of multiple S. aureus properties. Finally, a constitutive YFP reporter displayed stable, robust labeling of biofilm growth in a flow cell apparatus. This toolbox of fluorescent reporter plasmids will facilitate cell labeling for a variety of different experimental applications.
Keywords: Staphylococcus aureus, fluorescence, fluorescent reporters, GFP, YFP, mCherry
Introduction
Staphylococcus aureus is emerging as one of the most problematic pathogens facing our healthcare system (Jarvis et al., 2007, Klevens et al., 2007). With the growing interest in S. aureus pathogenesis mechanisms, there is an increasing need for tools to monitor gene expression, protein localization, and host-pathogen interactions. Green fluorescent protein (GFP), originally derived from the jellyfish Aequorea victoria, is one of the most generally useful biological reporters (Joo et al., 2008, Nienhaus, 2008, Tsien, 1998). Engineered GFP variants have been employed extensively to quanitfy S. aureus gene expression from a variety of different promoters (Cheung et al., 1998, Kahl et al., 2000, Malone et al., 2007), track intracellular invasion (Qazi et al., 2001, Shompole et al., 2003), examine protein localization (Grundling and Schneewind, 2006), and perform flow sorting experiments (Schneider et al., 2002, Yarwood et al., 2004).
One valuable application of GFP labeling is the monitoring of biofilm maturation and dispersal mechanisms (Boles and Horswill, 2008, Yarwood, et al., 2004, Yarwood et al., 2007). By labeling with GFP, live cells can be visualized in situ, bypassing potential complications with post-staining as an endpoint readout. Recently, we demonstrated that S. aureus biofilms can also be dual-labeled with both GFP and red fluorescent protein (RFP) constructs (Boles and Horswill, 2008), opening doors to simultaneous monitoring of multiple environmental cues.
In this report, we present a toolbox of fluorescent plasmids for S. aureus. We have employed three generally useful fluorescent reporters, including GFPuvr (Bateman et al., 2001), YFP10B (Yarwood, et al., 2004), and mCherry (Shaner et al., 2004). We placed these reporters under control of a series of different well-characterized promoters, and additionally, the constructs were built on both chloramphenicol and erythromycin resistance plasmids to enhance flexibility. We investigated the importance of the ribosome binding site for protein expression in S. aureus and further tested the general usefulness of the reporters in microscopy, FACS, and flow cell biofilm monitoring.
Materials and Methods
Bacterial strains and culture conditions
All bacterial strains and plasmids used in this report are listed in Table 1. Plasmids were maintained in Escherichia coli BW25141 in Luria-Bertani broth. Plasmids were transformed to S. aureus RN4220 by electroporation (Schenk and Laddaga, 1992). Antibiotics were used for maintenance of plasmids. Antibiotic concentrations for S. aureus were: erythromycin (Erm) 10 ug/ml, chloramphenicol (Cam) 10 ug/ml. E. coli antibiotic concentrations were: ampicillin (Amp) 100 ug/ml.
Table 1.
Strains and plasmids used in this study
| Strain/Plasmid | Genotype/Properties | Reference |
|---|---|---|
| E. coli BW25141 | Cloning strain | (Datsenko and Wanner, 2000) |
| S. aureus | ||
| RN4220 | Restriction deficient cloning host | (Novick, 1991) |
| SA502a | agr Type II strain | (Ji, et al., 1997) |
| SH1000 | NCTC8325-4 corrected for rsbU+ | (Horsburgh et al., 2002) |
| SH1001 | SH1000 / Δagr∷TetM | (Horsburgh, et al., 2002) |
| AH1012 | SH1000 / ΔsigB | (Lauderdale, et al., 2009) |
| AH1218 | SH1000 / pAH1 and pAH16 | This study |
| AH1219 | SH1001 / pAH1 and pAH16 | This study |
| Plasmid | ||
| pALC2084 | Plasmid with tetracycline-inducible GFPuvr expression, Camr | (Bateman, et al., 2001) |
| pCE-MCS | Empty plasmid, Ermr | This study |
| pDB59 | Plasmid for agr P3-dependent YFP10B expression, Camr | (Yarwood, et al., 2004) |
| pAH1 | Plasmid for agr P3-dependent mCherry expression, Camr | This study |
| pAH5 | Plasmid for SigB (asp23) dependent YFP10B expression, Camr | This study |
| pAH6 | Plasmid for SigB (asp23) dependent mCherry expression, Camr | This study |
| pAH7 | Plasmid for agr P3-dependent YFP10B expression, Ermr | This study |
| pAH8 | Plasmid for agr P3-dependent mCherry expression, Ermr | This study |
| pAH9 | Plasmid for sarAP1-dependent mCherry expression, Ermr | (Boles and Horswill, 2008) |
| pAH12 | Plasmid for SigB (asp23) dependent mCherry expression, Ermr | This study |
| pAH13 | Plasmid with tetracycline-inducible GFPuvr expression, Ermr | This study |
| pAH14 | Plasmid for sarAP1-dependent YFP10B expression, Ermr | This study |
| pAH15 | pAH14 with sarA ribosome binding site | This study |
| pAH16 | pAH14 with sod ribosome binding site | This study |
| pAH17 | pAH14 with hld ribosome binding site | This study |
Recombinant DNA techniques
Restriction enzymes were purchased from New England Biolabs (Beverly, MA) and were used according to the manufacturer’s instructions. All oligonucleotides were synthesized at Integrated DNA Technologies (Coralville, IA). All DNA manipulations were performed with E. coli strain BW25141 (Datsenko and Wanner, 2000). Nonradioactive sequencing was performed at the DNA sequencing facility at the University of Iowa.
Fluorescence measurements
For testing the fluorescent reporters, S. aureus RN4220 containing different plasmids were grown overnight in tryptic soy broth (TSB) plus antibiotic. Cultures were diluted 50-fold into 5 ml of fresh media, shaken at 200 rpm at 37°C in 18 × 100 mm tubes, and fluorescence was read at various time points as indicated in each figure. For each time point, both cell density (OD595) and green/yellow fluorescence (excitation at 485 nm, emission at 535 nm, gain setting of 60) were measured in a Tecan GENios (Research Triangle Park, NC) microtiter plate reader. For red fluorescence, a filter set with excitation at 580 nm and emission at 635 nm was used at gain setting of 100 in the Tecan plate reader. Fluorescent readings were obtained by removing 200 μL from each tube and assaying in a microtiter plate (Costar 6303 plates, Corning). For the tet promoter, cultures were grown to an optical density (OD) of 0.5 at 595 nm and induced with a range of anhydrotetracycline (aTet) concentrations. For the asp23 promoter, overnight cultures were diluted 200-fold and then fluorescence readings were taken at designated times. For the agr reporter experiments, AIP-II control samples were generated by preparing cell-free supernatant of S. aureus SA502a overnight culture. The supernatant containing AIP-II was diluted 10-fold into a culture containing the agr fluorescent reporter at the beginning of the growth.
Plasmid constructions
Plasmids pDB59 and pCE104 served as the base vectors for all constructions (Yarwood, et al., 2004). The relevant details of all plasmids are listed in Table 2. The pAH plasmid numbering does not reflect the order of construction. All constructs were verified by DNA sequencing.
Table 2.
Properties of fluorescent reporter plasmids
| Name | Shuttle Backbone | E. coli Resistance | S. aureus Resistance | Promoter | Reporter | RBS |
|---|---|---|---|---|---|---|
| pALC2084 | pSK236 | Amp | Cam | tet | GFPuvr | sarA |
| pDB59 | pUC18/pC194 | Amp | Cam | agr P3 | YFP10B | Gene 10 |
| pAH1 | pUC18/pC194 | Amp | Cam | agr P3 | mCherry | sarA |
| pAH5 | pUC18/pC194 | Amp | Cam | asp23 | YFP10B | Gene 10 |
| pAH6 | pUC18/pC194 | Amp | Cam | asp23 | mCherry | sarA |
| pAH7 | pUC18/pE194 | Amp | Erm | agr P3 | YFP10B | Gene 10 |
| pAH8 | pUC18/pE194 | Amp | Erm | agr P3 | mCherry | sarA |
| pAH9 | pUC18/pE194 | Amp | Erm | sarA P1 | mCherry | sarA |
| pAH12 | pUC18/pE194 | Amp | Erm | asp23 | mCherry | sarA |
| pAH13 | pUC18/pE194 | Amp | Erm | tet | GFPuvr | sarA |
| pAH14 | pUC18/pE194 | Amp | Erm | sarA P1 | YFP10B | Gene 10 |
| pAH15 | pUC18/pE194 | Amp | Erm | sarA P1 | YFP10B | sarA |
| pAH16 | pUC18/pE194 | Amp | Erm | sarA P1 | YFP10B | sod |
| pAH17 | pUC18/pE194 | Amp | Erm | sarA P1 | YFP10B | hld |
pAH1, pAH5, and pAH6 construction
To build pAH5, the asp23 promoter was amplified from S. aureus SH1000 genomic template using the oligonucleotides Asp-Bam (GTTGTTGGATCCTAGCGTTTCTATTAATCGCGATATTATTC) and Asp2 (5’-GTTGTTGGTACCAATAGATTCTCCTTTTACTTGTTAATTTTTATA-3’). A second PCR was performed using plasmid pDB59 as template with the oligonucleotides AspYFP (5’-GGAGAATCTATTGGTACCAACAACAAGAAGGAGATATACATATGAGTAAAGG-3’) and YFP EcoRI (5’-GTTGTTGAATTCTTATTTGTATAGTTCATCCATGCCA-3’). The two PCR products were fused with overlap extension PCR (Urban et al., 1997), digested with BamHI and EcoRI, and ligated into pDB59 digested by the same enzymes. The resulting plasmid has the asp23 promoter driving YFP10B and was called pAH5. To change fluorescent reporters, the mCherry gene was removed from pAH9 (Boles and Horswill, 2008) using KpnI and EcoRI and ligated into pAH5 cut with the same enzymes. The resulting plasmid has the asp23 promoter driving the mCherry reporter and was called pAH6. To build pAH1, the agr P3 promoter region was PCR amplified from SH1000 genomic DNA with oligonucleotides incorporating HindIII and KpnI sites (for 5’-GTTGTTAAGCTTCTGTCATTATACGATTTAGTACAATC-3’, rev 5’-GTTGTTGGTACCTTAAACAACTCATCAACTATTTTCC-3’). The PCR product was cloned into the vector pCR2.1 using the TOPO-TA cloning kit (Invitrogen) according to manufacturer’s instructions, generating an intermediate plasmid called pCR2.1-RNAIII. The promoter fragment was removed from this plasmid with BamHI and KpnI and ligated into pAH6 digested with the same enzymes. The resulting plasmid has the agr P3 promoter driving the mCherry reporter and was called pAH1.
pAH7 construction
Plasmid pDB59 was digested with KpnI and HindIII to remove the agr P3 (from strain MN8) fusion to YFP10B. The fragment was cloned into pCE104 cut with the same enzymes. The resulting plasmid has the agr P3 promoter driving YFP10B and was called pAH7.
pAH8 and pAH12 construction
A DNA fragment containing the agr P3 promoter was removed from the pCR2.1-RNAIII clone (described above) with HindIII and KpnI, and this fragment was cloned into pAH9 (Boles and Horswill, 2008) cut with the same enzymes. The resulting vector has the agr P3 promoter driving the mCherry reporter in an Erm-resistant plasmid and was called pAH8. For pAH12 plasmid construction, the asp23 promoter was PCR amplified with oligonucleotides Asp1 (5’GGGAAAAAGCTTTAGCGTTTCTATTAATCGCGATATTATTC-3’) and Asp2 (described above) and cloned into the vector pCR2.1 using the TOPO-TA cloning kit (Invitrogen). The promoter fragment was removed with HindIII and KpnI, and cloned into pAH9 cut with the same enzymes. The resulting plasmid has the asp23 promoter driving mCherry reporter in an Erm-resistant plasmid.
pAH13 construction
The promoter and reporter region on pAH9 were removed with a HindIII and EcoRI double digest. For subsequent cloning, a linker was added by hybridizing two oligonucleotides (CLM316, 5’-AGCTCAGATCTACGTTACGTAGCTAGCT-3’; CLM317, 5’-AATTAGCTAGCTACGTAACGTAGATCTG-3’) and ligating them into the digested pAH9. The cloning step resulted in plasmid pCE-MCS, which has BglII and NheI as two new cloning sites. The tetracycline repressor, tet promoter, and GFPuvr reporter were PCR amplified from pALC2084 (Bateman, et al., 2001) using the oligonucleotides CLM269 (5’-GTTGTTAGATCTGTCGACGGTATCGATAACTC-3’) and CLM271 (5’-GTTGTTGCTAGCTTATTTGTAGAGCTCATCCATG-3’). The PCR product was digested with BglII and NheI and ligated into pCE-MCS cut with the same enzymes. The resulting plasmid, called pAH13, allows for tetracycline-inducible GFPuvr expression on an Erm-resistant plasmid.
pAH14, pAH15, pAH16, and pAH17 construction
A series of plasmids with the sarA P1 promoter driving YFP10B were constructed using pAH9 as the destination vector. The YFP10B reporter from pAH5 was removed with KpnI and EcoRI and cloned into pAH9 digested with the same enzymes. This plasmid was called pAH14 and has the YFP10B reporter translated with an RBS region from gene 10 (Yarwood, et al., 2004). The gene 10 RBS was replaced with three other RBS sequences from the NCTC8325 genome, including the ones upstream of the sarA, superoxide dismutase (sod), and delta-toxin (hld) genes. The RBS regions were embedded in the flanking DNA on a 5’ oligonucleotide for YFP10B. The oligonucleotides were CLM275 (5’-GTTGTTGGTACCTAGGGAGGTTTTAAACATGAGTAAAGGAGAAGAACTTTTC-3’), CLM321:5’-GTTGTTGGTACCTTAGGAGGATGATTATTTATGAGTAAAGGAGAAGAACTTTTC-3’), and CLM322 (5’-GTTGTTGGTACCAAGGAAGGAGTGATTTCAATGAGTAAAGGAGAAGAACTTTTC-3’) for the sarA, sod, and hld RBS regions, respectively. Each PCR amplification was performed with one of the oligonucleotides described above and a downstream oligonucleotide for the YFP10B reporter called YFP EcoRI (see above). The PCR product were digested with KpnI and EcoRI and ligated into pAH14 cut with the same enzymes. The resulting plasmids, called pAH15, pAH16, and pAH17, had the sarA P1 promoter driving YFP10B with the sarA, sod, and hld RBS regions, respectively.
Microscopy
Strains SH1000 and SH1001 containing plasmids pAH1 and pAH16 were grown in TSB with Erm and Cam for 12 hr, and the cultures were processed for microscopy. Confocal fluorescence laser microscopy (CLSM) was performed using a Nikon Eclipse E600 microscope with a Radiance 2100 system (Biorad). A Nikon 60x (Plan Apo) objective lens was used for image acquisition. The excitation wavelength for GFP was 488 nm (argon laser) and the emission was collected at 515 ± 15 nm. To detect RFP fluorescence, the excitation wavelength was set to 637 nm and the emission was collected at wavelengths >660 nm using a 660LP filter. Images were initially processed with LaserSharp 2000 (Bio-Rad) software, and post-acquisition, images were processed using Volocity (Improvision, Lexington, Mass.) software.
Flow cytometry
The following five strains were used for fluorescence-activated cell sorting (FACS): SH1000 alone, SH1000 with pAH1, SH1000 with pAH16, SH1000 with both pAH1 and pAH16 (strain AH1218), and SH1001 with both pAH1 and pAH16 (strain AH1219). Strains were grown in TSB for 16 hr, and the media was supplemented with Erm and/or Cam as necessary for plasmid maintenance. Cell debris was removed using a 70 μM cell strainer (BD Falcon), and cell density was adjusted to 1 × 106 CFU/ml. To remove clumps, cell cultures were sonicated for 5 pulses at 50% duty using a Sonifier 450 (Branson, Danbury, CT). FACS was performed at the University of Iowa Flow Cytometry facility using a DiVa machine. The pAH16 YFP10B reporter was detected using an argon ion laser for excitation at 488 nM and an emission filter at 530 nM with a ±30 nm bandpass. The pAH1 mCherry reporter was detected using a dye laser for excitation at 600 nM and an emission filter at 630 nM with a ±22 nm bandpass.
Biofilm analysis
S. aureus biofilms were grown using a once-through continuous flow system that has been previously described (Yarwood, et al., 2004). Biofilm growth media, inoculum size, laminar flow rate, and CLSM analysis of the biofilm were all as previously described (Boles and Horswill, 2008). For growth of SH1000 biofilms containing plasmid pAH16, media was supplemented with 1 μg/ml Erm. CLSM images of the biofilm were processed using Volocity software (Improvision, Lexington, Mass.). Biofilm thickness and biomass density (bio-volume) was determined using COMSTAT (Heydorn et al., 2000).
Results and Discussion
Construction of fluorescent reporter plasmids
As a starting point for building reporter plasmids, we chose the E. coli - S. aureus shuttle vectors pDB59 and pCE104 (Yarwood, et al., 2004). Plasmid pDB59 was originally constructed by fusing E. coli vector pUC18 to S. aureus naturally-occurring plasmid pC194. The shuttle vector can be propagated in E. coli as an ampicillin-resistant, high-copy plasmid, and in S. aureus, it confers chloramphenicol (Cam) resistance. Plasmid pDB59 served as the base vector for all the S. aureus Cam resistant shuttle vectors constructed in this report (Fig. 1A). As an alternative to Cam, we constructed a series of plasmids based on pCE104, which is a similar shuttle vector with S. aureus plasmid pE194 fused to pUC18. The pE194 backbone confers erythromycin (Erm) resistance to S. aureus, and all Erm resistant constructs were built using pCE104 (Fig. 1B).
Figure 1.

Graphic maps of S. aureus fluorescent reporter plasmids. A. pC194-based plasmids that confer Amp resistance to E. coli and Cam resistance to S. aureus. B. pE194-based plasmids that confer Amp resistance to E. coli and Erm resistance to S. aureus. Relevant regions and restriction sites are shown on each plasmid for cloning. The reporters can be exchanged using KpnI and EcoRI sites. The restriction site upstream of the promoter regions is not compatible. The plasmid maps are not drawn to scale.
For fluorescent reporters, we constructed most of the plasmids using the genes encoding YFP10B (Yarwood, et al., 2004) or mCherry (Shaner, et al., 2004). The YFP10B marker was attractive since it is known to work in S. aureus and is detectable with common GFP or fluorescein filter sets (Yarwood, et al., 2004). The mCherry protein is a recently engineered monomeric variant with improved properties compared to the older RFP variants (Shaner et al., 2005). We discovered this protein fluoresces well in S. aureus with virtually no overlap to the GFP or YFP spectra (Boles and Horswill, 2008), making it attractive as an alternative reporter. As a third fluorescent reporter, we have employed a red-shifted variant (GFPuvr) of the original GFPuv construct (Cheung, et al., 1998). Details of all the plasmids constructed and others used for the experiments are outlined in Tables 1 and 2.
sarA P1 as a constitutive promoter
A goal of this work was to develop reporter plasmids that enable constitutive fluorescent labeling of S. aureus cells. To accomplish this task, we chose to use the sarA promoter region, one of the best studied S. aureus transcriptional elements. The region contains three promoters that span 800 bp upstream of the sarA coding sequence and yields three distinct transcripts (Bayer et al., 1996, Cheung and Manna, 2005, Cheung, et al., 1998). The promoter in closest proximity to sarA, called P1, is the strongest of these three promoters, and P1 is frequently used as a constitutive promoter in biofilm and host labeling experiments (Cheung, et al., 1998, Femling et al., 2005, Yarwood, et al., 2004). We constructed sarA P1 fusions to mCherry (pAH9) and YFP10B (pAH15) fluorescent reporters and tested the new constructs in time course experiments. In comparison to negative control plasmid pCE-MCS, both pAH9 (Fig. 2A) and pAH15 (Fig. 2B) conferred robust fluorescent response in late logarithmic growth and stationary phase. While the sarA P1 promoter is known to be induced by mid logarithmic growth (Cheung, et al., 1998), the fluorescent response with pAH9 and pAH15 in early and mid logarithmic growth phases was low relative to cell density (data not shown). The low fluorescence could be due to the reporter protein folding rate in S. aureus. These observations demonstrate the pAH9 and pAH15 plasmids could be used for S. aureus fluorescent labeling studies where an inducer compound is not desirable.
Figure 2.
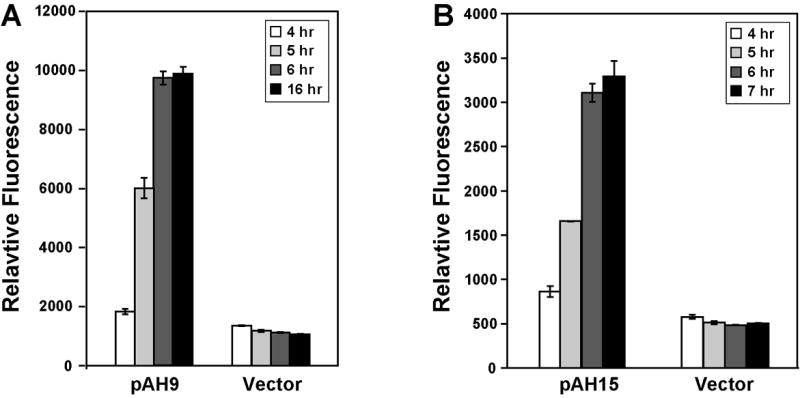
Constitutive fluorescent reporters for S. aureus. S. aureus RN4220 was transformed with plasmids pAH9, pAH15, and pCE-MCS. The transformed strains were grown in TSB with Erm, and samples were removed at 4, 5, 6, and 7 hr (pAH15) or 16 hr (pAH9). In the experimental setup, RN4220 reached late logarithmic growth by 4 hours. Fluorescence was measured using green or red filter sets, and the results were plotted relative to cell density at 595 nm. A. mCherry fluorescence of pAH9 compared to pCE-MCS (vector). B. YFP10B fluorescence of pAH15 compared to pCE-MCS (vector).
Sigma B and agr promoters
For physiological and pathogenic studies in S. aureus, promoter fusions were constructed to monitor the activity of sigma factor B (SigB) and the agr quorum-sensing system. SigB is an alternative sigma factor used by diverse Gram positive bacteria to coordinate gene expression in response to environmental stress, such as alkaline, heat, and high salt conditions (Senn et al., 2005). In S. aureus, SigB also regulates many factors related to virulence, such as carotenoid, hemolysins, extracellular invasive enzymes, and biofilm formation (Lauderdale et al., 2009). The asp23 promoter is a well characterized SigB-dependent promoter that has been utilized as an effective means to monitor SigB activity (Giachino et al., 2001). We constructed asp23 promoter fusions to YFP10B and mCherry reporters in a Cam resistant plasmid backbone, referring to them as pAH5 and pAH6, respectively. Additionally, we constructed an asp23 promoter fusion to mCherry fusion in the Erm backbone for increased flexibility, referred to as pAH12. All three plasmids were transformed into strains SH1000 (sigB+) and AH1012 (ΔsigB), and fluorescent readings were taken at early log and stationary phase (Fig. 3). For each plasmid, little difference in the asp23 promoter response between strains SH1000 and AH1012 was observed at low optical densities. However, under stationary phase inducing conditions, the asp23 promoter strongly induced the YFP10B and mCherry reporters in the wildtype SH1000 strain (Fig. 3). Importantly, no induction was observed in the ΔsigB mutant strain. Expression of YFP10B from plasmid pAH5 was low due to an inefficient RBS region, which will be outlined in more detail below.
Figure 3.
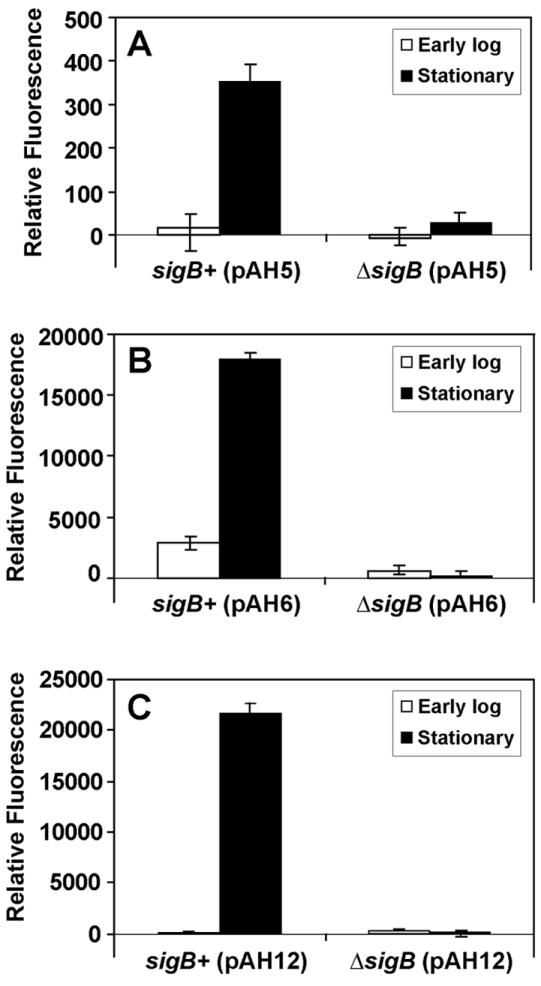
Monitoring S. aureus SigB activity using asp23 reporter plasmids. S. aureus strains SH1000 (sigB+) and AH1012 (ΔsigB) were transformed with plasmids pAH5, pAH6, and pAH12. The transformed strains were grown in TSB, and samples were removed at 4 hr (early log) or 16 hr (stationary phase). Fluorescence was measured using green or red filter sets, and the results were plotted relative to cell density at 595 nm. A. YFP10B fluorescence from pAH5. B. mCherry fluorescence from pAH6. C. mCherry fluorescence from pAH12.
The agr quorum-sensing system is regulator of virulence factor expression that responds to the extracellular concentration of a secreted peptide signal (Novick, 2003). Reporters for the agr system are available (Yarwood, et al., 2004), and to enhance flexibility, we constructed a suite of new agr responsive plasmids for various applications. Plasmid pDB59 serves as a positive control and has been successfully used in multiple reports (Boles and Horswill, 2008, Kavanaugh et al., 2007, Malone, et al., 2007, Yarwood, et al., 2004). As a Cam-resistant alternative, we constructed an agr P3 fusion to mCherry, referred to as plasmid pAH1. We also constructed YFP10B and mCherry fusion plasmids on an Erm-resistant plasmid backbone for additional flexibility. Each of the four plasmids were tested in strains SH1000 (agr+) and SH1001 (Δagr∷TetM) to compare activity of the Δagr system. Additionally, we tested agr interference by adding supernatant from an agr Type II strain SA502a. The AIP signal (AIP-II) made by SA502a will inhibit the agr Type I system of SH1000 (Ji et al., 1997). The agr P3 YFP10B plasmids, pDB59 and pAH7, both behaved as anticipated with the reporter expression inducing in late logarithmic phase of wildtype cells and not in the Δagr mutant (Fig. 4A). The addition of AIP-II containing supernatant inhibited the YFP10B fluorescence induction; demonstrating agr interference was detectable with both the pDB59 and pAH7 plasmids. Likewise, the two agr P3 mCherry plasmids, pAH1 and pAH8, functioned in a similar manner to the YFP10B plasmids (Fig. 4B). Altogether, these various plasmids provide a diverse set of tools to monitor the activities of the SigB and agr global regulatory systems.
Figure 4.
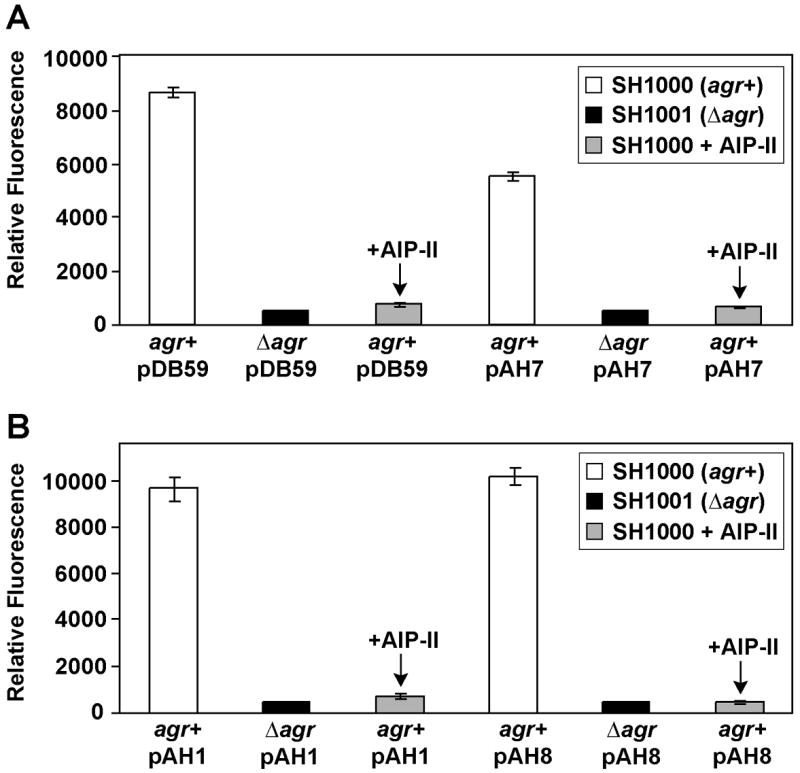
Monitoring S. aureus quorum-sensing using agr reporter plasmids. Strains S. aureus SH1000 (agr+) and SH1001 (Δagr) were transformed with pDB59, pAH1, pAH7, and pAH8 and grown in TSB with appropriate antibiotic. As indicated, transformed SH1000 strains were also treated with supernatant from AIP-II producing strain SA502a. After 9 hr growth, green or red fluorescence was measured in a plate reader and plotted relative to cell density at 595 nm. A.YFP10B fluorescence from pDB59 and pAH7. B. mCherry fluorescence from pAH1 and pAH8.
Inducible promoters
For some experimental applications, it may be necessary to have an inducible fluorescent reporter. Plasmid pALC2084 contains GFPuvr under control of the tet promoter (Bateman, et al., 2001), allowing induction with tetracycline or analogues, such as anhydrotetracycline (aTet). This plasmid has been successfully used for labeling experiments, but for some applications, the option of another antibiotic resistance may be beneficial. We moved the Ptet-GFPuvr cassette to an Erm-resistant backbone, called plasmid pAH13, and examined expression in strain RN4220. Using a range of aTet concentrations, we observed a dose response level of GFPuvr induction (Fig. 5), indicating the pAH13 plasmid was functional. In comparison to pALC2084, the overall fluorescence is roughly two-fold lower (data not shown). Plasmid pALC2084 is based on cloning vector pSK236 (Bateman, et al., 2001), which has a copy number ~90 in comparison to the pE194 copy number of ~55 (Novick, 1989), perhaps explaining the lower fluorescence of pAH13.
Figure 5.
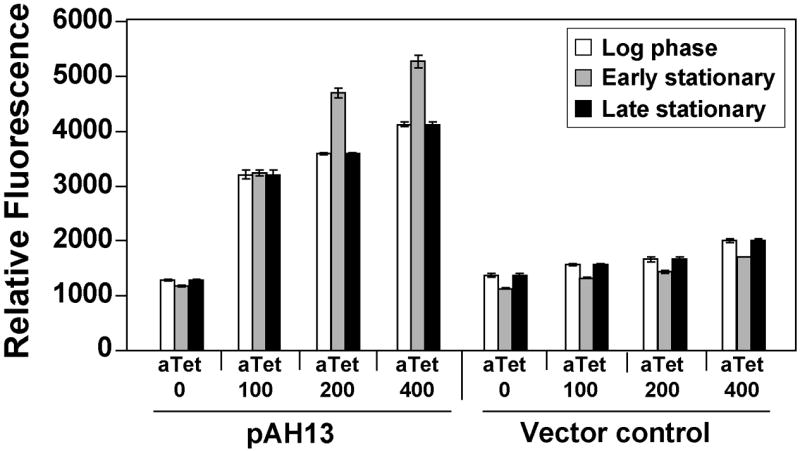
Fluorescent response of an inducible reporter plasmid. S. aureus RN4220 was transformed with pAH13 (tet, YFP10B) and an empty vector control (pCE-MCS), and cultures were grown for 2.5 hr (log phase), 5 hr (early stationary), and 16 hr (late stationary) with varying levels of aTet inducer. YFP10B fluorescence was measured in a plate reader and readings were plotted relative to cell density at 595 nm.
Comparing ribosome binding sites
When we compared asp23 reporter plasmids pAH5 and pAH6 (Fig. 3), we observed that pAH5 was considerably less effective at generating a fluorescent response. While these plasmids have different fluorescent reporters, the reporters are also translated using divergent RBS sequences (Table 2), with pAH5 containing a gene 10 RBS and pAH6 containing an RBS based on the sarA gene. These RBS differences, and a recent S. epidermidis report that protein translation is sensitive to the RBS region (Franke et al., 2007), prompted us to reexamine the importance of the RBS in fluorescent reporter expression. Interestingly, Franke et al. (2007) observed that the S. epidermidis sod and hld RBS regions were some of the most effective at protein expression, even better than the sarA RBS (Franke, et al., 2007). Using pAH15 (sarA RBS) as a base plasmid, we constructed three other RBS variants to compare the sarA RBS to those of gene 10, sod, and hld, resulting in plasmids pAH14, pAH16, and pAH17, respectively (Fig. 6A). A time course comparison of each plasmid in strain RN4220 was performed and YFP10B reporter levels were monitored. The gene 10 RBS was the worst performer (Fig. 6B), supporting our observations of the poor performance of pAH5. Interestingly, both the sod (pAH16) and hld (pAH17) RBS regions were improved over sarA RBS, with the sod RBS displaying the highest YFP10B levels at equivalent times. Franke et al. (2007) also observed that sod and hld RBS sequences were superior, although the hld RBS displayed the best response in S. epidermidis (Franke, et al., 2007). Altogether, these observations indicate that protein expression in Staphylococcus displays a strong dependence on the RBS sequence.
Figure 6.

Significance of the RBS region for S. aureus reporter expression. A. Graphic map of the pAH14-pAH17 plasmids and the different RBS sequences used. B. S. aureus RN4220 was transformed with each of the four plasmids, and cultures were grown for 4.5 hr (log phase) and 7 hr (stationary phase). YFP10B fluorescence was measured in a plate reader and readings were plotted relative to cell density at 595 nm.
Behavior of fluorescent reporters in S. aureus
With multiple different plasmid constructs available, we desired to know the properties of the fluorescent reporters when expressed in S. aureus. Currently, excitation and emission scans for the GFPuvr, YFP10B, and mCherry are not available for these reporters in S. aureus, and for some experimental applications, it is essential to know the excitation/emission behavior. As representatives of these three reporters, plasmids pAH9 (sarA P1, mCherry), pAH13 (tet, GFPuvr), and pAH15 (sarA P1, YFP10B) were transformed into RN4220, and overnight cultures were prepared with the appropriate antibiotic and inducer. For each reporter construct, excitation and emission scans were obtained (Fig. 7). GFPuvr displayed similar excitation/emission behavior to the reported values for EGFP, and YFP10B was similar to EYFP (Shaner, et al., 2005). The mCherry results were also similar to the reported values (Shaner, et al., 2004). In each case, we observed minimal fluorescence contamination from S. aureus cells. Importantly, by comparing these fluorescence scans, it is apparent that there is essentially no overlap between GFPuvr and mCherry, or YFP10B and mCherry, indicating either of these two combinations could be used for dual-labeling experiments.
Figure 7.

Excitation and emission scans of fluorescent reporters in S. aureus. Strain RN4220 was transformed with pAH9 (sarA P1, mCherry, red lines), pAH13 (tet, GFPuvr, green lines), and pAH15 (sarA P1, YFP10B, yellow lines). Cultures were grown for 16 hr in TSB with Erm, and additionally, 400 ng/ml aTet was added to induce GFPuvr expression from pAH13. Excitation and emission scans were obtained for each reporter, and an average of five scans was normalized and plotted.
Applications of fluorescent reporters
With a suite of fluorescent reporters now available for S. aureus cell labeling, we tested the reporters using a variety of experimental applications in order to gauge their effectiveness. For these tests, we used plasmid pAH16 (sarA P1, sod RBS, YFP10B) for constitutive labeling of S. aureus cells. As a secondary inducible reporter, we employed plasmid pAH1 (agr P3, sarA RBS, mCherry) for selected dual-labeling experiments. For initial cell imaging tests, strains SH1000 (agr+) and SH1001 (Δagr) were transformed with both pAH1 and pAH16 plasmids, resulting in strains AH1218 and AH1219, respectively. Fluorescence microscopy images were obtained using a green channel to visualize YFP10B in pAH16 and a red channel to visualize the agr-controlled mCherry in pAH1 (Fig. 8). As anticipated, the pAH1 plasmid allowed mCherry labeling only when a functional agr system was present. When the green and red channels were merged, the combined green and red fluorescence resulted in orangish-yellow colored cells in the agr+ background, similar as recently reported in dual-labeled biofilms (Boles and Horswill, 2008). Interestingly, the Δagr mutant repeatedly clumped under confocal visualization, an observation supported by the enhanced ability of agr mutants to attach to surfaces and develop into biofilms (Boles and Horswill, 2008). These findings demonstrate that S. aureus can be dual-labeled with fluorescent reporters to simultaneously monitor multiple phenotypes.
Figure 8.

Cell labeling of S. aureus with fluorescent reporters. Plasmids pAH1 (agr P3, mCherry) and pAH16 (sarA P1, sod RBS, YFP10B) were used for microscopy test. The top three panels are fluorescent images of SH1000 (agr+) cells transformed with both plasmid pAH1 and pAH16. The bottom three panels are SH1001 (Δagr) cells was transformed with the same two plasmids. In each case, overnight cultures were prepared with TSB supplemented with Cam and Erm, and fluorescence microscopy images were obtained. Representative images showing the green, red, and merged channels are shown.
To extend the functionality of the fluorescent reporters, we tested the separation of S. aureus populations using fluorescence-activated cell sorting (FACS). Strain SH1000 without a plasmid served as a negative gating control to gauge the yellow and red fluorescence intensity shifts (Fig. 9A & B). Again, strains AH1218 and AH1219 (described above) were used for this experiment. Comparing Figure 9C & E, both cell populations displayed near uniform expression of the YFP10B reporter encoded on pAH16. Using the background readings for setting the fluorescence cutoff gate (Fig. 9A), the AH1218 and AH1219 populations shifted 97% and 98%, respectively, towards yellow fluorescence, demonstrating the sarA P1 promoter and sod RBS enable robust, constitutive production of protein reporters. Similarly, the agr+ strain (AH1218) exhibited strong expression of the agr-controlled mCherry reporter encoded on plasmid pAH1 (Fig. 9D), with 93% of the population shifting towards red fluorescence compared to unlabeled control (Fig. 9B). As anticipated, the mCherry expression was eliminated in the AH1219 agr mutant strain (Fig. 9F). Altogether, the FACS test supported our observations from the microscopy images of fluorescent S. aureus cells (Fig. 8). The successful separation of S. aureus populations demonstrates the fluorescent reporters described herein could be used for a variety of FACS applications that require single or dual cell labeling.
Figure 9.

Flow sorting of S. aureus labeled with fluorescent reporters. SH1000 without a plasmid served as a negative gating control (histograms A & B). Plasmids pAH1 (agr P3, sarA RBS, mCherry) and pAH16 (sarA P1, sod RBS, YFP10B) were used for the FACS test. SH1000 (agr+) was transformed with pAH1 and pAH16, resulting in strain AH1218 (histograms C & D). Similarly, SH1001 (Δagr) was transformed with the same plasmids, resulting in strain AH1219 (histograms E & F). Each strain was grown overnight in TSB (with Cam and Erm as needed), cell density was adjusted to 106 CFU/mL, and populations were separated by FACS. Histograms A, C, and E display the yellow fluorescence channel and B, D, F display the red fluorescence channel. Units represent relative cell counts detected by the flow cytometer plotted at the measured fluorescence intensity.
One of the growing and most important applications of fluorescent reporters is visualization of S. aureus biofilm growth. Currently, most S. aureus biofilm observations are generated through post-staining in static and flow-cell format, or alternatively through direct visualization of the flow-cell biomass. The challenge with post-staining is that endpoint manipulations can complicate time course experiments. Fluorescent reporters have an advantage in facilitating the in situ monitoring of maturation and/or dispersal events of live biofilm growth. Constitutive reporters are attractive in that they bypass the need for inducers, such as tetracycline, which can complicate mixed-culture biofilms, eukaryotic cell interactions, and other biofilm applications. For reporter expression, the sarA P1 promoter has been utilized as an effective biofilm constitutive promoter (Yarwood, et al., 2004). Plasmid pAH16 (sarA P1, sod RBS, YFP10B) is the strongest constitutive reporter described herein and displayed robust expression in the microscopy and FACS experiments. To determine the potential of pAH16 for biofilm analysis, we tested the visualization of biofilm growth using this plasmid reporter in a flow cell apparatus. Biofilms of SH1000 with pAH16 were grown in a once-through, continuous flow cell system for 5 days, and growth and fluorescence labeling was monitored with CLSM. Images were captured at the 2 and 5 day time point, and representative top-down, cross-section, and three dimensional image reconstructions are shown in Figure 10. After two days, YFP labeling of the biomass was confluent across the entire surface and the biofilms have no noticeable growth defects while maintaining plasmid pAH16. Average thickness of the biofilm was ~19 μm, matching our previous observations of 2-day old SH1000 biofilms. Over the next three days, average biofilm thickness increased to ~32 μm and biomass density also increased from 9 to 24 μm3/μm2 (Fig. 10). Importantly, YFP labeling penetrated throughout the cell layers and no noticeable loss of fluorescence was observed, demonstrating stability of the plasmid. Thus, plasmid pAH16 represents an effective fluorescent reporter for in situ monitoring of biofilm growth, making it a valuable tool for diverse biofilm studies.
Figure 10.

Monitoring of flow cell biofilm growth using reporter plasmid pAH16. S. aureus SH1000 transformed with plasmid pAH16 (sarA P1, sod RBS, YFP10B) and grown in a once-through flow cell apparatus for 5 days. CLSM images were captured at 2 and 5 days. A. CLSM image reconstructions of a z series with each grid square representing 20 μM. B. CLSM top down (XY view) and cross-section (right, YZ view; bottom, XZ view) images of biofilm layers.
Conclusions
We have constructed a toolbox of fluorescent reporter plasmids for use in S. aureus. The reporters allow constitutive and inducible fluorescent labeling with different antibiotic selections for increased flexibility in diverse applications. Reporters for the agr quorum-sensing and SigB regulatory systems were also constructed and characterized to facilitate monitoring of the activity of these important global regulators. Through the course of this work, we identified a dependence on the RBS region for reporter expression and determined that the sod RBS was strong and effective, suggesting the RBS region should be a point of consideration for protein expression studies in S. aureus. To determine the general usefulness of these reporter plasmids, dual-labeling experiments were performed using cell microscopy and FACS, demonstrating multiple properties could be simultaneously monitored for different applications. Finally, the strongest, constitutive reporter constructed in this report displayed robust properties for monitoring flow-cell biofilm growth. Altogether, these new fluorescent reporter plasmids will enhance the molecular tools available for S. aureus labeling studies.
Acknowledgments
We thank A. Cheung for providing plasmid pALC2084. We thank D. Bartels for providing plasmids pDB59 and pCE104. K. J. Lauderdale was supported by an NIH Predoctoral Training Program in Biotechnology, Grant Number T32 GM08365-18. B. R. Boles was supported by NIH Training Grant Number T32 AI07511 and American Heart Association Midwest Postdoctoral Fellowship Grant Number 0825877G. The project was supported by Award Number R01AI078921 from the National Institute of Allergy and Infections Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infections Diseases or the National Institute of Health. The FACS data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine Core Research Facilities/Holden Comprehensive Cancer Center Core Laboratory at the University of Iowa. The Facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect Immun. 2001;69:7851–7857. doi: 10.1128/IAI.69.12.7851-7857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer MG, Heinrichs JH, Cheung AL. The molecular architecture of the sar locus in Staphylococcus aureus. J Bacteriol. 1996;178:4563–4570. doi: 10.1128/jb.178.15.4563-4570.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles BR, Horswill AR. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 2008;4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AL, Manna AC. Role of the distal sarA promoters in SarA expression in Staphylococcus aureus. Infect Immun. 2005;73:4391–4394. doi: 10.1128/IAI.73.7.4391-4394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AL, Nast CC, Bayer AS. Selective activation of sar promoters with the use of green fluorescent protein transcriptional fusions as the detection system in the rabbit endocarditis model. Infect Immun. 1998;66:5988–5993. doi: 10.1128/iai.66.12.5988-5993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femling JK, Nauseef WM, Weiss JP. Synergy between extracellular group IIA phospholipase A2 and phagocyte NADPH oxidase in digestion of phospholipids of Staphylococcus aureus ingested by human neutrophils. J Immunol. 2005;175:4653–4661. doi: 10.4049/jimmunol.175.7.4653. [DOI] [PubMed] [Google Scholar]
- Franke GC, Dobinsky S, Mack D, Wang CJ, Sobottka I, Christner M, Knobloch JK, Horstkotte MA, Aepfelbacher M, Rohde H. Expression and functional characterization of gfpmut3.1 and its unstable variants in Staphylococcus epidermidis. J Microbiol Methods. 2007;71:123–132. doi: 10.1016/j.mimet.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Giachino P, Engelmann S, Bischoff M. Sigma(B) activity depends on RsbU in Staphylococcus aureus. J Bacteriol. 2001;183:1843–1852. doi: 10.1128/JB.183.6.1843-1852.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundling A, Schneewind O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J Bacteriol. 2006;188:2463–2472. doi: 10.1128/JB.188.7.2463-2472.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, Molin S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology. 2000;146(Pt 10):2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. sigmaB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J Bacteriol. 2002;184:5457–5467. doi: 10.1128/JB.184.19.5457-5467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis WR, Schlosser J, Chinn RY, Tweeten S, Jackson M. National prevalence of methicillin-resistant Staphylococcus aureus in inpatients at US health care facilities, 2006. Am J Infect Control. 2007;35:631–637. doi: 10.1016/j.ajic.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Ji G, Beavis R, Novick RP. Bacterial interference caused by autoinducing peptide variants. Science. 1997;276:2027–2030. doi: 10.1126/science.276.5321.2027. [DOI] [PubMed] [Google Scholar]
- Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Advances in single-molecule fluorescence methods for molecular biology. Annu Rev Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, Peters G, Cheung AL. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect Immun. 2000;68:5385–5392. doi: 10.1128/iai.68.9.5385-5392.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh JS, Thoendel M, Horswill AR. A role for type I signal peptidase in Staphylococcus aureus quorum-sensing. Mol Microbiol. 2007;65:780–798. doi: 10.1111/j.1365-2958.2007.05830.x. [DOI] [PubMed] [Google Scholar]
- Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- Lauderdale KJ, Boles BR, Cheung AL, Horswill AR. Interconnections between Sigma B, agr, and proteolytic activity in Staphylococcus aureus biofilm maturation. Infect Immun. 2009 doi: 10.1128/IAI.01036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CL, Boles BR, Horswill AR. Biosynthesis of Staphylococcus aureus autoinducing peptides using the Synechocystis DnaB mini-intein. Appl Environ Microbiol. 2007 doi: 10.1128/AEM.00912-07. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhaus GU. The green fluorescent protein: a key tool to study chemical processes in living cells. Angew Chem Int Ed Engl. 2008;47:8992–8994. doi: 10.1002/anie.200804998. [DOI] [PubMed] [Google Scholar]
- Novick RP. Staphylococcal plasmids and their replication. Annu Rev Microbiol. 1989;43:537–565. doi: 10.1146/annurev.mi.43.100189.002541. [DOI] [PubMed] [Google Scholar]
- Novick RP. Genetic systems in staphylococci. Methods Enzymol. 1991;204:587–636. doi: 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol. 2003;48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- Qazi SN, Counil E, Morrissey J, Rees CE, Cockayne A, Winzer K, Chan WC, Williams P, Hill PJ. agr expression precedes escape of internalized Staphylococcus aureus from the host endosome. Infect Immun. 2001;69:7074–7082. doi: 10.1128/IAI.69.11.7074-7082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk S, Laddaga RA. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol Lett. 1992;73:133–138. doi: 10.1016/0378-1097(92)90596-g. [DOI] [PubMed] [Google Scholar]
- Schneider WP, Ho SK, Christine J, Yao M, Marra A, Hromockyj AE. Virulence gene identification by differential fluorescence induction analysis of Staphylococcus aureus gene expression during infection-simulating culture. Infect Immun. 2002;70:1326–1333. doi: 10.1128/IAI.70.3.1326-1333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn MM, Giachino P, Homerova D, Steinhuber A, Strassner J, Kormanec J, Fluckiger U, Berger-Bachi B, Bischoff M. Molecular analysis and organization of the sigmaB operon in Staphylococcus aureus. J Bacteriol. 2005;187:8006–8019. doi: 10.1128/JB.187.23.8006-8019.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Shompole S, Henon KT, Liou LE, Dziewanowska K, Bohach GA, Bayles KW. Biphasic intracellular expression of Staphylococcus aureus virulence factors and evidence for Agr-mediated diffusion sensing. Mol Microbiol. 2003;49:919–927. doi: 10.1046/j.1365-2958.2003.03618.x. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Urban A, Neukirchen S, Jaeger KE. A rapid and efficient method for site-directed mutagenesis using one-step overlap extension PCR. Nucleic Acids Res. 1997;25:2227–2228. doi: 10.1093/nar/25.11.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarwood JM, Bartels DJ, Volper EM, Greenberg EP. Quorum sensing in Staphylococcus aureus biofilms. J Bacteriol. 2004;186:1838–1850. doi: 10.1128/JB.186.6.1838-1850.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarwood JM, Paquette KM, Tikh IB, Volper EM, Greenberg EP. Generation of virulence factor variants in Staphylococcus aureus biofilms. J Bacteriol. 2007 doi: 10.1128/JB.00789-07. [DOI] [PMC free article] [PubMed] [Google Scholar]