

Abstract
Participation of RAS, RAF, and mitogen-activated protein kinase (MAPK) in learning and memory has been demonstrated in a number of studies, but the molecular events requisite for cascade activation and regulation have not been explored. We demonstrate that the adapter protein DRK (downstream of receptor kinase) which is essential for signaling to RAS in developmental contexts, is preferentially distributed in the adult mushroom bodies, centers for olfactory learning and memory. We demonstrate that drk mutant heterozygotes exhibit deficits in olfactory learning and memory, apparent under limited training conditions, but are not impaired in sensory responses requisite for the association of the stimuli, or brain neuroanatomy. Furthermore, we demonstrate that the protein is required acutely within mushroom body neurons to mediate efficient learning, a process that requires RAF activation. Importantly, 90 min memory remained impaired, even after differential training yielding equivalent learning in animals with compromised DRK levels and controls and did not require RAF. Sustained MAPK activation is compromised in drk mutants and surprisingly is negatively regulated by constitutive RAF activity. The data establish a role for DRK in Drosophila behavioral neuroplasticity and suggest a dual role for the protein, first in RAF activation-dependent learning and additionally in RAF-inhibition dependent sustained MAPK activation essential for memory formation or stability.
Keywords: learning, memory, DRK, RAS, RAF, signaling
Introduction
Accumulating evidence established the essential role of signaling through the typically oncogenic RAS/RAF/mitogen-activated protein kinase (MAPK) cascade in vertebrate (Kelleher et al., 2004; Sweatt, 2004; Chen et al., 2006; Davis and Laroche, 2006; Shalin et al., 2006; Chwang et al., 2007) and invertebrate (Bailey et al., 2004; Sharma and Carew, 2004) learning and memory. Cascade regulation is relevant for human cognition as loss of the RAS regulator Neurofibromin (NF) results in the mentally retarding condition Neurofibromatosis 1 (Weeber and Sweatt, 2002). However, although RAS/RAF/MAPK signaling is clearly essential for neuroplasticity, mechanisms that activate and regulate this pathway in neurons are largely unclear.
Neuronal RAS can be activated via receptor-tyrosine kinases (RTKs), neurotransmitter-activated G-protein-coupled or NMDA-glutamate receptors, voltage-gated calcium channels and cell adhesion molecules (Grewal et al., 1999; Mazzucchelli and Brambilla, 2000; Weeber et al., 2002; Krapivinsky et al., 2003). Adapter proteins like GRB2/DRK link RAS to receptors (Pawson and Saxton, 1999) and confer some signaling selectivity and specificity (Pawson and Saxton, 1999; Luschnig et al., 2000). Typically, activated RAS activates a MEK-kinase, one of which is RAF, at the plasma membrane. Among other actions, RAF activates another kinase MEK, which activates yet another kinase MAPK. MAPK modulates transcription factor activity in the nucleus, but also targets cytoplasmic and membrane proteins (Martin et al., 1997; Sweatt, 2001). Recent findings suggest that neurons use these molecules in alternative context-specific signaling routes to achieve different specific end results (Kolch, 2005; Douziech et al., 2006; Shalin et al., 2006), adding complexity to this cascade and its regulation.
Drosophila has been instrumental in identifying genes involved in learning and memory (Skoulakis and Grammenoudi, 2006). It offers a powerful system to explore RAS/RAF/MAPK signaling and its regulation in neuroplasticity as was done for eye and embryonic development (Dickson et al., 1996; Baek and Lee, 1999). Unlike vertebrates, however, and except for the potential RAF-regulating 14-3-3ζ protein LEO (Skoulakis and Davis, 1996; Li et al., 1997; Philip et al., 2001), and possibly dNF1 (Guo et al., 2000), typical cascade members have not been implicated in Drosophila learning and memory. However, the α-integrin VOL (Grotewiel et al., 1998) and the adhesion molecule FASII (Cheng et al., 2001) are required for olfactory learning and memory and may be involved in RAS/RAF/MAPK signaling (Martin and Kandel, 1996; Ruoslahti, 1999; Stork, 2003). Significantly, these proteins and LEO accumulate preferentially in the mushroom bodies (MBs), centers for olfactory learning and memory (Heisenberg, 2003; Davis, 2004). The MBs are bilateral neuronal clusters in the dorsal posterior brain cortex, extending dendrites ventrally forming the calyces and fasciculated axons anteriorly, bifurcating to form β, β' and γ medial and α, α' vertical lobes (Crittenden et al., 1998; Heisenberg, 2003).
To systematically study RAS/RAF/MAPK signaling in Drosophila learning and memory, we aimed to identify cascade members with roles in these processes based on their presence in the MBs (Skoulakis and Grammenoudi, 2006). Here, we report on DRK, a protein with striking distribution in the MBs, which is essential for transmission of RTK signals to RAS/RAF/MAPK (Perrimon et al., 1995; Raabe et al., 1995).
Materials and Methods
Drosophila culture, strains, and genetics.
Drosophila were cultured as described previously (Mershin et al., 2004). The transposon insertions mutant alleles drk10626 (hereafter drkP1) (Simon et al., 1993), and drkk02401 (hereafter drkP2) (Roch et al., 1998), were obtained from the Bloomington Stock Center. The deletion allele drkΔP24 was obtained from N. Perrimon (Harvard Medical School, Boston, MA) (Hou et al., 1995). The two point mutations within the SH2 domain drkE0A (amino acid 106 H to Y) and drkR1 (amino acid 67 A to H), reported to yield nonfunctional protein with respect to its ability to bind and support signaling originating from a receptor tyrosine kinase, were obtained from M. Simon (Stanford University, Stanford, CA) and E. Hafen (Universitaet Zurich, Zurich, Switzerland), respectively (Simon et al., 1991; Olivier et al., 1993; Simon et al., 1993). All strains were normalized to an isogenic ry506 strain (Skoulakis et al., 1993; Skoulakis and Davis, 1996) as follows: For the drkP1 transposon allele carrying the ry+ marker, free recombination was allowed with the ry506 second chromosome and ry+ animals were selected each generation for 6 generations. Point mutations and the w+-bearing transposon allele drkP2 were crossed to a strain carrying second chromosomal balancers in a ry506 genetic background and individual females carrying the CyO balancer over the mutant allele were used to establish three lines after six further backcrosses to the isogenic balancer strain. One backcrossed line was used for detailed behavioral analyses after pilot experiments demonstrating equivalent learning performance among the lines. UAS-Rafgof was obtained from A. Brand (Cambridge University, Cambridge, UK) (Brand and Perrimon, 1994). The eyaph strain (Choi and Benzer, 1994) was obtained from the Choi laboratory (Baylor College of Medicine, Houston, TX). c739, TubGAL80ts were obtained from G. Roman (University of Houston, Houston, TX).
Germline transformants for behavioral rescue were generated from a full-length drk cDNA (Simon et al., 1993), placed under heat shock promoter control in a ry+-bearing vector (Roman et al., 1999) and injected into ry506 isogenic embryos. Two independent third chromosome lines, T1and T5.3 were selected for ease of genetic manipulations. A transgene capable of generating double stranded drk RNA was generated by cloning a BamHI – EcoRI fragment from the full-length cDNA (Simon et al., 1993) into the BglII – EcoRI sites of the sympUAST vector (Giordano et al., 2002) and the resulting construct was injected into w1118 embryos carrying isogenized Canton S-derived chromosomes. Two independent lines were selected, drkRNAi1.2 (drkR-1.2) on the second and drkRNAi2 (drkR-2) on the third chromosome. Because, the genetic background of the drkRNAi-generating transformants is w1118, all relevant drk mutant alleles were introduced to that genetic background. To generate UAS-RafWT a HindIII/XbaI fragment containing the entire Raf ORF (Rommel et al., 1997) was subcloned into pUAST and germline transformants were obtained in the Canton S-w1118 genetic background using standard methods. drkΔP24 mutant heterozygotes harboring UAS-Rafgof, UAS-RafWT, or UAS-drkRNAi transgenes and flies harboring both drkR-1.2 and drkR-2 were generated by standard genetic crosses. The strain carrying the UAS-RasV12S35 effector loop mutant was obtained from Dr. V. Budnik (University of Massachusetts Medical School, Worcester, MA) (Koh et al., 2002) and the transgene was introduced into the w1118 genetic background with repeated backrosses and then recombined onto the second chromosome bearing the drkΔP24 mutation in the same genetic background.
For behavioral experiments, nonbalancer-bearing progeny from crosses of CyO-balanced drk males to ry506 females were used. To generate flies for behavioral rescue, balanced drk mutant flies, homozygous for the transgene on the third chromosome were crossed to homozygous transgene bearing stocks and nonbalanced progeny were selected for testing. For conditional behavioral rescue, the transgenes were induced by three 20 min heat shocks delivered every 6 h, followed by a 4 h rest period at 25°C before training and testing. To abrogate DRK levels, ElavGAL4, c772GAL4, c739GAL4, 201YGAL4, MB247GAL4, c232GAL4 females were crossed en masse to drkR-1.2 and drkR-2 males. Progeny of the cross between w1118 females and drkR-1.2 and drkR-2 males were used as controls. Flies were placed at 29°C for 24 h for maximal transgene induction and allowed 45 min recovery at 25°C before training. For rescue experiments with UAS-Rafgof transgenes, c772GAL4;TubGAL80ts (McGuire et al., 2003) and c739GAL4,TubGAL80ts (Ferris et al., 2006) and control w1118 females were crossed en masse to drkΔP24/CyO;UAS-Rafgof, or UAS-Rafgof males. Progeny was reared at 18°C and resultant nonbalanced 4–6-d-old flies were used for behavioral experiments. To maximally induce the transgenes, flies were moved from 18°C to 31°C for 16–20 h followed with recovery at 25°C for 45 min before conditioning. Because UAS-RafWT resides on the first chromosome, males carrying UAS-RafWT in wild type and drk mutant background were crossed to c772GAL4 and w1118 females and nonbalanced females only, were used for rescue experiments.
Immunohistochemistry.
Immunohistochemistry and histology were performed as described previously (Skoulakis and Davis, 1996; Crittenden et al., 1998). The DRK distribution pattern was determined using two different rabbit polyclonal sera, one from M. Simon (Crittenden et al., 1998) and one generated for this study. The antibody we generated, was raised in rabbits against bacterially expressed full-length DRK protein fused at its N terminus with glutathione-s-transferase (GST). Its specificity was initially tested against homozygous embryos, which were found to lack signal (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Both antibodies were used at 1:1500, while the secondary at 1:1000. For DRK detection in heads of mutant flies and flies expressing drkRNAi under c772GAL4, 6 μm paraffin sections of control and mutant animals mounted on the same slide were obtained and challenged with a-DRK antibody used at 1:2000 dilution and secondary a-rabbit HRP at 1:2000 to increase the sensitivity of the assay. The HRP reaction was allowed to proceed for 30 s for all slides. Images from control and experimental head sections were obtained in the same session using the same acquisition settings for each photograph.
RT-PCR and Western blot analysis.
For detection of transgene specific transcripts, RNA was extracted within 30 min after the last induction as previously described (Philip et al., 2001; Mershin et al., 2004). Transgene specific transcripts were selected with a reverse primer specific for the unique SV40 derived sequence at the 3′ of the vector (Roman et al., 1999) and a forward primer specific to the drk cDNA. 10% of each RT was subjected to PCR. As a qualitative control of the RT, 200 ng of either rp49 or leo6.2 reverse primer was used, followed by PCR with rp49, or leo-specific primers respectively (Philip et al., 2001). For detection of DRK in Western blots, total protein extract equivalent of ¼ of an adult fly head was loaded per lane of 10% acrylamide gels, transferred to PVDF membranes and probed with the a-DRK antibody at 1:4000 and mouse anti-syntaxin (DSHB) at 1:2500. For detection of pMAPK and total MAPK levels, extract equivalent to 1 adult head was loaded per lane and the primary antibodies were used at 1:500 for mouse a-pMAPK (Sigma), and 1:2000 for rabbit a-MAPK (Cell Signaling Technology). Mouse a-tubulin (DSHB) at 1:2500 was used as an internal loading control. Four independent experiments were scanned, the band intensities were determined using ImageQuant 5.0 (Molecular Dynamics) and used to calculate ratios of pMAPK/MAPK. To assay the level of p-MAPK after training, w1118, drkΔP24, and drke0A heterozygotes were trained with 6 US/CS pairings as detailed below and immediately frozen on dry ice in prechilled 15 ml tubes. The tubes were rapidly vortexed to separate fly body parts and five heads were quickly selected and homogenized in Laemmli buffer containing protease inhibitor mix (Roche) and 1 mm Sodium orthovanadate. Blots to probe the level of p-MAPK after induction of transgenes in control and mutant backgrounds used extracts from dissected brains. Brains were dissected in cold PBS supplemented with the protease mix (Roche) and 1 mm Sodium orthovanadate and then transferred and lysed in similarly supplemented Laemmli buffer.
Behavioral analyses.
All experiments were performed in a balanced design, where all genotypes involved in an experiment were tested per day. The experimenter was blind to the genotype in experiments testing learning of mutant heterozygotes (see Fig. 2). Behavioral experiments were replicated at least once with flies from different crosses and a different time period (biological replicates). Although the absolute performance scores varied somewhat between biological replicates, the relative difference in the performance of mutants and controls remained the same.
Figure 2.
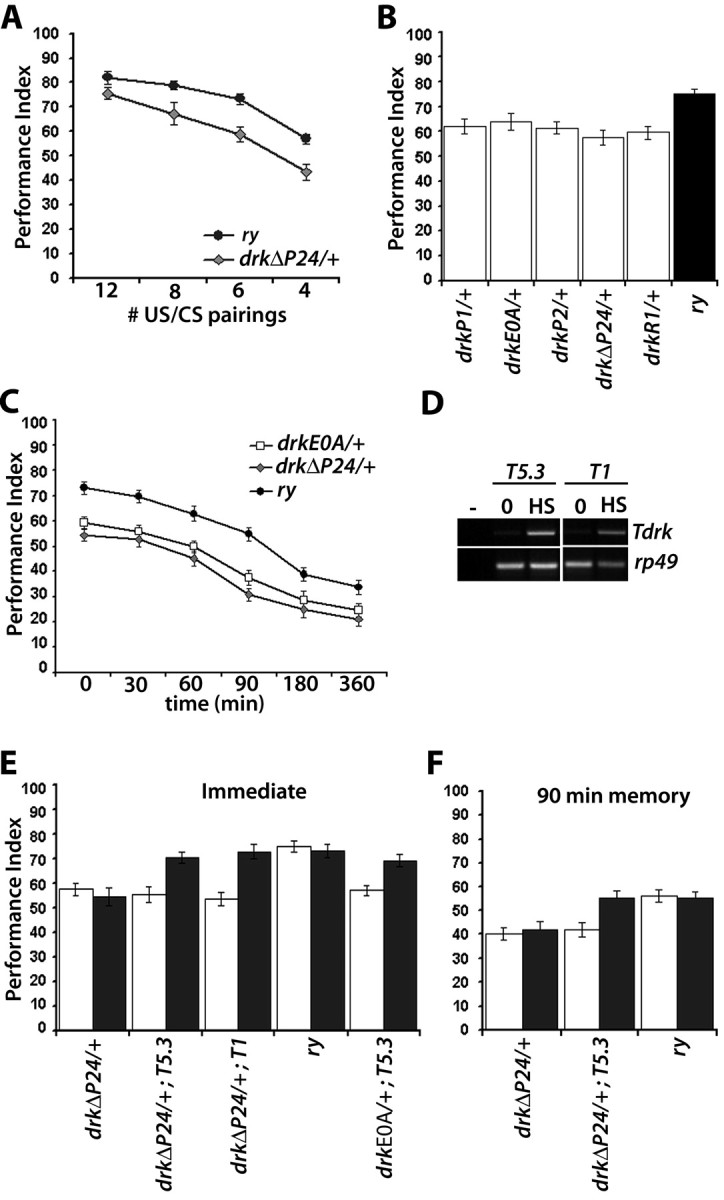
Learning and memory deficits of drk mutant heterozygotes. Mean performance indices and their SEMs (PI ± SEM) are shown for all experiments. A, Performance immediately after conditioned odor avoidance after training with the indicated number of pairings. n = 10 for all points. The performance of ry 506 flies was significantly different from that of and drkΔP24/+ after 6 and 8 pairings (p < 0.001, Student's t test), but not after 12 pairings (p < 0.02). B, Learning after training with 6 CS/US pairings of drk mutant alleles and controls. n ≥ 12. ANOVA indicated significant effects of genotype (F(5,83) = 13.956, p < 0.001) and subsequently confirmed for each allele against the control with Dunnett's tests. C, Memory of 6 CS/US training for two representative drk alleles. n ≥ 12 for each time point. Time 0 represents learning. Two-way ANOVA revealed significant effects of genotype (F(2,218) = 11.627, p < 0.001) and time (F(5,218) = 18.732, p < 0.001). Subsequent planned comparisons revealed significant differences (p < 0.001) between the drkΔP24 and drkE0A heterozygotes and controls at all time intervals, except at 360 min where the differences were significant at the p < 0.05 level (actual values, p = 0.016 and p = 0.028 for drkΔP24 and drkE0A, respectively). D, RT-PCR from heterozygous drk deletion mutants carrying either the drkT5.3 or the drkT1 transgenes subjected to three heat shock inductions, or left untreated (0) before RNA isolation. Amplification of rp49 transcripts served as a semiquantitative control. The empty lane displays PCR amplification products from nonreverse transcribed RNA. E, Learning of drk mutant heterozygotes bearing the drkT5.3, drkT1 transgenes and controls after induction (black bars), or without induction (open bars). n ≥ 10. ANOVA indicated significant effects of treatment (F(1,104) = 15.376, p < 0.001) and genotype (F(9,104) = 21.327, p < 0.001). Subsequent Tukey–Kramer test (α = 0.001) indicated significant differences between induced and uninduced drkΔP24/+; T5.3, drkΔP24/+; T1 and drkE0A/+; T5.3, but not between heat-shocked and nonshocked ry506 and drkΔP24/+. In addition, the performance of induced drkΔP24/+; T5.3, drkΔP24/+; T1 and drkE0A/+; T5.3 was not different from that of controls under the same conditions. F, Conditional rescue of the 90 min memory deficit of drk deletion heterozygotes. All strains were treated before conditioning as described above, trained, stored at the training temperature (24–25°C) and tested 90 min post-training. Group mean PIs ± SEM are shown for n ≥ 10. ANOVA indicated significant effects of treatment (F(1,64) = 21.368, p < 0.001) and genotype (F(5,64) = 14.521, p < 0.001). Subsequent Dunnett's tests confirmed significant differences in performance of heat shocked ry and drkΔP24/+ and non-heat-shocked drkΔP24/+; T5.3 (p < 0.001). However, the performance of heat-shocked drkΔP24/+; T5.3 was statistically indistinguishable from that of similarly treated ry and untreated ry flies.
Olfactory learning and memory in the negatively reinforced paradigm coupling aversive odors as conditioned stimuli (CS+ and CS−) with the electric shock unconditioned stimulus (US) (Tully and Quinn, 1985) was performed essentially as described previously (Philip et al., 2001; Mershin et al., 2004). The timing of stimulus delivery was kept proportional to that for the full 12 CS/US pairing protocol, such that 3 shocks were delivered in 15 s of continuous CS+ presentation, 6 pairings within 30 s and so on. Conditioning with discrete stimuli was performed according to Beck et al. (2000). Olfactory and shock avoidance assays and the nonassociative odor/shock pre-exposure (Preat, 1998) experiments were performed as described (Mershin et al., 2004; Acevedo et al., 2007a). We refer to the “3 min memory” earliest post-training performance assessment as learning, or immediate memory (IM) (Skoulakis and Davis, 1996). Data were analyzed parametrically with the JMP statistical package (SAS Institute Inc., Cary, NC) as described before (Philip et al., 2001; Mershin et al., 2004) and detailed in the text or figure legends.
Results
Preferential expression of drk in mushroom body neurons
The drk gene encodes a protein of 211 aa consisting entirely of a central RTK-binding SRC-Homology domain 2 (SH2) flanked by two SH3 domains (Pawson and Gish, 1992) and is the ortholog of the vertebrate GRB2 (Olivier et al., 1993; Simon et al., 1993; Raabe et al., 1995). The protein binds to the guanine exchange factor SOS via its N-terminal SH3 domain forming a complex essential for RAS activation. The C-terminal SH3 domain binds to DISABLED, which may link SRC-like tyrosine kinase signaling to the RAS/RAF/MAPK cascade (Le and Simon, 1998). Therefore, DRK plays an essential role in the initiation of multiple signaling cascades that potentially lead to MAPK activation.
Investigation of the role of DRK in olfactory learning and memory were initiated by the observation that reporter β-galactosidase in drkP1 heterozygotes accumulated preferentially in Kenyon cells (data not shown). We confirmed this observation and examined in detail the distribution of DRK in adult brain with the polyclonal antibody we generated. DRK was found preferentially distributed in MB axons, the pedunculus and the α, β and γ lobes (Fig. 1). The protein was undetectable in cell bodies (Fig. 1A), modest in the R4 neurons of the ellipsoid body (Fig. 1B), antennal lobe glomeruli (Fig. 1C,D,E,F) and very low in the calyces (Fig. 1A,E,F) and α′ and β′ lobes (Fig. 1C,E,F). This distribution is in agreement with previously published results using a different antibody (Crittenden et al., 1998). In addition, sections challenged with preimmune sera did not yield any staining and staining was significantly reduced in head lysates from mutants and animals with RNA interference (RNAi)-mediated reduction of the protein (see below). These results demonstrate the specificity of the antibody and its usefulness as a highly specific marker for axons of α, β, and γ MB neurons. Similar to DRK, GRB2 accumulates in the hippocampus and amygdala (Jin et al., 2001; Lamprecht et al., 2002), centers of learning in vertebrates.
Figure 1.
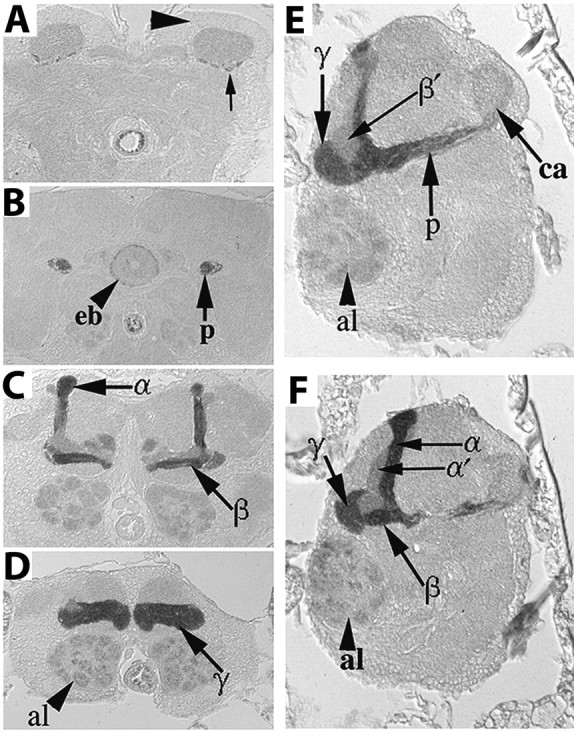
Distribution of DRK in the adult brain. Immunohistochemical detection of DRK in 5 μm frontal (A–D) and sagittal (E, F) paraffin sections through the adult brain. Dorsal is up in all photographs. A, DRK is absent from MB cell bodies (large arrowhead), while low levels appear in the calyces (ca) barely above general neuropil staining. Arrow points to four positively staining axonal fascicles. B, DRK accumulates within the pedunculus (p), and R4 neurons of the ellipsoid body (eb, arrowhead). C, Preferential DRK distribution within the α, β and γ (D) lobes. Low level of the protein is detectable in glomeruli of the antennal lobe (al, arrowhead in D). E, F, Saggital sections demonstrate the differential distribution of DRK within the pedunculus, α, β and γ lobes as indicated by the arrows. Anterior is to the left.
drk heterozygous mutants learn inefficiently
The striking distribution in the MBs suggested that as with other proteins highly enriched in these neurons (Heisenberg, 2003; Davis, 2004, 2005; Skoulakis and Grammenoudi, 2006; Berry et al., 2008), DRK may be required for olfactory learning and memory. However, homozygotes for drk mutations die at the larval or pupal stages (Olivier et al., 1993; Simon et al., 1993; Hou et al., 1995). Nevertheless, we reasoned that since the protein does not possess enzymatic activity, yet is essential for signal transduction of RTK-initiated signals (Lowenstein et al., 1992; Hou et al., 1995; Cheng et al., 1998; Feller et al., 2002), reduction in its amount may affect learning and memory detectably.
To determine whether reduction in DRK affects behavioral neuroplasticity, we subjected the deletion heterozygotes drkΔP24 expected to harbor 50% of the protein, to olfactory associative conditioning. Initial performance measurements are possible ∼3 min after initial association of the stimuli, but we will call this immediate memory “learning,” for consistency with published olfactory conditioning experiments. Initially we used the standard 12 CS/US pairings for training (Tully and Quinn, 1985), but although learning of drkΔP24/+ animals was lower than that of controls (Fig. 2A), it was not different statistically. However, reducing the number of pairings to 8, 6, or 4 (see Materials and Methods) revealed highly significant learning deficits in the mutants. Similar results were obtained in multiple independent experiments with drkΔP24/+ heterozygotes in both ry506 and w1118 genetic backgrounds (see below) and drkP1/+ animals (data not shown). These results demonstrate that the 20% decrease in learning after training with 6 or less pairings, represents a bona fide learning impairment reflecting the reduction in DRK dosage. This pairing-specific effect is independent of genetic background and drk allele and represents a highly significant reduction in learning, considering the multiple biochemical pathways subserving learning and memory (Skoulakis and Grammenoudi, 2006). Thus, it appears that under our experimental conditions, 12 CS/US pairings train control flies to “saturation,” or a performance plateau. These apparently over-training conditions, allow drk mutant heterozygotes to nearly overcome the limitation presented by reduced DRK and “catch up” with controls, yielding nearly similar performance indices. In contrast, reducing the pairings such that robust, but subplateau learning was attained by controls, revealed the deficit in the heterozygotes, suggesting that reduction in DRK affects the efficiency of learning, but not learning ability per se. We used the six-pairing protocol for all subsequent experiments because it consistently yielded maximal differences between mutants and controls.
To confirm the importance of DRK levels for efficient learning, we examined the performance of a collection of transposon insertions and point mutant heterozygotes (see Materials and Methods). Again, compared with ry506 controls, all mutant heterozygotes exhibited consistent and highly significant reductions in learning of the same magnitude as that of drkΔP24/+ animals (Fig. 2B). This ascertained that the deficit maps genetically to the drk gene and was not associated only with the deficiency-bearing chromosome. Because the learning deficit was similar for all heterozygotes, we selected the point mutant drkE0A, in addition to drkΔP24 for subsequent experiments. Furthermore, the inefficient learning phenotype of drk heterozygotes was independently verified using a discrete stimulus conditioning paradigm (Beck et al., 2000; Cheng et al., 2001; Mershin et al., 2004). Mutants performed significantly lower than equivalently trained controls, but importantly and in support of the inefficient learning notion, improved their performance upon additional pairings nearly the same as controls (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Nevertheless, mutants always appeared to perform similar to control animals trained with one less pairing. These results are consistent with the interpretation that reduction in functional DRK results in inefficient learning, which is exaggerated upon reduced training.
To determine how inefficient learning affects memory, retention of 6 CS/US training was assessed up to 360 min later. Mutant heterozygotes exhibited significantly lower performance 30, 60, and 90 min after training (p < 0.001). Maximal performance difference from controls was consistently observed 90 min post-training, while at 180 and 360 min the difference was smaller and significant at the p < 0.05 level (Fig. 2C). Interestingly, 90 min memory after 12 pairings, which produced learning approximately equivalent to ry506 and 8 CS/US, were significantly different from that of controls (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). These data suggest that although overtraining can effectively rescue the learning deficit of drk heterozygotes it does not affect memory of the association.
The learning deficits of drk mutant heterozygotes may reflect a reduction in the efficiency of DRK mediated signaling, or developmental deficits that disallow performance equivalent to that of controls. However, histological and immunohistochemical examination of the heads of mutant heterozygotes with multiple antigenic markers did not reveal any gross structural anomalies in the brain (supplemental Fig. 2, 1–6, available at www.jneurosci.org as supplemental material). Moreover, avoidance of the odors used as CS and electroshock (US) were not different from that of controls (Table 1), indicating that compromised perception of the CS and/or US stimuli could not account for the learning and memory deficits. In addition, the heterozygous mutants exhibited normal odor avoidance after pre-exposure to electric shock (Acevedo et al., 2007c) (supplemental Table 1, available at www.jneurosci.org as supplemental material), indicating that differential response to odor-shock exposure does not underlie the learning and memory deficits. Collectively, the results of the behavioral and histological analyses indicate that reduced functional DRK does not alter normal brain development or result in reduced sensory perception of the stimuli used for conditioning. Thus, the behavioral deficits of the mutant heterozygotes are bona fide learning and memory deficits.
Table 1.
Task-relevant sensory behaviors
| Genotype | Benzaldehyde | Octanol | Electric shock |
|---|---|---|---|
| drkP1/+ | 68.27 ± 2.95 | 54.82 ± 3.27 | 76.23 ± 1.94 |
| drkE0A/+ | 66.22 ± 3.16 | 58.26 ± 3.07 | 78.74 ± 2.55 |
| drkP2/+ | 71.09 ± 3.36 | 57.94 ± 2.78 | 73.93 ± 2.36 |
| drkΔP24/+ | 69.43 ± 2.76 | 60.12 ± 2.97 | 75.43 ± 1.94 |
| drkR1/+ | 64.98 ± 3.87 | 58.29 ± 3.28 | 74.29 ± 2.86 |
| +; T5.3 | 64.65 ± 2.85 | 52.82 ± 4.06 | 72.92 ± 2.74 |
| drkE0A/+; T5.3 | 62.28 ± 3.73 | 59.46 ± 4.16 | 74.05 ± 2.56 |
| drkΔP24/+; T5.3 | 70.14 ± 3.65 | 59.36 ± 3.76 | 73.24 ± 2.32 |
| drkΔP24/+; T1 | 69.32 ± 3.46 | 61.08 ± 4.17 | 76.48 ± 2.05 |
| ry506 | 65.71 ± 2.89 | 57.32 ± 2.98 | 74.61 ± 2.25 |
| UAS-drkR-2/+ | 73.40 ± 5.47 | 67.30 ± 7.60 | 77.98 ± 3.38 |
| UAS-drkR-2/c772 | 68.27 ± 7.51 | 59.91 ± 3.36 | 82.21 ± 3.45 |
| UAS-drkR-2/c739 | 70.30 ± 7.42 | 66.19 ± 2.52 | 83.20 ± 3.23 |
| UAS-drkR-2/c232 | 74.67 ± 8.65 | 68.88 ± 6.30 | 77.88 ± 4.19 |
| UAS-drkR-1.2/+ | 63.71 ± 3.51 | 50.32 ± 1.80 | 83.78 ± 3.20 |
| UAS-drkR-1.2/c772 | 72.36 ± 6.85 | 44.62 ± 4.54 | 85.66 ± 2.87 |
| UAS-drkR-1.2/c739 | 56.88 ± 7.69 | 50.86 ± 3.47 | 81.96 ± 1.08 |
| UAS-drkR-1.2/+;drkR-2/+ | 83.45 ± 5.18 | 60.17 ± 5.22 | 77.25 ± 2.64 |
| UAS-drkR-1.2/c772;drkR-2/+ | 84.46 ± 1.20 | 55.17 ± 5.17 | 75.27 ± 3.97 |
| UAS-RasV12S35/+ | 72.35 ± 3.29 | 57.29 ± 2.80 | 76.87 ± 2.93 |
| drkΔP24,UAS-RasV12S35/+ | 68.84 ± 3.47 | 62.54 ± 4.13 | 75.58 ± 3.26 |
| UAS-RasV12S35/c772 | 64.24 ± 4.69 | 59.63 ± 3.88 | 78.35 ± 3.89 |
| c772/+; TubGAL80ts/+ | 70.36 ± 3.82 | 65.62 ± 2.96 | 81.29 ± 2.53 |
| drkΔP24,UAS-RasV12S35/c772;TubGAL80ts/+ | 72.67 ± 3.27 | 64.82 ± 4.81 | 78.43 ± 3.16 |
| drkΔP24,UAS-RasV12S35/+;TubGAL80ts/+ | 69.64 ± 4.72 | 66.40 ± 3.95 | 73.62 ± 3.68 |
| UAS-Rafgof/+ | 64.34 ± 3.94 | 79.02 ± 6.11 | 84.26 ± 3.65 |
| drkΔP24/+;UAS-Rafgof/+ | 61.50 ± 3.64 | 81.71 ± 5.38 | 76.48 ± 4.44 |
| drkΔP24/c772;UAS-Rafgof/TubGAL80ts | 69.02 ± 5.68 | 87.28 ± 5.46 | 78.98 ± 6.02 |
| drkΔP24/c739,TubGAL80ts;UASRafgof/+ | 64.75 ± 7.71 | 82.20 ± 5.55 | 89.29 ± 3.89 |
| c772;UAS-Rafgof/TubGAL80ts | 62.09 ± 6.07 | 75.05 ± 4.55 | 90.76 ± 3.76 |
| c739,TubGAL80ts;UAS-Rafgof/+ | 58.02 ± 4.02 | 85.60 ± 5.44 | 78.20 ± 3.72 |
| UAS-Rafwt/+ | 81.52 ± 3.37 | 50.29 ± 3.31 | 81.89 ± 4.19 |
| UAS- Rafwt /+;drkΔP24/+ | 85.41 ± 1.92 | 54.82 ± 7.08 | 88.82 ± 2.83 |
| UAS- Rafwt /+;c772/+ | 84.28 ± 5.79 | 47.75 ± 6.91 | 88.59 ± 4.86 |
| UAS- Rafwt/+;drkΔP24/c772 | 84.24 ± 2.09 | 55.41 ± 5.17 | 77.60 ± 6.18 |
Avoidance of the aversive odor stimuli (CS) and electric shock (US) is shown for all relevant strains. The performance of strains with ry506 genetic background (upper group) was compared to that of their genetic control (ry506), and ANOVA did not indicate differences (n ≥ 7 for all). Similarly, the performance of strains expressing the RNAi-mediating transgenes in the second group was compared with that of their heterozygous transgene controls, and significant differences were not uncovered (n >6). Avoidance of strains in the third group was compared to that of their proper control UAS-RasV12S35/+, but differences were not significant (n ≥ 6). The performance of strains in the fourth group was also compared to that their proper control UAS-Raf gof/+ or UAS-Raf wt/+, respectively, and significant differences were not revealed (n ≥ 7). All strains were not tested simultaneously, although all strains within a group were. Thus, statistical analyses for performance differences were performed exclusively within each group.
Conditional rescue of the learning and memory deficits of drk heterozygotes
We attempted conditional rescue of the behavioral deficit by regulated expression of drk transgenes to unequivocally demonstrate that the inefficient learning and memory of mutants does not have a developmental origin and to investigate whether the protein is required acutely for these processes. Two independent heat inducible drk transgenes, T1 and T5.3, were crossed into the drkΔP24 and drkE0A mutant backgrounds and upon induction, transgene-specific mRNAs were detected in heads of animals that bear them (Fig. 2D). Accumulation of T1 or T5.3 transcripts in wild-type animals did not result in enhanced learning or memory (data not shown). We concentrated on attempting to reverse the learning and the 90 min deficits because memory at this interval appeared affected maximally in drkΔP24 heterozygotes relative to controls (Fig. 2C). Learning (Fig. 2E) and 90 min memory (Fig. 2F) of transgene-bearing mutant heterozygotes were indistinguishable from that of controls after induction. The effect was observed with both transgenes and both drkΔP24 and drkE0A heterozygotes. In contrast, similarly treated mutant heterozygotes or uninduced transgenes in the mutant background did not rescue the learning and memory deficits. Transgene induction did not result in deficits or improvements in learning, or changes in sensorimotor behaviors (Table 1). The data strongly indicate that the associative learning and memory deficits do not originate in aberrant development below histological detection and DRK is required acutely for learning and memory formation.
DRK is required within the MBs for normal learning
The preferential distribution of DRK within the MBs suggested that the learning and memory deficits of the mutants are precipitated by reduced levels of the protein within these neurons. To address this issue directly and independently demonstrate that neuroplasticity deficits are not the outcome of altered development, we adopted a strategy of spatial-specific, RNAi-mediated abrogation of the protein. Thus, we generated independent strains (drkR-1.2 and drkR-2, see Materials and Methods) harboring transgenes able to generate double stranded interfering RNAs by symmetric transcription (Giordano et al., 2002).
Pan-neuronal expression with Elav of either independent RNAi-mediating transgene, or both simultaneously, did not result in lethality. Simultaneous pan-neuronal expression of drkR-1.2 and drkR-2 resulted in highly significant reduction of DRK, equivalent to that in heterozygotes of the null allele drkΔP24. Further reduction in DRK was achieved by expressing one of the silencing transgenes in the presence of drkΔP24 (Fig. 3.A). These results suggest that expression of the silencing transgenes yields an effect on DRK approximately equivalent to a heterozygote for a hypomorphic mutation, while expression of both transgenes appeared equivalent to a null heterozygote. The reduction of DRK in drkΔP24 heterozygote head lysates was in fact reflected in the level of the protein within the α/β and γ lobes of the MBs (Fig. 3B, 1 and 2 vs 3 and 4). To drive the transgenes within the MBs, we used the c772 driver because it is expressed developmentally late in pupal MBs (Armstrong et al., 1998) and because it is expressed in α, β and γ lobes (Mershin et al., 2004), in a pattern closely resembling that of DRK. In addition, we used the α/β lobe specific driver c739. A significant reduction of DRK in the MBs, particularly evident in the α/β lobes, was detectable upon expression of drkR-2 with c772 in otherwise normal (Fig. 3B, 5 and 6) or drkΔP24 heterozygous mutant background (Fig. 3B, 7 and 8). Similar results were obtained using whole mount dissected brain preparations and confocal microscopy (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Therefore, the notable difference between the null heterozygotes and animals with RNAi-mediated abrogation of DRK is that only MB neurons where the protein accumulates preferentially are affected in the latter, instead of the entire brain in the former.
Figure 3.
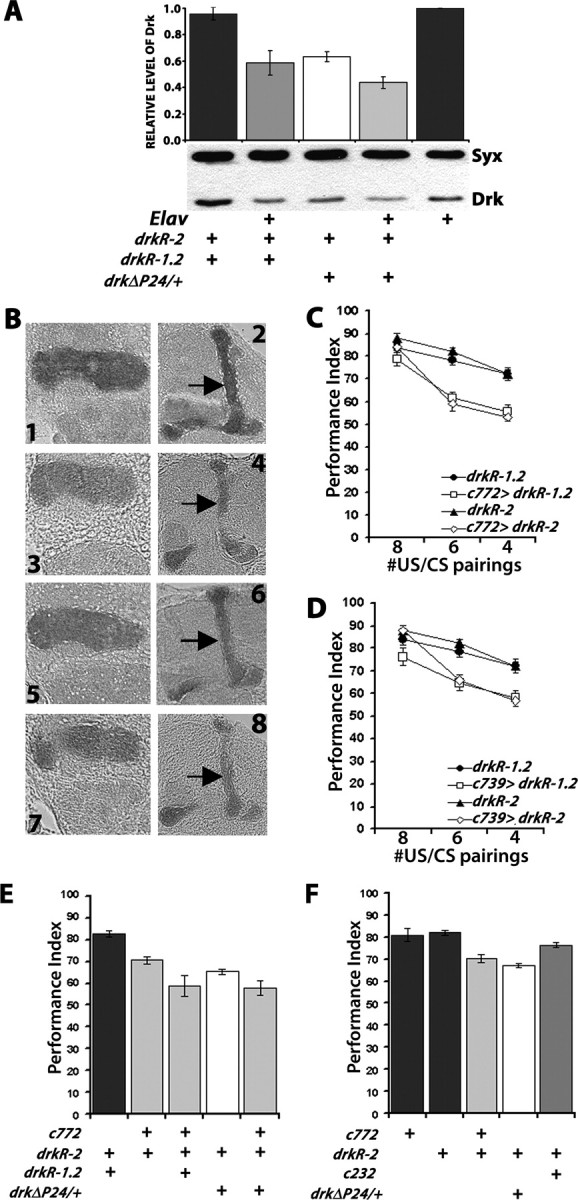
RNAi-mediated abrogation of DRK within the MBs phenocopies the drk mutant phenotype. A, Accumulation of DRK in head lysates from controls, drkΔP24/+ and animals driving pan-neuronal expression of drkR-1.2 and drkR-2 simultaneously with Elav. A representative semiquantitative Western blot of such lysates from the indicated genotypes challenged with a-DRK and a-SYNTAXIN as loading control is shown below, while quantification of five independent such experiments is shown above. Means ± SEM are shown. The level of DRK normalized over the level of SYNTAXIN in control strains was arbitrarily set to 1 (Philip et al., 2001). DRK in extracts from heterozygous mutants and Elav driven transgenes was statistically significant from that in controls (p < 0.001, planned comparisons). No difference was observed between drkR-1.2/+; drkR-2/+ animals controlling for potential effects of the insertions on DRK levels or Elav/+ controls (p > 0.3). B, Reduction of DRK levels in the MBs upon RNAi transgene expression illustrated in 6 μm paraffin sections challenged with a DRK antibody. 1,2: drkR-2/+ control animals. 3,4: drkΔP24/+; drkR-2/+ mutant heterozygotes. 5,6: c772/+; drkR-2/+. 7,8: drkΔP24/c772; drkR-2/+. Sections from all genotypes were processed in parallel and each slide contained control animals and all experimental genotypes. Representative images at the levels of the γ-lobe (1, 3, 5, 7) and α/β lobes (2, 4, 6, 8) are shown. Arrows point to the α lobes on the respective genotypes where the reduction in DRK is most obvious. C, Abrogation of DRK in α/β and γ MB lobes with c772 impairs learning and phenocopies the drk mutant phenotype. Group mean PI ± SEMs are shown, n ≥ 10. ANOVA showed significant effects of genotype (F(3,122) = 16.006, p < 0.0001) and number of CS/US pairings (F(2,122) = 19.185, p < 0.0001). The performance after 8 CS/US was not significantly different in experimental animals from their respective undriven heterozygous transgene controls (p > 0.53). However, with reduced pairings the effects were significant (planned comparisons, p < 0.0001). D, Abrogation of DRK in α/β MB lobes with c739 impairs learning and phenocopies the drk mutant phenotype. Group mean performance indices and their SEMs (PI ± SEM) are shown, n ≥ 7. ANOVA showed significant effects of genotype (F(3,88) = 13.282, p < 0.0001) and number of pairings (F(2,88) = 19.168, p < 0.0001). The performance after 8 CS/US was not significantly different in experimental animals with their respective undriven heterozygous transgene controls (p > 0.56). However, with reduced pairings the effects were significant (planned comparisons, p < 0.0001). E, The learning impairment is dependent on the amount of DRK protein within the MBs. PI ± SEMs are shown, n ≥ 9. ANOVA indicated significant effects of genotype (F(4,56) = 14.878, p < 0.0001). Subsequent contrast analysis showed significant differences in the performances of drkR-1.2/+; drkR-2/+ control animals (black bars) and c772/+; drkR-2/+ (p < 0.0005). The performance of c772/+; drkR-2/+ was also significantly different from that of drkΔP24/c772; drkR-2/+ (p < 0.001), and from drkR-1.2/c772; drkR-2/+ (p < 0.05). F, Abrogation of DRK only within the MBs impairs learning. PI ± SEMs are shown, n ≥ 9. ANOVA revealed significant effects of genotype (F(4,57) = 17.480, p < 0.0001). Planned comparisons did not reveal any differences between drkR-1.2/+; drkR-2/+ controls 2/+, 772/+ and drkR-2/c232 (p > 0.06). The differences between controls and c772/+; drkR-2/+ and drkΔP24/+; drkR-2/+ remained significant p < 0.0001).
Reduction of DRK within the MBs with expression of either RNAi-mediating transgene under c772 or c739 did not alter sensorimotor responses to electric shock and aversive odors (Table 1), task-relevant nonassociative behaviors (supplemental Table 1, available at www.jneurosci.org as supplemental material) or the gross structure of the MBs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), in accord with results obtained from the mutant heterozygotes. In contrast, DRK abrogation within the MBs resulted in a CS/US pairing-specific reduction in learning. Whereas learning of animals expressing either transgene was indistinguishable from that of controls after 8 CS/US pairings, significant differences were uncovered after limiting the pairings to 6 or 4 (Fig. 3C,D). Similar results were obtained with animals expressing drkR-1.2 under the MB-specific MB247 and the more broadly expressed, but enriched within a subset of α and γ lobe neurons 201Y GAL4 driver (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). These results indicate that DRK is required within the MBs, especially the αβγ neurons, for normal learning and are consistent with the notion that reduction of the protein within these neurons underlies the inefficient learning of the heterozygous mutants. The relatively milder reduction of DRK upon expression of either transgene compared with the 50% reduction in null heterozygotes, is the most likely explanation for the manifestation of learning deficits after training with 6 or less pairings (Fig. 3C,D) and not with 8 as with drkΔP24 heterozygotes (Fig. 2A). Consistent with this, two copies of the RNAi-mediating transgenes yielded a larger deficit than that precipitated by either transgene, or the heterozygous null mutation and similar to that of the null heterozygote combined with one transgene (Fig. 3E). Thus, the inefficient learning phenotype is proportional to the level of DRK reduction within the MBs in consequence to the number of RNAi-mediating transgenes expressed in these neurons.
In contrast, DRK abrogation in ellipsoid body neurons where low levels of the protein were observed did not affect learning (Fig. 3F). This result supports the specificity of the learning deficit to DRK abrogation within the MBs. The negligible contribution of DRK in neurons outside the MBs in the learning and memory impairments is also supported by the fact that although null heterozygotes expressing an RNAi-mediating transgene in the MBs apparently harbor the least DRK (Fig. 3A), their learning deficit was not more severe than animals simultaneously expressing drkR-1.2 and drkR-2 in the MBs (Fig. 3E). Therefore, we have revealed a similar learning deficit in drk mutant heterozygotes and by limiting the amount of DRK specifically within the MBs, demonstrating a dose-dependent essential role of protein for normal olfactory learning.
A role for DRK in memory
Heterozygous drk mutants exhibited compromised 90-min memory proportional to their learning deficits (Fig. 1C), which were rescued by conditional transgene expression (Fig. 2F). To determine whether a similar phenotype was exhibited by animals with abrogated DRK within MBs, we examined 90-min memory in flies expressing the RNAi-mediating transgenes. To get robust memory, we trained animals with 8 CS/US instead of the usual 6 used for assessment of learning (Fig. 3). As anticipated, learning was not significantly different in controls and animals expressing the drkR-2 or drkR-1.2 transgenes (Fig. 4A; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In contrast, 90-min memory of the association was significantly different in animals with RNAi-mediated DRK abrogation than equivalently trained controls and the difference was consistently more pronounced under c772 (Fig. 4A; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). A similar phenotype was observed with both drkΔP24/+ and drkE0A/+ animals trained with 12 CS/US (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Therefore, although intensive training largely eliminated the differences in learning/immediate performance, 90-min memory remained deficient. This may indicate that although performances were not statistically different, training with 8 CS/US did not actually yield equal learning in controls and mutants. This learning difference became apparent as memory deficits because this measure afforded better resolution. Alternatively, 8 CS/US training resulted in equivalent learning as suggested by the immediate performance scores, but the memory deficit was revealed because DRK is required for memory independent of its role in learning and memory is more sensitive to abrogation of the protein in the MBs than learning is.
Figure 4.
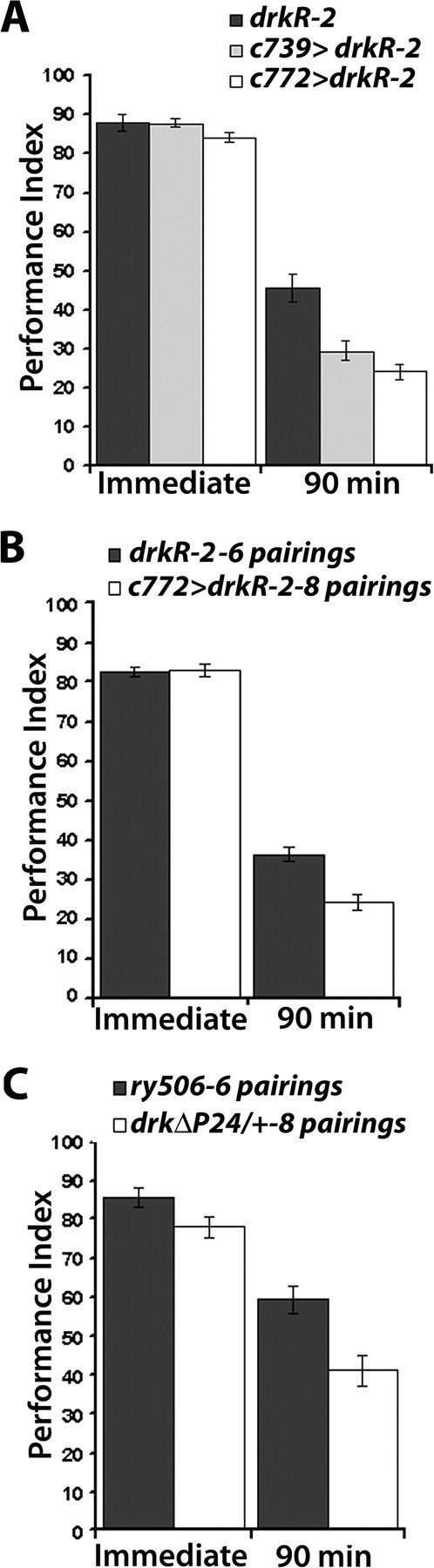
Differential effects of DRK reduction on 90 min memory. Mean performance indices and SEMs (PI ± SEM) are shown for the indicated genotypes. A, Abrogating DRK in the MBs impairs 90 min memory. Initial training with 8 CS/US produced equal immediate performance between control animals and animals expressing drkRNAi (F(2,15) = 1.761, p > 0.2), n = 5. ANOVA for 90 min memory of the training revealed significant effects of genotype (F(2,50) = 13.490, p < 0.0001). n ≥ 15. B, Training RNAi transgene expressing animals with increased number of pairings does not improve the memory impairment, revealing a specific effect of DRK abrogation in the MBs on memory. Learning was equal between under-trained controls (6 pairings) and drkR-2 expressing animals trained with 8 CS/US. (F(2,12) = 0.029, p > 0.86, n = 6). In contrast, 90 min memory remained impaired in RNAi-expressing flies compared with under-trained controls (F(1,46) = 18.057, p < 0.0001, n ≥20). C, The 90 min memory impairment remained unaltered by more intensive training of drkΔP24/+ heterozygotes indicating a memory specific impairment. ANOVA did not reveal differences between undertrained controls and drkΔP24/+ heterozygotes trained with eight pairings (F(1,12) = 2.327, p > 0.16, n = 6). Ninety-minute memory was significantly impaired in drkΔP24/+ flies compared with under-trained controls (F(1,16) = 5.978, p < 0.05, n ≥7).
To investigate whether the differential effect on 90-min memory resulted from decreased learning in animals with reduced DRK, we trained control animals with 6 CS/US, while animals with lower amounts of DRK in the MBs were trained more intensely with 8 CS/US. Although this differential training regimen clearly yielded equal learning, 90-min memory of the more intense training was still significantly reduced in drkR-2-expressing animals compared with that of lesser trained controls (Fig. 4B). This phenotype was not particular to RNAi-mediated abrogation of DRK because we applied differential training to drkΔP24 heterozygotes in their original ry506 genetic background with similar results (Fig. 4C). In congruence, mutant heterozygotes trained with three discrete pairings displayed reduced 30 min memory from that of control flies trained to approximately equivalent levels with 2 CS/US (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). These results indicate that in addition to its role in learning, DRK appears to independently affect 90 min memory formation or stability within the MBs.
DRK engages RAF for learning, but not for 90 min memory
Signals that engage DRK are known to eventually activate MAPK (Perrimon et al., 1995), and pivotal members of the classical RAS/RAF cascade including MAPK are expressed in the adult central brain (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Hence, to investigate whether DRK reduction affects signaling through this pathway, we determined the level of activated, diphosphorylated MAPK (pMAPK) in head lysates of mutant heterozygotes and animals expressing drkR-2 and drkR-1.2. Compared with total MAPK, the phosphorylated protein was reduced ∼50% in all animals with reduced DRK levels (Fig. 5A). Therefore, reduction in DRK appears to affect the levels of phosphorylated MAPK, but not the amount of total MAPK, suggesting impairment in signaling possibly through RAS and RAF.
Figure 5.
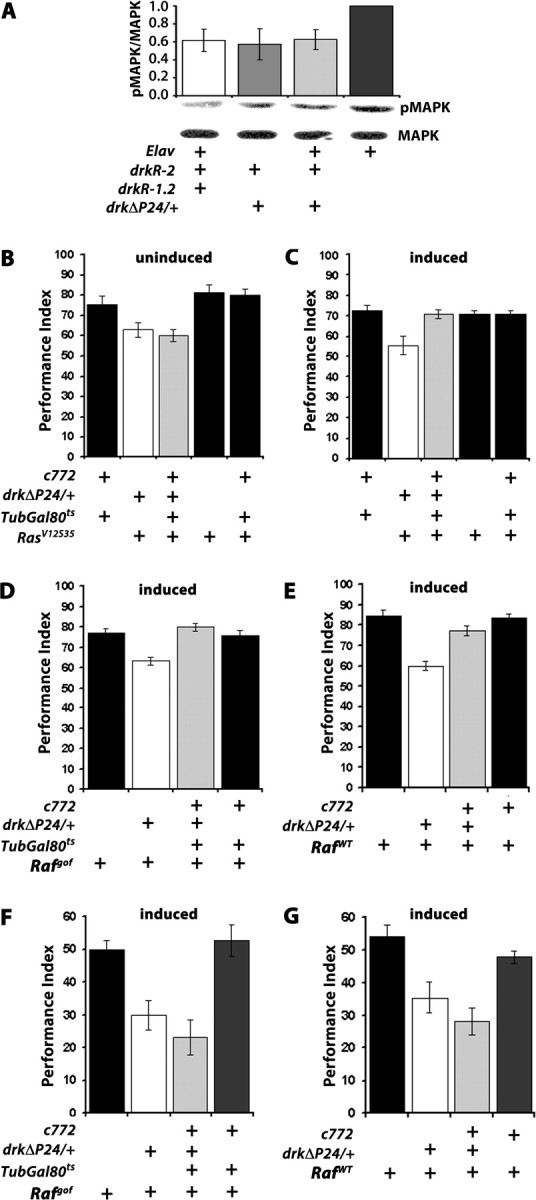
Rescue of the learning, but not the 90 min memory deficit of drkΔP24 heterozygotes by conditional expression of Ras and Raf transgenes in the MBs. A, MAPK phosphorylation is impaired upon abrogation of DRK in the adult nervous system. A representative semiquantitative Western blot is shown below the quantitative data from four independent blots. The ratio of pMAPK/MAPK in the control Elav/+ strain was arbitrarily set to 1. The mean ± SE are shown. ANOVA indicated significant differences between genotypes (F(3,19) = 3.853, p < 0.05). Compared with control, phosphorylated MAPK was significantly reduced in Elav/+; drkR-1.2/+; drkR-2/+ (p < 0.01) and drkΔP24/+; drkR-2/+ (p = 0.01) while the difference from Elav/+; drkΔP24/+; drkR-2/+ was significant at p = 0.02 (Student's t test). B, Performance after 6 CS/US training in flies carrying a RasV12S35 effector loop mutant transgene in a mutant background (gray bar) kept inactive before training (uninduced), did not perform better than mutant animals without it (open bars). Mean performance indices and SEMs (PI ± SEM) are shown as in all experiments below. ANOVA indicated genotype effects (F(4,30) = 5.991, p < 0.0001, n = 6). However, subsequent contrast analysis demonstrated that the differences were among control groups (black bars) and flies carrying the drkΔP24 mutation (p < 0.001), and the differences between the later two groups were not significant (p < 0.57). C, Expression of the RasV12S35 transgene in the MBs (induced) by inactivation of the Gal80ts rescued the learning deficit of drkΔP24 animals (open bar) as indicated by ANOVA (F(4,42) = 6.407, p < 0.0001, n ≥ 8). Contrast analysis demonstrated highly significant differences (p < 0.0003) in the performance of animals carrying the drkΔP24 mutation, but expressing the transgene (gray bar) and those that do not (open bar), while the performance of the former was not different from that of controls (p < 0.84). D, Induction of constitutively active RAF (Rafgof) in the adult MBs rescues learning after 6 CS/US training. Performance only after induction of the transgene is shown, as uninduced transgene did not have any effect on the mutant phenotype as shown above for the RasV12S35 transgene. ANOVA revealed significant effect of genotype (F(3,57) = 8.441, p < 0.0001, n ≥ 10) and treatment (F(1,87) = 34.02, p < 0.0001). Contrast analysis indicated significant differences between drkΔP24/+; Rafgof/+ animals and flies expressing the transgene (p < 0.0002), indicating full rescue of the defect. No differences were observed between flies expressing the transgene and control groups (p < 0.14). E, Partial rescue of drkΔP24heterozygote learning deficits upon conditional accumulation of wild-type RAF protein in the MBs. Performances only after transgene induction is shown. ANOVA revealed differences among RAF accumulating strains (F(3,35) = 20.816, p < 0.0001, n ≥ 7). Subsequent contrast analysis showed significant differences (p < 0.0001) between drk mutant heterozygotes not expressing the Rafwt transgene and those expressing it under c772. However, the performance of Rafwt/+; drkΔP24/+ (gray bar) was different (p < 0.05) from that of Rafwt/+and Rafwt/+; c772/+ animals. F, The drkΔP24/+ 90 min memory impairment is not reversed upon conditional expression of constitutively active RAF in the MBs. Only the performance of animals after induction of the transgenes and 6 CS/US training is shown. ANOVA suggested significant differences (F(3,37) = 10.449, p < 0.0001, n ≥ 8). Subsequent Dunnett's tests using the performance of Rafgof/+ as control indicated significant differences with drkΔP24/+; Rafgof/+ (p < 0.005) and c772/drkΔP24; Rafgof /TubGAL80ts (p < 0.0001) suggesting a lack of rescue. (The difference with c772/+; Rafgof/TubGAL80ts was not significant, p > 0.7). G, The drkΔP24/+ 90 min memory impairment is not reversed upon expression of wild-type RAF in the MBs after 6 CS/US training. ANOVA indicated significant differences (F(3,27) = 9.926, p < 0.0002, n ≥ 7), and Dunnett's tests indicated that compared with the performance of Rafwt/+, that of Rafwt/+; drkΔP24/+ was significantly different (p < 0.007) and importantly so was that of Rafwt/+; c772/drkΔP24 (p < 0.0002) indicating lack of rescue. Performances after transgene induction are shown.
Since RAS and RAF have been reported essential for learning in vertebrates (Costa et al., 2002; Weeber et al., 2002; Chen et al., 2006), we aimed to determine whether these molecules are engaged by the DRK-initiated signal within the MBs. To that end, we investigated whether the learning and memory deficits of drk mutant heterozygotes could be ameliorated or reversed by temporally controlled expression of constitutively active ras and raf transgenes within the MBs. Tissue and temporal specific expression of such transgenes was achieved with the TARGET system (McGuire et al., 2003, 2004b). We used c772 and c739 because they were the most restrictively expressed MB Gal4 drivers that yielded an effect with the drkRNAi transgenes. Initially, we attempted rescue with the constitutively active RASV12 protein (Szüts et al., 1997). However, its conditional accumulation within MBs of adult control animals and drkΔP24 heterozygotes resulted in severe learning deficits, without apparent structural changes in these neurons (A. Moressis and E. M. C. Skoulakis, unpublished observations). Although the results suggest that constitutive activation of RAS in the MBs perturbs normal learning, they disallow conclusions regarding potential DRK/RAS interactions within these neurons. However, RASV12 has been reported to constitutively activate not only RAF, but also PI3-kinase and Ral-GDS (Karim and Rubin, 1998; Koh et al., 2002), and thus the above-mentioned learning deficit could result either from simultaneous perturbation of all three signaling pathways, or one of them alone. To specifically probe the effect of Ras on MAPK phosphorylation, which is decreased in drk mutants and RNAi-expressing animals, we used animals bearing the RasV12S35 double mutant transgene, which activates constitutively only RAF (Karim and Rubin, 1998; Koh et al., 2002). The transgene insertion alone, or combined with the c772 driver but kept silent by GAL80ts (McGuire et al., 2004a) did not rescue the deficit of drkΔP24/+ animals, or affect learning adversely (Fig. 5B). In contrast, expression of the RasV12S35 transgene by inactivation of the GAL80ts suppressor restored normal learning to drkΔP24 heterozygotes, while it did not appear to affect learning in control animals (Fig. 5C). These results suggest that impaired DRK signaling to RAF via RAS likely underlies the learning deficits of drk heterozygotes.
To examine the potential involvement of RAF in the DRK mediated signal directly, we used a transgene encoding a constitutively active RAF kinase (Rafgof) because it lacked the N-terminal regulatory domain (Brand and Perrimon, 1994). While keeping the transgene silent did not alter the learning deficit of drkΔP24/+ (supplemental Fig. 4D, available at www.jneurosci.org as supplemental material), conditional expression of Rafgof in adult MBs reversed the learning deficit of drkΔP24 heterozygotes (c772/drkΔP24; UAS-Rafgof/TubGAL80ts), while expression of the transgene in control animals (c772/+; UAS-Rafgof/TubGAL80ts) did not result in impaired or enhanced learning (Fig. 5D). Similar results were obtained under c739 (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). Thus, rescue of the drkΔP24 heterozygote impairment was not a consequence of enhanced learning upon Rafgof expression resulting in a performance plateau before that of control animals. This interpretation was confirmed by limiting CS/US pairings to three. This would be expected to exaggerate any potential enhanced learning of Rafgof expressing animals, thus explaining the apparent “rescue,” but yielded similar performances for both strains (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). In addition, we attempted to rescue the learning impairment of drkΔP24 heterozygotes with a full-length Raf transgene (UAS-RafWT). Unlike Rafgof, paneuronal or MB-restricted expression of the wild-type protein did not result in lethality, thus it was not necessary to use the TARGET system to restrict its expression temporally. Rather, we drove it in the MBs with the late-expressing c772 (Armstrong et al., 1998; Mershin et al., 2004). Accumulation of the wild-type RAF in MB neurons improved immediate performance of drkΔP24 heterozygotes significantly, but not to the level of equivalently trained controls (Fig. 5E). It appears then that although it accumulates nearly equivalently with RAFgof (data not shown), activation of the wild-type protein was limited, likely due to the 50% DRK reduction in drkΔP24 heterozygotes. In contrast, elevation of the constitutively active kinase yielded full rescue because it does not require DRK for activation. Similarly, MB-specific accumulation under c772 of both Rafgof and RafWT resulted in full reversal of the learning deficit of drkE0A/+ flies (supplemental Fig. 4E, available at www.jneurosci.org as supplemental material). Full rescue of the drkE0A/+ learning deficit with RafWT expression is consistent with the mutant being a strong hypomorph compared with the more demanding rescue of the drkΔP24 null allele.
Surprisingly, neither RAFgof, nor RAFWT accumulation in the MBs rescued 90 min memory (Fig. 5F,G). Since the transgenic proteins accumulate in the MBs in addition to the endogenous kinase, it is unlikely that activated RAF, at least in the case of the constitutively active protein, does not suffice to support 90 min memory, unless it is unstable and falls below a critical threshold quickly after training. Moreover, the full rescue of the drkΔP24/+ learning defects with RAFgof predicts that memory would be normal if it simply was a consequence of learning. However, our results support the alternative hypothesis that RAF activation via DRK is essential for normal learning only, and that parallel or subsequent engagement of signaling molecules other than RAF is required for memory. This hypothesis is further supported by the ability of DRK to engage different molecules by its two SH3 domains (Rubin, 1991; Raabe et al., 1996; Le and Simon, 1998; Feller et al., 2002).
Deficient MAPK activation in drk mutant heterozygotes
To substantiate independently the results of the behavioral analyses and explore the potential mechanism(s) underlying the apparent dual effects of DRK reduction on learning and memory, we investigated pMAPK levels in adult brains for two reasons. First, they were significantly reduced in animals with reduced DRK (Fig. 5A). Second, we hypothesized that memory, but not learning may be affected by the altered pMAPK levels since persistent MAPK activation has been linked to memory formation (Weeber and Sweatt, 2002; Sweatt, 2004; Thomas and Huganir, 2004).
The dynamics of MAPK activation in the brains of adult flies were investigated at three particular time points after 6 US/CS associative training (Fig. 6A). At 2 min post-training, roughly coincident with behavioral assessment of learning, pMAPK levels were substantially elevated in control animals and drk mutant heterozygotes. However, pMAPK levels in controls were not significantly different from those in drkΔP24/+ and drkE0A/+ animals (Fig. 6B). In contrast, 15 min post-training, pMAPK remained elevated in controls, but was not different from the level in naive animals for drkΔP24 and drkE0A heterozygotes. Furthermore, although pMAPK appeared to return to naive levels 90 min post-training, in drkΔP24/+ and drkE0A/+ it was significantly further reduced not only compared with controls, but even below the level in naive animals (Fig. 6B). These results indicate that 50% reduction in DRK does not affect MAPK activation acutely after training, but rather impairs significantly the sustained activation of the kinase, particularly evident at 90 min. This suggests that in agreement with previous reports (Hoeffer et al., 2003; Sweatt, 2004; Thomas and Huganir, 2004), failure to sustain pMAPK levels likely underlies the 90 min memory deficit. Furthermore, considering the behavioral rescue of learning but not 90 min memory by raf tranagenes in the MBs, these results further support the notion that RAF activity is involved in acute elevation of pMAPK post-training, but not for its sustained activation. Therefore, the 90 min memory deficit of drk heterozygotes is likely independent of RAF as indicated by the behavioral analyses.
Figure 6.

Impaired MAPK activation in drk mutant heterozygotes. A, Representative Western blots probing the level of pMAPK compared with total levels of the protein (MAPK) after associative conditioning as detailed in Materials and Methods. The level of Tubulin in each of the indicated control and mutant head lysates was used to normalize protein levels in each lane and for quantification. Lysate from a single head equivalent was loaded per lane. Naive denotes lysates from w1118 animals that underwent all manipulations in parallel except for associative learning. Blots from animals allowed to rest 2 min, 15 min or 90 min post-training (as indicated in the graph below in B), followed by lysate preparation are shown left to right. B, The mean ± SEM from at least 3 independent determinations of the relative amount of pMAPK to that of MAPK in the samples is shown. The levels of pMAPK and MAPK were determined relative to Tubulin to normalize loading differences. This ratio for naive animals was fixed to 1. ANOVA did not indicate significant differences among the samples obtained 2 min after training (F(3,19) = 1.350, p < 0.29, n = 4). In contrast, differences among the samples obtained 15 min post-training were significant (F(3,15) = 3.998, p < 0.03, n = 3). Subsequent pairwise t tests showed significant difference from naive only for wild-type flies (p < 0.009), but not for drkΔP24/+ or drkE0A/+ (p < 0.45 and p < 0.29, respectively). Contrast analysis showed that w1118 pMAPK levels were significantly different from drkΔP24/+ and drkE0A/+ (p < 0.02). Ninety-minute post-training ANOVA indicated significant effects of genotype (F(3,17) = 7.638 p < 0.003, n = 4). Pairwise t tests showed that wild-type pMAPK levels were not different from that in naive animals (p < 0.16), indicating that MAPK phosphorylation returned to naive levels. In contrast, pMAPK levels in drkΔP24/+ and drkE0A/+ were different (lower) than that in naive flies (p < 0.04 and p < 0.01, respectively). Contrast analysis also showed that pMAPK levels in w1118 were significantly higher than those in drkΔP24/+ and drkE0A/+ (p < 0.004). C, Western blots probing the level of pMAPK in single dissected brains of naive flies carrying Rafgof (left side), or RafWT (right side) transgenes in control (w1118), or drkΔP24/+ mutant background, with or without transgene induction by incubation at 30°C as indicated by the crosses. The pMAPK level is elevated upon Rafgof and to a lesser degree upon RafWT expression in w1118, but not when the flies are heterozygous for the drkΔP24 null allele. Quantification of the levels revealed highly significant differences between controls and drkΔP24/+ upon Rafgof (F(1,6) = 354, p < 0.001, n = 3) and upon RafWT (F(1,6) = 7.85, p < 0.04, n = 3) expression.
To further support these conclusions, we aimed to determine pMAPK levels in naive control and drkΔP24/+ animals after induction of the Raf transgenes. As expected, conditional pan-neuronal accumulation of RAFgof resulted in substantial elevation of pMAPK in the brains of control animals while total MAPK levels appeared unchanged (Fig. 6C). Similarly, pan-neuronal accumulation of RAFWT, elevated pMAPK levels, albeit as expected to a somewhat lesser degree compared with the effect of the constitutively active RAFgof. Surprisingly, pan-neuronal accumulation of RAFgof and RAFWT in drkΔP24 drastically decreased pMAPK levels, without altering MAPK (Fig. 6C). In fact, quantification of multiple blots demonstrated that in drkΔP24 heterozygotes accumulating RAFgof, pMAPK levels were 15% of those in control flies. Similarly, pMAPK levels were reduced 50–55% in drkΔP24/+ accumulating RAFWT (Fig. 6C). Therefore, the reduction in pMAPK levels was more prominent upon accumulation of the constitutively active RAFgof. These results indicate that prolonged RAF activation suppresses MAPK activation, or sustained pMAPK levels when DRK is reduced by 50%. Because pMAPK levels in drkΔP24/+ and drkE0A/+ were not different from controls after behavioral training, normal DRK levels do not appear essential for MAPK activation, but rather to maintain sustained pMAPK levels, perhaps by suppressing RAF activity.
Therefore, consistent with our behavioral observations, learning depends on RAF activity, but MAPK activation does not appear essential for this process. In contrast, sustained MAPK activation appears requisite for 90 min memory formation, but in drk mutant background it is further suppressed by transgenic RAF over-accumulation. This then is unlikely to result in RAF-dependent rescue of the 90 min memory deficit in the heterozygous mutants. Collectively our data suggest that DRK plays a dual role, one dependent on RAF activation for learning and an independent role in memory, which depends on sustaining pMAPK levels, possibly by antagonizing RAF activity. Furthermore, our results extend the known functions of DRK to postdevelopmental neuronal processes which underlie MB-mediated olfactory learning and memory.
Discussion
Analyses of their roles in the development of the Drosophila embryonic body plan (Perrimon et al., 1995; Gelb and Tartaglia, 2006) and the compound eye (Wassarman et al., 1995; Doroquez and Rebay, 2006) have been essential for understanding the in vivo functional relationship and signaling among members of the RAS/RAF/MAPK cascade. We extend this analysis to olfactory learning and memory with this initial demonstration that DRK accumulates in the MBs with a highly specific pattern and is required within these neurons for learning/acquisition and 90 min memory. These results support the proposed role of 14-3-3ζ/LEO, a protein with acute role(s) in olfactory learning and memory (Skoulakis and Davis, 1996; Philip et al., 2001) in regulation of RAF activity (Skoulakis and Davis, 1998). Consistently, a role in neuroplasticity has also been reported for the DRK rat ortholog GRB2, which is involved in long-term fear memory formation in the lateral amygdala (Lamprecht et al., 2002).
Our data suggest that DRK likely engages distinct signaling molecules and cascades required for learning and memory, perhaps because as in developmental contexts (Hou et al., 1995; Le and Simon, 1998; Feller et al., 2002), it interacts with different molecules with each SH3 domain. Clearly, the DRK mediated signal engages RAS1 and surprisingly signals specifically to RAF for efficient learning. Congruently, conditional knockout of B-RAF in the murine hippocampus resulted in deficient spatial and contextual learning (Chen et al., 2006), in accord with the learning deficits of other members of the cascade in mice (Brambilla et al., 1997; Giese et al., 2001; Ohno et al., 2001; Costa et al., 2002). However, as in the murine model, the molecular mechanism(s) used by RAF for learning is currently unclear and will require identification and characterization of additional molecules involved in the process. Since in adult MBs no RTK has been identified to date, the nature of the extracellular signal transduced via DRK is unknown. However, since both DRK-SH2 domain mutants drkR1 and drk E0A, exhibited deficits in learning, involvement of at least one RTK in learning and memory is probable. Interestingly, the tyrosine-phosphorylated, tyrosine phosphatase CSW and notably both SRC family members have been identified as DRK interactors in genetic screens (Cooper et al., 1996; Zhang et al., 1999; Firth et al., 2000). SRC could mediate an integrin-originated signal to RAF (Kinbara et al., 2003; Stork, 2003) via DRK. RAF activation via SRC has been recently demonstrated in Drosophila embryogenesis (Xia et al., 2008) and may be operant in the MBs as well. Interestingly, the integrin VOLADO accumulates preferentially in the MBs and mutants exhibit learning and memory deficits (Grotewiel et al., 1998). This provides an experimentally tractable indirect alternate route for the DRK-mediated signal to RAF.
DRK-mediated signals appear to be required for 90 min memory even under training conditions yielding normal learning, underscoring the proposed dual role of DRK in these processes. This signal(s) is required for maintenance of MAPK activation, known to be essential for memory formation (Adams and Sweatt, 2002; Weeber and Sweatt, 2002; Bozon et al., 2003; Shalin et al., 2004), as illustrated by post-training pMAPK levels in the Drosophila brain. Surprisingly, prolonged RAF activity such as yielded by the constitutively active enzyme appears inhibitory to sustained MAPK activation. Interestingly, these results suggest that RAF activation does not inexorably lead to MAPK activation. Importantly, DRK appears to be required for inhibition of prolonged RAF activity and this is at least one likely role of the protein in 90 min memory formation, independent of its RAF-activation-dependent role in learning. The precise mechanism of DRK-dependent RAF inhibition is unknown, but currently under investigation.
Multiple roles in learning and memory have been described for protein kinase A in Drosophila (Skoulakis et al., 1993; Li et al., 1996) and other invertebrates (Müller and Carew, 1998; Müller, 2000), but not yet for a protein without enzymatic activity like DRK. However, GRB2 is required for long term fear memory in the rat, which requires the RhoGAP, RhoA and Rho-Kinase-dependent pathway (Lamprecht et al., 2002). Whether an equivalent pathway is used for DRK-dependent 90 min memory formation in Drosophila warrants further investigation. However, a Drosophila p190-RhoGAP is expressed in the MBs and is essential for repression of axonal retraction and is negatively regulated by the integrin-MYOSPHEROID and SRC64 (Billuart et al., 2001). The latter, suggests a potential link with cytoskeletal dynamics in congruence with the vertebrate data (Lamprecht et al., 2002), but potential roles for p190-RhoGAP and MYOSPHEROID in MB-mediated learning and memory have not been evaluated yet.
The distribution for most RAS/RAF/MAPK cascade member proteins in the adult MBs is currently unknown. However, roles for some signaling pathway members have been described for physiological and structural plasticity at the larval neuromuscular junction (Guo et al., 1997; Koh et al., 2002; Hoeffer et al., 2003), but not for behavioral neuroplasticity. An exception is dNF1, a negative RAS regulating GTPase-activating protein (GAP), loss of which also results in olfactory learning and memory deficits (Guo et al., 2000). The protein is known to engage RAS and signaling via MAPK is involved in regulation of circadian behaviors (Williams et al., 2001). However, other reports suggest that the function of dNF1 in learning is to directly or indirectly regulate the RUTABAGA adenylyl cyclase or PKA activity (Guo et al., 2000; Ho et al., 2007) and to engage RAS only for long term memory formation (Ho et al., 2007). Because our data suggest that RAF in the MBs is involved in the DRK-mediated signal that contributes to learning but not memory formation, it is tempting to speculate that dNF1-regulated species of RAS mediate the latter, independently of RAF. However, a potential conflict with involvement of dNF1 in these pathways is presented by its apparent absence in the MBs (Walker et al., 2006). Therefore, it is presently unclear whether DRK-mediated signaling requires dNF1 activity in MBs for normal learning and memory.
Interestingly, our data demonstrate that drk mutant heterozygotes and animals with abrogated DRK in the MBs learn inefficiently, but do nevertheless learn to the same level as controls upon overtraining. Thus, learning ability is not compromised in the mutant heterozygotes, or c772/drkΔP24; drkR-2/+ animals which retain even less protein in their MBs (Fig. 3A,B,C). This pairing-specific learning deficit may reflect the proposed engagement of the molecule in multiple processes and, therefore, explain the demonstrated dosage sensitive phenotypes. Upon extended training, DRK molecules liberated from signaling complexes formed during the initial pairings may become re-engaged, thus overcoming the limitation of reduced protein and yielding near normal performances. Similar association between pairings and the dynamics of enzymatic activities relative to behavioral output have been described for associative conditioning in invertebrates (Müller and Carew, 1998; Müller, 2000; Friedrich et al., 2004). Significantly, a similar under-training protocol was necessary to uncover fear conditioning defects in mice conditionally lacking Kinase Suppressor of Ras1 (KSR), a scaffolding protein that could compartmentalize RAS/RAF/MAPK signaling within hippocampal neurons (Shalin et al., 2006). Thus, it is possible that DRK is involved in signaling that regulates the rate of learning per CS/US. A similar phenotype was observed by manipulation of the RDL GABAA receptor in the MBs. Over-expression of this receptor resulted in a pairing-specific decrease in learning efficiency, which also reached control levels with overtraining. The learning rate was increased respectively upon RNAi mediated abrogation of the protein in the same neurons (Liu et al., 2007).
Collectively, these observations lead to the interesting hypothesis that signaling pathways which regulate learning efficiency operate within the MBs in addition to molecules and pathways regulating learning ability. Mutations in the former may be manifested as small, perhaps not significant changes in learning exaggerated upon reduced training. In contrast, we predict that mutations in the latter are likely to result in larger overall learning defects, which could not be eliminated by overtraining. Because a lot of studies and screens for olfactory learning mutants have been conducted using intensive training protocols of at least 12 CS/US pairings, it is possible that mutations affecting learning efficiency and not learning ability have been missed. Interestingly, RUT adenylyl cyclase null mutants exhibit large learning deficits (Han et al., 1992; Skoulakis and Grammenoudi, 2006) and although they improve their performance upon overtraining, they do not reach control levels (Sideri and Skoulakis unpublished), suggesting a compromise in learning altogether. Ongoing, careful re-evaluation of extant learning and memory mutants is likely to reveal additional members for these categories and support this hypothesis. Finally, the nature of the signals that DRK mediates and the identities and roles of additional molecules engaged in mediating it remain currently unknown. Nevertheless, our results suggest that they will likely be involved in learning efficiency and provide a platform and several testable hypotheses to elucidate them in the future.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS19904 and a Welch Foundation Chair to R.L.D., and NIH Grant MH59851 to E.M.C.S. Support for work in Europe was provided by Marie Curie IRG 3570 to E.M.C.S. A.M. was supported by a grant from the Greek State Scholarships Foundation (IKY). We thank B. Schroeder, A. Sacharidou, and T. Azizi-Babane for work in early phases of this project, and A. Karkali and members of the Skoulakis laboratory for help with experiments, discussions, ideas, and advice. We are indebted to the Laboratory Animal Resources and Research at Texas A&M University for generation of antibodies, M. Simon for antibodies and mutant stocks, and N. Perrimon, E. Hafen, K. W. Choi, V. Budnik, G. Roman, the Bloomington Drosophila Stock Center for mutant stocks, and the Developmental Studies Hybridoma Bank (University of Iowa) for antibodies.
References
- Acevedo SF, Froudarakis EI, Tsiorva AA, Skoulakis EM. Distinct neuronal circuits mediate experience-dependent, non-associative osmotactic responses in Drosophila. Mol Cell Neurosci. 2007a;34:378–389. doi: 10.1016/j.mcn.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Acevedo SF, Tsigkari KK, Grammenoudi S, Skoulakis EMC. In vivo functional specificity and homeostasis of Drosophila 14-3-3 proteins. Genetics. 2007b;177:239–253. doi: 10.1534/genetics.107.072280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo SF, Froudarakis EI, Kanellopoulos A, Skoulakis EM. Protection from premature habituation requires functional mushroom bodies in Drosophila. Learn Mem. 2007c;14:376–384. doi: 10.1101/lm.566007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JP, Sweatt JD. Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- Armstrong JD, de Belle JS, Wang Z, Kaiser K. Metamorphosis of the mushroom bodies; large scale rearrangements of the neural substrates for associative lerning and memory in Drosophila. Learn Mem. 1998;5:102–114. [PMC free article] [PubMed] [Google Scholar]
- Baek KH, Lee KY. Signal transduction pathway for anterior-posterior development in Drosophila. J Biomed Sci. 1999;6:314–319. doi: 10.1007/BF02253520. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER, Si K. The persistence of long-term memory: a molecular approach to self-sustaining changes in learning-induced synaptic growth. Neuron. 2004;44:49–57. doi: 10.1016/j.neuron.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Beck CD, Schroeder B, Davis RL. Learning performance of Normal and Mutant Drosophila after repeated conditioning trials with discrete stimuli. J Neurosci. 2000;20:2944–2953. doi: 10.1523/JNEUROSCI.20-08-02944.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry J, Krause WC, Davis RL. Olfactory memory traces in Drosophila. Prog Brain Res. 2008;169:293–304. doi: 10.1016/S0079-6123(07)00018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billuart P, Winter CG, Maresh A, Zhao X, Luo L. Regulating axon branch stability: The role of p190 RhoGAP in repressing a retraction signaling pathway. Cell. 2001;107:195–207. doi: 10.1016/s0092-8674(01)00522-0. [DOI] [PubMed] [Google Scholar]
- Bozon B, Kelly A, Josselyn SA, Silva AJ, Davis S, Laroche S. MAPK, CREB and zif268 are all required for the consolidation of recognition memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:805–814. doi: 10.1098/rstb.2002.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Rossi-Arnaud C, Grant SG, Chapman PF, Lipp HP, Sturani E, Klein R. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Raf acts downstream of the EGF receptor to determine dorsoventral polarity during Drosophila oogenesis. Genes Dev. 1994;8:629–639. doi: 10.1101/gad.8.5.629. [DOI] [PubMed] [Google Scholar]
- Chen AP, Ohno M, Giese KP, Kühn R, Chen RL, Silva AJ. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J Neurosci Res. 2006;83:28–38. doi: 10.1002/jnr.20703. [DOI] [PubMed] [Google Scholar]
- Cheng AM, Saxton TM, Sakai R, Kulkarni S, Mbamalu G, Vogel W, Tortorice CG, Cardiff RD, Cross JC, Muller WJ, Pawson T. Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell. 1998;95:793–803. doi: 10.1016/s0092-8674(00)81702-x. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Endo K, Wu K, Rodan AR, Heberlein U, Davis RL. Drosophila fasciclinII is required for the formation of odor memories and for normal sensitivity to alcohol. Cell. 2001;105:757–768. doi: 10.1016/s0092-8674(01)00386-5. [DOI] [PubMed] [Google Scholar]
- Choi KW, Benzer S. Migration of glia along photoreceptor axons in the developing Drosophila eye. Neuron. 1994;12:423–431. doi: 10.1016/0896-6273(94)90282-8. [DOI] [PubMed] [Google Scholar]
- Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci. 2007;27:12732–12742. doi: 10.1523/JNEUROSCI.2522-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Simon MA, Kussick SJ. Signaling by ectopically expressed Drosophila Src64 requires the protein-tyrosine phosphatase corkscrew and the adapter downstream of receptor kinases. Cell Growth Differ. 1996;7:1435–1441. [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Crittenden JR, Skoulakis EM, Han KA, Kalderon D, Davis RL. Tripartite mushroom body architecture revealed by antigenic markers. Learn Mem. 1998;5:38–51. [PMC free article] [PubMed] [Google Scholar]
- Davis RL. Olfactory learning. Neuron. 2004;44:31–48. doi: 10.1016/j.neuron.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Davis RL. Olfactory memory formation in Drosophila: from molecular to systems neuroscience. Annu Rev Neurosci. 2005;28:275–302. doi: 10.1146/annurev.neuro.28.061604.135651. [DOI] [PubMed] [Google Scholar]
- Davis S, Laroche S. Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes Brain Behav. 2006;5:61–72. doi: 10.1111/j.1601-183X.2006.00230.x. [DOI] [PubMed] [Google Scholar]
- Dickson BJ, van der Straten A, Dominguez M, Hafen E. Mutations modulating Raf signaling in Drosophila eye development. Genetics. 1996;142:163–171. doi: 10.1093/genetics/142.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroquez DB, Rebay I. Signal integration during development: mechanisms of EGFR and Notch pathway function and cross-talk. Crit Rev Biochem Mol Biol. 2006;41:339–385. doi: 10.1080/10409230600914344. [DOI] [PubMed] [Google Scholar]
- Douziech M, Sahmi M, Laberge G, Therrien M. A KSR/CNK complex mediated by HYP, a novel SAM domain-containing protein, regulates RAS-dependent RAF activation in Drosophila. Genes Dev. 2006;20:807–819. doi: 10.1101/gad.1390406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller SM, Wecklein H, Lewitzky M, Kibler E, Raabe T. SH3 domain-mediated binding of the Drk protein to Dos is an important step in signaling of Drosophila receptor tyrosine kinases. Mech Dev. 2002;116:129–139. doi: 10.1016/s0925-4773(02)00147-8. [DOI] [PubMed] [Google Scholar]
- Ferris J, Ge H, Liu L, Roman G. G(o) signaling is required for Drosophila associative learning. Nat Neurosci. 2006;9:1036–1040. doi: 10.1038/nn1738. [DOI] [PubMed] [Google Scholar]
- Firth L, Manchester J, Lorenzen JA, Baron M, Perkins LA. Identification of genomic regions that interact with a viable allele of the Drosophila protein tyrosine phosphatase corkscrew. Genetics. 2000;156:733–748. doi: 10.1093/genetics/156.2.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich A, Thomas U, Müller U. Learning at different satiation levels reveals parallel functions for the cAMP-protein kinase A cascade in formation of long-term memory. J Neurosci. 2004;24:4460–4468. doi: 10.1523/JNEUROSCI.0669-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb BD, Tartaglia M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet 15 Spec No. 2006;2:R220–R226. doi: 10.1093/hmg/ddl197. [DOI] [PubMed] [Google Scholar]
- Giese KP, Friedman E, Telliez JB, Fedorov NB, Wines M, Feig LA, Silva AJ. Hippocampus-dependent learning and memory is impaired in mice lacking the Ras-guanine-nucleotide releasing factor 1 (Ras-GRF1) Neuropharmacology. 2001;41:791–800. doi: 10.1016/s0028-3908(01)00096-x. [DOI] [PubMed] [Google Scholar]
- Giordano E, Rendina R, Peluso I, Furia M. RNAi triggered by symmetrically transcribed transgenes in Drosophila melanogaster. Genetics. 2002;160:637–648. doi: 10.1093/genetics/160.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signaling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Grotewiel MS, Beck CD, Wu KH, Zhu XR, Davis RL. Integrin-mediated short-term memory in Drosophila. Nature. 1998;391:455–460. doi: 10.1038/35079. [DOI] [PubMed] [Google Scholar]
- Guo HF, The I, Hannan F, Bernards A, Zhong Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- Guo HF, Tong J, Hannan F, Luo L, Zhong Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature. 2000;403:895–898. doi: 10.1038/35002593. [DOI] [PubMed] [Google Scholar]
- Han PL, Levin LR, Reed RR, Davis RL. Preferential expression of the Drosophila rutabaga gene in mushroom bodies, neural centers for learning in insects. Neuron. 1992;9:619–627. doi: 10.1016/0896-6273(92)90026-a. [DOI] [PubMed] [Google Scholar]
- Heisenberg M. Mushroom body memoir: from maps to models. Nat Rev Neurosci. 2003;4:266–275. doi: 10.1038/nrn1074. [DOI] [PubMed] [Google Scholar]
- Ho IS, Hannan F, Guo HF, Hakker I, Zhong Y. Distinct functional domains of neurofibromatosis type 1 regulate immediate versus long-term memory formation. J Neurosci. 2007;27:6852–6857. doi: 10.1523/JNEUROSCI.0933-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Sanyal S, Ramaswami M. Acute induction of conserved synaptic signaling pathways in Drosophila melanogaster. J Neurosci. 2003;23:6362–6372. doi: 10.1523/JNEUROSCI.23-15-06362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou XS, Chou TB, Melnick MB, Perrimon N. The torso receptor tyrosine kinase can activate Raf in a Ras-independent pathway. Cell. 1995;81:63–71. doi: 10.1016/0092-8674(95)90371-2. [DOI] [PubMed] [Google Scholar]
- Jin K, Mao XO, Eshoo MW, Nagayama T, Minami M, Simon RP, Greenberg DA. Microarray analysis of hippocampal gene expression in global cerebral ischemia. Ann Neurol. 2001;50:93–103. doi: 10.1002/ana.1073. [DOI] [PubMed] [Google Scholar]
- Karim FD, Rubin GM. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development. 1998;125:1–9. doi: 10.1242/dev.125.1.1. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kinbara K, Goldfinger LE, Hansen M, Chou FL, Ginsberg MH. RasGTPases: integrins' friends or foes? Nat Rev Mol Cell Biol. 2003;4:767–776. doi: 10.1038/nrm1229. [DOI] [PubMed] [Google Scholar]
- Koh YH, Ruiz-Canada C, Gorczyca M, Budnik V. The Ras1-mitogen-activated protein kinase signal transduction pathway regulates synaptic plasticity through fasciclin II-mediated cell adhesion. J Neurosci. 2002;22:2496–2504. doi: 10.1523/JNEUROSCI.22-07-02496.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, Farb CR, LeDoux JE. Fear memory formation involves p190RhoGAP and ROCK proteins through a GRB2-mediated complex. Neuron. 2002;36:727–738. doi: 10.1016/s0896-6273(02)01047-4. [DOI] [PubMed] [Google Scholar]
- Le N, Simon MA. Disabled is a putative adaptor protein that functions during signaling by the sevenless receptor tyrosine kinase. Mol Cell Biol. 1998;18:4844–4854. doi: 10.1128/mcb.18.8.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Hariharan IK, Chen F, Huang Y, Fischer JA. Genetic interactions with Rap1 and Ras1 reveal a second function for the fat facets deubiquitinating enzyme in Drosophila eye development. Proc Natl Acad Sci U S A. 1997;94:12515–12520. doi: 10.1073/pnas.94.23.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Tully T, Kalderon D. Effects of a conditional Drosophila PKA mutant on olfactory learning and memory. Learn Mem. 1996;2:320–333. doi: 10.1101/lm.2.6.320. [DOI] [PubMed] [Google Scholar]
- Liu X, Krause WC, Davis RL. GABAA receptor RDL inhibits Drosophila olfactory associative learning. Neuron. 2007;56:1090–1102. doi: 10.1016/j.neuron.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D, Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70:431–442. doi: 10.1016/0092-8674(92)90167-b. [DOI] [PubMed] [Google Scholar]
- Luschnig S, Krauss J, Bohmann K, Desjeux I, Nüsslein-Volhard C. The Drosophila SHC adaptor protein is required for signaling by a subset of receptor tyrosine kinases. Mol Cell. 2000;5:231–241. doi: 10.1016/s1097-2765(00)80419-0. [DOI] [PubMed] [Google Scholar]
- Martin KC, Kandel ER. Cell adhesion molecules, CREB, and the formation of new synaptic connections during development and learning. Neuron. 1996;17:567–570. doi: 10.1016/s0896-6273(00)80188-9. [DOI] [PubMed] [Google Scholar]
- Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for Long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Brambilla R. Ras-related and MAPK signalling in neuronal plasticity and memory formation. Cell Mol Life Sci. 2000;57:604–611. doi: 10.1007/PL00000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL. Spatiotemporal rescue of memory dysfunction in Drosophila. Science. 2003;302:1765–1768. doi: 10.1126/science.1089035. [DOI] [PubMed] [Google Scholar]
- McGuire SE, Mao Z, Davis RL. Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci STKE. 2004a;2004:pl6. doi: 10.1126/stke.2202004pl6. [DOI] [PubMed] [Google Scholar]
- McGuire SE, Roman G, Davis RL. Gene expression systems in Drosophila: a synthesis of time and space. Trends Genet. 2004b;20:384–391. doi: 10.1016/j.tig.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Mershin A, Pavlopoulos E, Fitch O, Braden BC, Nanopoulos DV, Skoulakis EMC. Learning and Memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem. 2004;11:277–287. doi: 10.1101/lm.70804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U. Prolonged activation of cAMP-dependent protein kinase during conditioning induces long-term memory in honeybees. Neuron. 2000;27:159–168. doi: 10.1016/s0896-6273(00)00017-9. [DOI] [PubMed] [Google Scholar]
- Müller U, Carew TJ. Serotonin induces temporally and mechanistically distinct phases of persistent PKA activity in Aplysia sensory neurons. Neuron. 1998;21:1423–1434. doi: 10.1016/s0896-6273(00)80660-1. [DOI] [PubMed] [Google Scholar]
- Ohno M, Frankland PW, Chen AP, Costa RM, Silva AJ. Inducible, pharmacogenetic approaches to the study of learning and memory. Nat Neurosci. 2001;4:1238–1243. doi: 10.1038/nn771. [DOI] [PubMed] [Google Scholar]
- Olivier JP, Raabe T, Henkemeyer M, Dickson B, Mbamalu G, Margolis B, Schlessinger J, Hafen E, Pawson T. A Drosophila SH2-SH3 adaptor protein implicated in coupling the Sevenless tyrosine kinase to an activator of Ras guanine exchange, Sos. Cell. 1993;73:179–191. doi: 10.1016/0092-8674(93)90170-u. [DOI] [PubMed] [Google Scholar]
- Pawson T, Gish GD. SH2 and SH3 Domains: from structure to function. Cell. 1992;71:359–362. doi: 10.1016/0092-8674(92)90504-6. [DOI] [PubMed] [Google Scholar]
- Pawson T, Saxton TM. Signaling networks–do all roads lead to the same genes? Cell. 1999;97:675–678. doi: 10.1016/s0092-8674(00)80779-5. [DOI] [PubMed] [Google Scholar]
- Perrimon N, Lu X, Hou XS, Hsu JC, Melnick MB, Chou TB, Perkins LA. Dissection of the torso signal transduction pathway in Drosophila. Mol Reprod Dev. 1995;42:515–522. doi: 10.1002/mrd.1080420421. [DOI] [PubMed] [Google Scholar]
- Philip N, Acevedo SF, Skoulakis EM. Conditional rescue of olfactory learning and memory defects in mutants of the 14-3-3z gene leonardo. J Neurosci. 2001;21:8417–8425. doi: 10.1523/JNEUROSCI.21-21-08417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preat T. Decreased odor avoidance after electric shock in Drosophila mutants biases learning and memory tests. J Neurosci. 1998;18:8534–8538. doi: 10.1523/JNEUROSCI.18-20-08534.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe T, Olivier JP, Dickson B, Liu X, Gish GD, Pawson T, Hafen E. Biochemical and genetic analysis of the Drk SH2/SH3 adaptor protein of Drosophila. EMBO J. 1995;14:2509–2518. doi: 10.1002/j.1460-2075.1995.tb07248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe T, Riesgo-Escovar J, Liu X, Bausenwein BS, Deak P, Maröy P, Hafen E. DOS, a novel Plekstrin homology domain-containing protein required for signal transduction between sevenless and Ras1 in Drosophila. Cell. 1996;85:911–920. doi: 10.1016/s0092-8674(00)81274-x. [DOI] [PubMed] [Google Scholar]
- Roch F, Serras F, Cifuentes FJ, Corominas M, Alsina B, Amorós M, López-Varea A, Hernández R, Guerra D, Cavicchi S, Baguñá J, García-Bellido A. Screening of larval/pupal P-element induced lethals on the second chromosome in Drosophila melanogaster: clonal analysis and morphology of imaginal discs. Mol Gen Genet. 1998;257:103–112. doi: 10.1007/pl00008620. [DOI] [PubMed] [Google Scholar]
- Roman G, He J, Davis RL. New series of Drosophila expression vectors suitable for behavioral rescue. Biotechniques. 1999;27:54–56. doi: 10.2144/99271bm09. [DOI] [PubMed] [Google Scholar]
- Rommel C, Radziwill G, Moelling K, Hafen E. Negative regulation of Raf activity by binding of 14-3-3 to the aminoterminus of Raf in vivo. Mech Dev. 1997;64:95–104. doi: 10.1016/s0925-4773(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Rubin GM. Signal transduction and the fate of R7 photoreceptor in Drosophila. Trends Genet. 1991;7:372–377. doi: 10.1016/0168-9525(91)90258-r. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Fibronectin and its integrin receptors in cancer. Adv Cancer Res. 1999;76:1–20. doi: 10.1016/s0065-230x(08)60772-1. [DOI] [PubMed] [Google Scholar]
- Shalin SC, Zirrgiebel U, Honsa KJ, Julien JP, Miller FD, Kaplan DR, Sweatt JD. Neuronal MEK is important for normal fear conditioning in mice. J Neurosci Res. 2004;75:760–770. doi: 10.1002/jnr.20052. [DOI] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Carew TJ. The roles of MAPK cascades in synaptic plasticity and memory in Aplysia: facilitatory effects and inhibitory contraints. Learn Mem. 2004;11:373–378. doi: 10.1101/lm.81104. [DOI] [PubMed] [Google Scholar]
- Simon MA, Dodson GS, Rubin GM. An SH3-SH2-SH3 Protein is required for p21Ras1 activation and binds to Sevenless and Sos proteins in vitro. Cell. 1993;73:169–177. doi: 10.1016/0092-8674(93)90169-q. [DOI] [PubMed] [Google Scholar]
- Simon MA, Bowtell DD, Dodson GS, Laverty TR, Rubin GM. Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell. 1991;67:701–716. doi: 10.1016/0092-8674(91)90065-7. [DOI] [PubMed] [Google Scholar]
- Skoulakis EM, Davis RL. 14-3-3 proteins in neuronal development and function. Mol Neurobiol. 1998;16:269–284. doi: 10.1007/BF02741386. [DOI] [PubMed] [Google Scholar]
- Skoulakis EM, Grammenoudi S. Dunces and da Vincis: the genetics of learning and memory in Drosophila. Cell Mol Life Sci. 2006;63:975–988. doi: 10.1007/s00018-006-6023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulakis EM, Kalderon D, Davis RL. Preferential expression in mushroom bodies of the catalytic subunit of protein kinase A and its role in learning and memory. Neuron. 1993;11:197–208. doi: 10.1016/0896-6273(93)90178-t. [DOI] [PubMed] [Google Scholar]
- Stork PJ. Does Rap1 deserve a bad Rap? Trends Biochem Sci. 2003;28:267–275. doi: 10.1016/S0968-0004(03)00087-2. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Protooncogenes subserve memory formation in the adult CNS. Neuron. 2001;31:671–674. doi: 10.1016/s0896-6273(01)00419-6. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Szüts D, Freeman M, Bienz M. Antagonism between EGFR and Wingless signalling in the larval cuticle of Drosophila. Development. 1997;124:3209–3219. doi: 10.1242/dev.124.16.3209. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol. 1985;157:263–277. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- Walker JA, Tchoudakova AV, McKenney PT, Brill S, Wu D, Cowley GS, Hariharan IK, Bernards A. Reduced growth of Drosophila neurofibromatosis 1 mutants reflects a non-cell-autonomous requirement for GTPase-Activating Protein activity in larval neurons. Genes Dev. 2006;20:3311–3323. doi: 10.1101/gad.1466806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman DA, Therrien M, Rubin GM. The Ras signaling pathway in Drosophila. Curr Opin Genet Dev. 1995;5:44–50. doi: 10.1016/s0959-437x(95)90052-7. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Sweatt JD. Molecular neurobiology of human cognition. Neuron. 2002;33:845–848. doi: 10.1016/s0896-6273(02)00634-7. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Levenson JM, Sweatt JD. Molecular genetics of human cognition. Mol Interv. 2002;2:376–391. 339. doi: 10.1124/mi.2.6.376. [DOI] [PubMed] [Google Scholar]
- Williams JA, Su HS, Bernards A, Field J, Sehgal A. A circadian output in Drosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science. 2001;293:2251–2256. doi: 10.1126/science.1063097. [DOI] [PubMed] [Google Scholar]
- Xia F, Li J, Hickey GW, Tsurumi A, Larson K, Guo D, Yan SJ, Silver-Morse L, Li WX. Raf activation is regulated by Tyrosine 510 phosphorylation in Drosophila. PLoS Biol. 2008;6:e128. doi: 10.1371/journal.pbio.0060128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zheng Q, Lu X. A genetic screen for modifiers of Drosophila Src42A identifies mutations in Egfr, rolled and a novel signaling gene. Genetics. 1999;151:697–711. doi: 10.1093/genetics/151.2.697. [DOI] [PMC free article] [PubMed] [Google Scholar]