

Abstract
Aldehyde dehydrogenase (ALDH) isozymes are critically important in the metabolism of acetaldehyde, thus preventing its accumulation after ethanol exposure. We previously reported that mitochondrial ALDH2 could be inactivated via -nitrosylation in ethanol-exposed rats. This study was aimed at investigating whether cytosolic ALDH1, with a relatively low- value (11–18 μM) for acetaldehyde, could be also inhibited in ethanol-exposed rats. Chronic or binge ethanol exposure significantly decreased ALDH1 activity, which was restored by addition of dithiothreitol. Immunoblot analysis with the anti--nitroso-Cys antibody showed one immunoreactive band in the immunoprecipiated ALDH1 only from ethanol-exposed rats, but not from pair-fed controls, suggesting -nitrosylation of ALDH1. Therefore inactivation of ALDH1 via -nitrosylation can result in accumulation of acetaldehyde upon ethanol exposure.
Keywords: Cytosolic aldehyde dehydrogenase (ALDH1), Ethanol, S-nitrosylation, reversible inhibition, acetaldehyde metabolism
1. Introduction
Various aldehydes including acetaldehyde and 4-hydroxynonenal are metabolized to corresponding carboxylic acids by aldehyde dehydrogenase (ALDH)1 isozymes [1–3]. Since many aldehydes are highly reactive and potentially toxic, ALDH isozymes are important in cellular detoxification [2,3]. The ALDH superfamily members represent NAD(P)+-dependent enzymes with broad substrate specificities and different subcellular locations: class1 (cytosolic, ALDH1), class 2 (mitochondrial, ALDH2), class 3 (tumor-associated, ALDH3), succinate semialdehyde dehydrogenase (ALDH5), etc [2,3]. In addition, it is known that a genetic polymorphism in the human gene plays a key role in alcohol-associated tissue injury and carcinogenesis [4–7].
Mammalian liver class 1 and class 2 ALDH enzymes were purified in the 1970s, and have been used extensively as models for studying their kinetic and structural characterizations. Both ALDH1 and ALDH2 are tetrameric enzymes with individual subunits composed of 500–501 amino acids. They share 68% sequence identity to each other [7,8] and have very similar three-dimensional structures [9–11]. Mitochondrial ALDH2 in humans is the major acetaldehyde-oxidizing enzyme with a very low m value (≤1.0 μM range) for acetaldehyde [12–14]. In contrast, both ALDH1 and ALDH2 in rodents are involved in the metabolism of acetaldehyde at physiological concentrations because rodent ALDH1 isozymes exhibit relatively-low m values (11–18 μM range) for acetaldehyde [14]. Isse [15] reported that acetaldehyde concentrations in blood and many tissues upon ethanol exposure were markedly elevated in −/− knockout mice compared to those concentrations in +/+ wild type mice. Our previous results with the transgenic mice containing the inactive human ALDH2-2 variant also showed that acute ethanol exposure significantly increased the hepatic acetaldehyde levels without changing the ethanol levels [16]. Furthermore, we recently identified cytosolic ALDH1 among the oxidatively-modified proteins in ethanol-fed mouse livers [17]. All these results strongly suggest that ALDH1 could be inhibited through oxidative/nitrosative modifications in ethanol-exposed rat livers. Consistent with this view, Cys residues of mitochondrial ALDH2 was -nitrosylated in nitric oxide-exposed hepatoma cells [18] and ethanol-exposed rats [19], leading to inactivation of ALDH2. However, it is still unknown whether ALDH1 activity can be altered by ethanol exposure and whether the suppressed ALDH1 activity can be restored by reducing agents such as DTT. Based on this information, we hypothesized that ALDH1 can be inactivated by -nitrosylation of Cys residue(s) upon ethanol-exposure. The aims of this study were to test this hypothesis and to examine whether DTT can reverse the suppressed ALDH1 activity in ethanol-exposed rats.
2. Materials and Methods
2.1. Chemicals and other materials
DTT, anti--nitroso-cysteine (-NO-Cys) antibody, biotin--maleimide (biotin-NM), propionaldehyde, and pyrazole were obtained from Sigma Chemical (St. Louis, MO, USA) in highest purity. The specific goat anti-ALDH1 antibody, (catalog number: sc-22588) which does not recognize mitochondrial ALDH2, and secondary antibodies conjugated with horse radish peroxidase (HRP) were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA).
2.2 Animal maintenance and pair-feeding treatment
Young male Sprague-Dawley rats (n ≥ 10 from Taconic Farms, Rockville, MD, USA) were maintained in accordance with the NIH guidelines and acutely treated with ethanol for 4 consecutive days (binge treatment), as recently described [20]. Control rats were treated in the same manner except for treatment with isocaloric dextrose-containing diet (dextrose control). Young adult Wistar rats (n= 6 per group from Charles River, Raleigh, NC, USA) were individually housed and fed a Lieber-DeCarli alcohol liquid diet (with 35% daily calories derived from ethanol) or an isocaloric dextrose control diet for 4 weeks, as described [21].
2.3 Labeling of oxidized cytosolic proteins with biotin-NM
Livers, freshly isolated from rats in different treatment groups, were pooled, rinsed with cold phosphate-buffered saline to remove blood, and homogenized with 3 volumes of STE buffer (250 mM sucrose, 50 mM Tris-Cl, pH 7.5, and 1 mM EDTA with protease inhibitor cocktails). Cytosolic and mitochondrial fractions were prepared by differential centrifugation, as described [17–19]. Concentration of the cytosolic proteins was determined using the BioRad protein assay kit, as described [19,22]. Labeling of oxidized proteins with biotin-NM was performed using a previously detailed method [19].
2.4 Measurement of ALDH1 activities
The activity of cytosolic ALDH1 was measured by increased production of NADH at 340 nm by following the method of Tank [23]. The reaction mixture contained: 60 mM Na-pyrophosphate buffer (pH 8.5), 2 mM NAD+, 5 mM pyrazole, 1 mM EDTA and cytosolic proteins (0.5 mg/assay). After the reaction mixture was kept for 2 min at room temperature, the enzyme reaction was initiated by adding the substrate (15 or 150 μM propionaldehyde). The absorbance change was monitored for 3 min to calculate the rate of NADH production. Specific activity of ALDH1 was calculated by using the molar extinction coefficient of reduced NAD(P) of 6.22 × 106 cm2 at 340 nm (Merck Index), where 1 unit represents a reduction of 1 μmole NAD+/min/mg protein at room temperature.
2.5 Immunoprecipitation of ALDH1 proteins and immunoblot analysis
Cytosolic proteins (2 mg/sample) were incubated with 5 μg of the specific antibody to ALDH1 for 2 h with constant agitation followed by addition of protein A/G-agarose for an additional 1 h to facilitate immunoprecipitation. Proteins bound to protein A/G-agarose beads were washed three times with 1 × phosphate-buffered saline and 1% CHAPS to remove non-specifically bound proteins. After the centrifugation, the proteins bound to the antibody and agarose beads were dissolved in 1-D Laemmli buffer for immunoblot analysis. The unpurified cytosolic proteins and immunoprecipitated ALDH1 proteins were resolved on SDS-polyacrylamide gels and subjected to immunoblot analysis with the specific anti-ALDH1 antibody or anti--NO-Cys antibody with enhanced chemiluminescence detection.
2.6 Data processing and statistical analysis
The data represent the results from at least three separate experiments, unless stated otherwise. Statistical analyses were performed using the Student’s test and <0.05 was considered statistically significant. Other materials and methods not described here were performed as previously described [17–19].
3. Results
3.1 Increased oxidative modification of ALDH1 after ethanol exposure
Chronic or binge ethanol treatment caused increased levels and activities of CYP2E1 and NOS, surrogate marker proteins of oxidative/nitrosative stress, indicating increased oxidative/nitrosative stress in ethanol-exposed tissues [17,19]. To directly demonstrate the increased levels of oxidized proteins after ethanol exposure, cytosolic proteins from the respective treatment groups were labeled with biotin-NM and subjected to analysis. Fig. 1A shows a typical pattern of biotin-NM labeled proteins stained with Coomassie blue, verifying that similar amounts of oxidized protein were analyzed in our study. Only a small number of biotin-NM labeled oxidized proteins were recognized with streptavidin-HRP in the dextrose-treated control samples (Fig. 1B, lanes 1 and 3). However, the intensity and the number of oxidized proteins detected by streptavidin-HRP were greatly increased in chronic and binge ethanol-treated rat livers, respectively (Fig. 1B, lanes 2 and 4). To further demonstrate oxidative modification of ALDH1 protein, biotin-NM labeled cytosolic proteins were analyzed before and after purification with streptavidin-agarose. Immunoblot analysis using the specific anti-ALDH1 antibody revealed that similar amounts of ALDH1 protein were detected in all treatment groups before the purification (Fig. 1C). After the streptavidin-mediated purification, a single band was recognized only in ethanol-exposed groups but not in dextrose-treated control groups (Fig. 1D). These immunoblot data clearly support that ALDH1 was oxidatively-modified after both chronic and binge ethanol treatments.
Fig. 1.

Presence of ALDH1 protein in the oxidized cytosolic proteins after ethanol exposure. Oxidized cytosolic proteins from dextrose-treated controls (−) or ethanol-exposed (+) rat livers were labeled with biotin-NM. Biotin-NM labeled proteins (20 μg/well) were then separated on 12% SDS-PAGE, transferred to PVDF-Immobilon membranes, and stained with Coomassie blue (A) or subjected to immunoblot analysis using streptavidin-HRP antibodies (B). Presence of ALDH1 protein in oxidized cytosolic proteins was confirmed by immunoblot analysis. Biotin-NM-labeled cytosolic proteins before (C) or after (D) purification with streptavidin-agarose were analyzed by 1D SDS-PAGE, and then subjected to immunoblot analysis (IB) using the specific anti-ALDH1 antibody. This figure represents a typical result from at least two different experiments.
3.2 Reversible inactivation of hepatic ALDH1 in ethanol-exposed rats
Keung and colleagues [14] reported that rat ALDH1 exhibits a relatively-low m value (15 ± 3 μM) for acetaldehyde. Based on this report, hepatic ALDH1 activity in different groups was determined by using 15 or 150 μM propionaldehyde as a substrate. As shown in Fig. 2A, ALDH1 activity using 15 μM propionaldehyde was significantly inhibited (by 61%) in chronically ethanol-fed rats compared to that of pair-fed rats. In contrast, ALDH1 activity in ethanol-fed rats was not inhibited when the activity was determined using 150 μM propionaldehyde. These data suggest that only ALDH1 with a relatively low-m for acetaldehyde is likely to be inhibited by oxidative modifications. Other cytosolic ALDH isozymes with high-m values for acetaldehyde may not be inactivated. Hepatic ALDH1 activities were 1.54 ± 0.24 and 2.14 ± 0.25 units in chronic and binge ethanol-exposed rats, respectively. These values represent approximately 61% and 42% reduction, respectively, of the ALDH1 activity in chronic and binge ethanol-treated rats, compared with those of the corresponding dextrose-treated controls (Fig. 2B). Furthermore, the suppressed ALDH1 activities were completely recovered by the addition of DTT in both chronic and binge ethanol-treated groups.
Fig. 2.
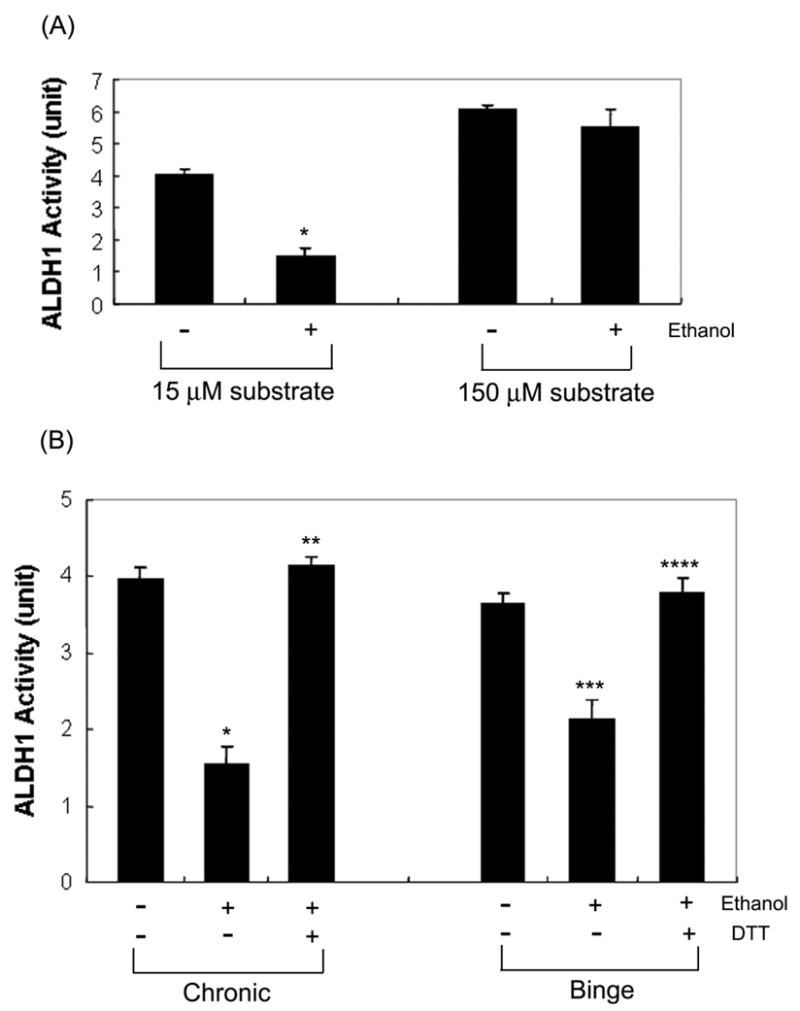
Reversible inactivation of ALDH1 in ethanol-exposed rat livers. (A) Hepatic ALDH1 activities in pair-fed controls (−) or chronically ethanol-fed rats (+) were determined by using 15 or 150 μM propionaldehyde, as indicated. (B) ALDH1 activities in different groups were measured with 15 μM propionaldehyde before and after incubation with 12 mM DTT. Each point represents the average ± S.D. of three determinations. *p<0.005, significantly different from the pair-fed controls; **p<0.001, significantly different from the ethanol-fed rats; ***p<0.005, significantly different from the dextrose-treated controls; ****p<0.005, significantly different from the binge ethanol-exposed rats.
To further elucidate the mechanism for ethanol-mediated ALDH1 inhibition, the hepatic ALDH1 proteins in the pair-fed controls or ethanol-fed rats were immunoprecipitated with the anti-ALDH1 antibody before or after incubation with DTT. Immunoblot analysis with the anti-ALDH1 antibody verified that similar amounts of ALDH1 protein (55 kDa) were immunoprecipitated from different treatment groups (Fig. 3A, left panel). Immunoblot analysis with the anti--NO-Cys antibody showed that one immunoreactive band (55 kDa) was recognized in the ethanol-fed rats (right panel, lane 2) but not in the pair-fed controls (lane 1). However, the immunoreactive -NO-Cys band disappeared (lane 3) when DTT was added prior to immunoprecipitation of ALDH1 in the ethanol-fed group. These results of -nitrosylated ALDH1 protein correlated with complete recovery of ALDH1 activity in the presence of DTT (Fig. 2B). These data strongly suggest that inhibition of ALDH1 by -nitrosylation in ethanol-exposed rats is reversible and that critical Cys residues including the active site Cys of ALDH1 could be -nitrosylated or subjected to other oxidative modifications following exposure to ethanol.
Fig. 3.

Reversible -nitrosylation of ALDH1. Liver cytosolic proteins (2 mg/analysis) from pair-fed control (−) or ethanol-fed rats (+) were used to immunoprecipitate (IP) ALDH1 with the specific anti-ALDH1 antibody before and after incubation with 12 mM DTT. Immunoprecipitated ALDH1 proteins were divided equally and subjected to immunoblot analysis using the anti-ALDH1 antibody (left panel) or the anti--NO-Cys antibody (right panel). This figure represents a typical result from two different experiments.
4. Discussion
It is well established that heavy ethanol consumption for extended periods of time can damage various cells and organs, mainly through increased oxidative/nitrosative stress, mitochondrial dysfunction, production of inflammatory cytokines, activation of the cell-death signaling pathways, and decreased levels of anti-oxidant molecules [24–27]. In fact, ethanol exposure can activate CYP2E1, NADPH oxidase, and NOS while it suppresses the trans-sulfuration and glutathione pathways [27] as well as the mitochondrial electron transport chain, resulting in increased production of reactive oxygen/nitrogen species (ROS/RNS). These elevated ROS/RNS can interact with DNA, proteins, and lipids, negatively affecting their cellular functions. In addition, recent results from this laboratory indicated that peroxiredoxin, involved in removal of peroxides [17], and mitochondrial ALDH2, a major enzyme in the metabolism of toxic acetaldehyde and lipid peroxides such as 4-hydroxynoenal, were inhibited after chronic and binge ethanol treatments [19]. Recent data with knockout mice [15] and transgenic mice containing the inactive human ALDH2-2 variant [16] showed that acetaldehyde was significantly elevated after acute ethanol exposure. Moreover, our previous results [17] clearly showed that ALDH1 was oxidatively-modified in ethanol-fed mouse livers. These data suggest that cytosolic ALDH1 with low-m values for acetaldehyde must be inactivated during and after ethanol treatment. Potential inhibition of ALDH1 and its implication on acetaldehyde accumulation following ethanol exposure have not been characterized systematically, although many reports showed inhibition of mitochondrial ALDH2 by toxic chemicals [19,28,29] and in pathological conditions [4–6,30]. Therefore, this study was aimed at investigating whether hepatic ALDH1 activity could be inhibited in ethanol-exposed rats and whether oxidative modification of its Cys residue(s) could play a role in the ALDH1 inhibition.
It is known that Cys residues in many proteins can be modified by any of the following modifications: oxidation to disulfides [31], sulfenic acid [32], sulfinic acid and sulfonic acid [33], glutathione-dependent -glutathionylation [34], NO-dependent -nitrosylation [35], NO-independent ADP-ribosylation [36], etc. Using site-directed mutagenesis followed by biochemical characterizations, Weiner and coworkers [37] demonstrated that Cys302 of ALDH2 is the active site Cys, which is highly conserved among the ALDH2 superfamily members. To further identify the active site of ALDH1, we aligned the peptide sequences around the putative active site Cys of major ALDH isozymes and compared those with Cys302 of ALDH2 [37]. As listed in Table 1, all of these ALDH isozymes including ALDH1 have similar peptide sequences around the active site Cys, which is 100% identical among all the ALDH isozymes we compared. Based on this structural comparison, Cys303 is considered the active site of ALDH1 and that this Cys was likely to be oxidatively modified after ethanol exposure, resulting in its inactivation. It is conceivable that other Cys residues and/or other amino acid residues of ALDH1 may be also oxidatively modified, contributing to inactivation of its activity. In fact, our preliminary results show that tyrosine residue(s) of cytosolic ALDH1 could be nitrated in ethanol-treated rats. Furthermore, Cys303 and other Cys residues of ALDH1 can be modified after exposure to toxic chemicals or under pathophysiological conditions, similar to those present to modify ALDH2 [19,28,29].
Table 1.
Comparison of the peptide sequences at the active site cysteine of rat ALDH isozymes.
| Name | Peptide sequence | * | Swiss-prot | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALDH1 | 291A | H | H | G | V | F | Y | H | Q | G | Q | C | C | V | A | A | S | R308 | P51647 |
| ALDH2 | 290A | H | F | A | L | F | F | N | Q | G | Q | C | C | C | A | G | S | R307 | P11884 |
| ALDH3 | 232I | A | W | G | K | F | M | N | S | G | Q | T | C | V | A | P | D | Y249 | P11883 |
| ALDH5 | 281A | M | A | S | K | F | R | N | A | G | Q | T | C | V | C | S | N | R298 | P51650 |
| ALDH6 | 302A | H | Q | G | V | F | F | N | Q | G | Q | C | C | T | A | A | S | R319 | Q8K4D8 |
| ALDH9 | 276A | L | L | A | N | F | L | T | Q | G | Q | V | C | C | N | G | T | R293 | Q9JLJ3 |
| RALDH2 | 308A | H | Q | G | V | F | F | N | Q | G | Q | C | C | T | A | G | S | R325 | Q63639 |
| FTHFDH | 695G | M | S | S | V | F | F | N | K | G | E | N | C | I | A | A | G | R712 | P28037 |
active site cysteine
Dark gray boxes show 100% identical amino acids and light gray boxes show highly conserved amino acids. RALDH2, retinal aldehyde dehydrogenase; FTHFDH, 10-formyltetrahydrofolate dehydrogenase (aldehyde dehydrogenase 1 family member L1).
Our current results show that ALDH1 is inhibited by ethanol-mediated -nitrosylation based on the following facts: (1) identification of ALDH1 in oxidized cytosolic proteins after ethanol exposure; (2) inhibition of ALDH1 activity; (3) -nitrosylation of Cys in immunoprecipitated ALDH1 recognized with the anti--NO-Cys antibody; (4) restoration of the suppressed ALDH1 activity by DTT; (5) disappearance of the -nitrosylated ALDH1 band in the presence of DTT. To our knowledge, our results represent the first report to show the inactivation of ALDH1 in rat livers via ethanol-mediated -nitrosylation. Other ALDH isozymes, with high-m values for acetaldehyde but low-m values for their specific substrates, do not seem to be inhibited when 150 μM propionaldehyde was used in the current study. Since other ALDH isozymes such as ALDH3, ALDH5, ALDH6, ALDH9, RALDH2, and 10-formyltetrahydrofolate dehydrogenase (FTHFDH) also have similar structures at their active sites (Table 1), the activities of these ALDH isozymes is likely to be inhibited, if measured with their specific substrates, by similar mechanisms upon exposure to ethanol or toxic chemicals that produce reactive intermediates and/or ROS/RNS. For instance, FTHFDH, involved in the metabolism of tetrahydrofolate, is another member of NADP+-dependent ALDH family. This enzyme contains an active site Cys (Cys707) with a C-terminal domain (amino acids 420–902) similar to that of NAD+-dependent ALDH2 [38,39]. The activity of FTHFDH was significantly inhibited in the ethanol-fed rat liver, although the inhibitory mechanism was not characterized [40]. In addition, the activity of FTHFDH was inhibited after acute treatment with a hepatotoxin acetaminophen [41]. The structural comparison (Table 1) and biochemical results [40,41] can support our view about potential inhibition of other ALDH isozymes via oxidative modifications, especially when their activities are determined with their specific substrates. Furthermore, since ALDH isozymes are important in the metabolism of potentially toxic aldehydes, inactivation of ALDH isozymes by various toxic chemicals is likely to cause marked accumulation of toxic aldehydes, leading to increased susceptibility towards irreversible damage. Our current results not only explain the accumulation of acetaldehyde in ethanol-treated knockout mice but also indicate that knockout mice might still be used as a model to study the role of elevated acetaldehyde in ethanol preference [42], acetaldehyde accumulation [15], and acetaldehyde-mediated DNA damage [43] or tissue damage [44] after long-term exposure to ethanol or acetaldehyde.
In conclusion, we have studied the mechanism of ALDH1 inhibition after ethanol exposure. Our results show that hepatic ALDH1 can be -nitrosylated in ethanol-exposed rats, leading to inhibition of its activity. Our current results may help explain the underlying mechanisms of ALDH1 inhibition and accumulation of acetaldehyde or lipid peroxides following exposure to ethanol or other toxic agents.
Acknowledgments
This study was supported by the Intramural Research Program of the National Institute on Alcohol Abuse and Alcoholism. We thank Dr. Robert L. Eskay for providing rat tissues and Dr. Norman Salem, Jr. for supporting our experiments.
Footnotes
Abbreviations used: ALDH1, cytosolic aldehyde dehydrogenase; ALDH2, mitochondrial aldehyde dehydrogenase; DTT, dithiothreitol;-NO-Cys, -nitroso-cysteine; biotin-NM, biotin--maleimide; HRP, horse-radish peroxidase; ROS/RNS, reactive oxygen/nitrogen species; FTHFDH, 10-formyltetrahydrofolate dehydrogenase.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yoshida A, Rzhetsky A, Hsu LC, Chang C. Human aldehyde dehydrogenase gene family. 1998;251:549–57. doi: 10.1046/j.1432-1327.1998.2510549.x. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl R. Aldehyde dehydrogenases and their role in carcinogenesis. 1992;27:283–335. doi: 10.3109/10409239209082565. [DOI] [PubMed] [Google Scholar]
- 3.Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. 2005;2:138–43. doi: 10.1186/1479-7364-2-2-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day CP, Bashir R, James OF, Bassendine MF, Crabb DW, Thomasson HR, Li TK, Edenberg HJ. Investigation of the role of polymorphisms at the alcohol and aldehyde dehydrogenase loci in genetic predisposition to alcohol-related end-organ damage. 1991;14:798–801. doi: 10.1002/hep.1840140509. [DOI] [PubMed] [Google Scholar]
- 5.Yokoyama A, Muramatsu T, Omori T, Yokoyama T, Matsushita S, Higuchi S, Maruyama K, Ishii H. Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. 2001;22:433–9. doi: 10.1093/carcin/22.3.433. [DOI] [PubMed] [Google Scholar]
- 6.Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, Terashima M, Tsutsumi Y, Suzuki S, Morii I, Hanai S, Ono K, Baba S, Tomoike H, Kawamura A, Miyazaki S, Nonogi H, Goto Y. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. 2002;25:677–81. doi: 10.1291/hypres.25.677. [DOI] [PubMed] [Google Scholar]
- 7.Ehrig T, Bosron WF, Li TK. Alcohol and aldehyde dehydrogenase. 1990;25:105–16. doi: 10.1093/oxfordjournals.alcalc.a044985. [DOI] [PubMed] [Google Scholar]
- 8.Hempel J, Nicholas H, Lindahl R. Aldehyde dehydrogenases: widespread structural and functional diversity within a shared framework. 1993;2:1890–900. doi: 10.1002/pro.5560021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinmetz CG, Xie P, Weiner H, Hurley TD. Structure of mitochondrial aldehyde dehydrogenase: the genetic component of ethanol aversion. 1997;5:701–11. doi: 10.1016/s0969-2126(97)00224-4. [DOI] [PubMed] [Google Scholar]
- 10.Moore SA, Baker HM, Blythe TJ, Kitson KE, Kitson TM, Baker EN. Sheep liver cytosolic aldehyde dehydrogenase: the structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. 1998;6:1541–51. doi: 10.1016/s0969-2126(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 11.Ni L, Zhou J, Hurley TD, Weiner H. Human liver mitochondrial aldehyde dehydrogenase: three-dimensional structure and the restoration of solubility and activity of chimeric forms. 1999;8:2784–90. doi: 10.1110/ps.8.12.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eriksson CJ. Ethanol and acetaldehyde metabolism in rat strains genetically selected for their ethanol preference. 1973;22:2283–92. doi: 10.1016/0006-2952(73)90009-9. [DOI] [PubMed] [Google Scholar]
- 13.Svanas GW, Weiner H. Aldehyde dehydrogenase activity as the rate-limiting factor for acetaldehyde metabolism in rat liver. 1985;236:36–46. doi: 10.1016/0003-9861(85)90603-4. [DOI] [PubMed] [Google Scholar]
- 14.Klyosov AA, Rashkovetsky LG, Tahir MK, Keung WM. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. 1996;35:4445–56. doi: 10.1021/bi9521093. [DOI] [PubMed] [Google Scholar]
- 15.Isse T, Matsuno K, Oyama T, Kitagawa K, Kawamoto T. Aldehyde dehydrogenase 2 gene targeting mouse lacking enzyme activity shows high acetaldehyde level in blood, brain, and liver after ethanol gavages. 2005;29:1959–64. doi: 10.1097/01.alc.0000187161.07820.21. [DOI] [PubMed] [Google Scholar]
- 16.Jeng J, Goldman D, Eriksson CJP, Kallarakal AT, Wang TTY, Weiner H, Song BJ. Enzymology and Molecular Biology of Carbonyl Metabolism. Vol. 12. Purdue University Press; West Lafayette, IN: 2005. Accumulation of hepatic acetaldehyde and reduced alcohol drinking in transgenic mice carrying the oriental variant of the human aldehyde dehydrogenase 2 gene; pp. 42–49. [Google Scholar]
- 17.Kim BJ, Hood BL, Aragon RA, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. 2006;6:1250–60. doi: 10.1002/pmic.200500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon KH, Kim BJ, Song BJ. Inhibition of mitochondrial aldehyde dehydrogenase by nitric oxide-mediated S-nitrosylation. 2005;579:6115–20. doi: 10.1016/j.febslet.2005.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. 2006;44:1218–30. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 20.Hamelink C, Hampson A, Wink DA, Eiden LE, Eskay RL. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. 2005;314:780–8. doi: 10.1124/jpet.105.085779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeong KS, Soh Y, Jeng J, Felder MR, Hardwick JP, Song BJ. Cytochrome P450 2E1 (CYP2E1)-dependent production of a 37-kDa acetaldehyde-protein adduct in the rat liver. 2000;384:81–7. doi: 10.1006/abbi.2000.2119. [DOI] [PubMed] [Google Scholar]
- 22.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. 2006;281:21256–65. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 23.Tank AW, Weiner H, Thurman JA. Enzymology and subcellular localization of aldehyde oxidation in rat liver. Oxidation of 3,4-dihydroxyphenylacetaldehyde derived from dopamine to 3,4-dihydroxyphenylacetic acid. 1981;30:3265–75. doi: 10.1016/0006-2952(81)90598-0. [DOI] [PubMed] [Google Scholar]
- 24.Lieber CS. Metabolism of alcohol. 2005;9:1–35. doi: 10.1016/j.cld.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Pessayre D, Mansouri A, Haouzi D, Fromenty B. Hepatotoxicity due to mitochondrial dysfunction. 1999;15:367–73. doi: 10.1023/a:1007649815992. [DOI] [PubMed] [Google Scholar]
- 26.Hoek JB, Pastorino JG. Cellular signaling mechanisms in alcohol-induced liver damage. 2004;24:257–72. doi: 10.1055/s-2004-832939. [DOI] [PubMed] [Google Scholar]
- 27.Villanueva JA, Halsted CH. Hepatic transmethylation reactions in micropigs with alcoholic liver disease. 2004;39:1303–10. doi: 10.1002/hep.20168. [DOI] [PubMed] [Google Scholar]
- 28.Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. 2006;19:102–10. doi: 10.1021/tx0501839. [DOI] [PubMed] [Google Scholar]
- 29.Hjelle JJ, Grubbs JH, Beer DG, Petersen DR. Time course of the carbon tetrachloride-induced decrease in mitochondrial aldehyde dehydrogenase activity. 1983;67:159–65. doi: 10.1016/0041-008x(83)90220-x. [DOI] [PubMed] [Google Scholar]
- 30.Park KS, Cho SY, Kim H, Paik YK. Proteomic alterations of the variants of human aldehyde dehydrogenase isozymes correlate with hepatocellular carcinoma. 2002;97:261–5. doi: 10.1002/ijc.1585. [DOI] [PubMed] [Google Scholar]
- 31.Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. 2004;279:21749–58. doi: 10.1074/jbc.M312267200. [DOI] [PubMed] [Google Scholar]
- 32.DeMaster EG, Quast BJ, Redfern B, Nagasawa HT. Reaction of nitric oxide with the free sulfhydryl group of human serum albumin yields a sulfenic acid and nitrous oxide. 1995;34:11494–9. doi: 10.1021/bi00036a023. [DOI] [PubMed] [Google Scholar]
- 33.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. 2005;280:3125–8. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 34.Mohr S, Hallak H, de Boitte A, Lapetina EG, Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. 1999;274:9427–30. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 35.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 36.McDonald LJ, Moss J. Nitric oxide-independent, thiol-associated ADP-ribosylation inactivates aldehyde dehydrogenase. 1993;268:17878–82. [PubMed] [Google Scholar]
- 37.Farres J, Wang TT, Cunningham SJ, Weiner H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. 1995;34:2592–8. doi: 10.1021/bi00008a025. [DOI] [PubMed] [Google Scholar]
- 38.Krupenko SA, Wagner C, Cook RJ. Cysteine 707 is involved in the dehydrogenase activity site of rat 10-formyltetrahydrofolate dehydrogenase. 1995;270:519–22. doi: 10.1074/jbc.270.2.519. [DOI] [PubMed] [Google Scholar]
- 39.Krupenko SA, Wagner C, Cook RJ. Expression, purification, and properties of the aldehyde dehydrogenase homologous carboxyl-terminal domain of rat 10-formyltetrahydrofolate dehydrogenase. 1997;272:10266–72. doi: 10.1074/jbc.272.15.10266. [DOI] [PubMed] [Google Scholar]
- 40.Min H, Im ES, Seo JS, Mun JA, Burri BJ. Effects of chronic ethanol ingestion and folate deficiency on the activity of 10-formyltetrahydrofolate dehydrogenase in rat liver. 2005;29:2188–93. doi: 10.1097/01.alc.0000191756.02856.a8. [DOI] [PubMed] [Google Scholar]
- 41.Pumford NR, Halmes NC, Martin BM, Cook RJ, Wagner C, Hinson JA. Covalent binding of acetaminophen to N-10-formyltetrahydrofolate dehydrogenase in mice. 1997;280:501–5. [PubMed] [Google Scholar]
- 42.Isse T, Oyama T, Kitagawa K, Matsuno K, Matsumoto A, Yoshida A, Nakayama K, Nakayama K, Kawamoto T. Diminished alcohol preference in transgenic mice lacking aldehyde dehydrogenase activity. 2002;12:621–6. doi: 10.1097/00008571-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Matsuda T, Matsumoto A, Uchida M, Kanaly R, Misaki K, Shibutani S, Kawamoto T, Kitagawa K, Nakayama KI, Tomokuni K, Ichiba M. Increased formation of hepatic N2-ethylidene-2′-deoxyguanosine DNA adducts in aldehyde dehydrogenase 2 knockout mice treated with ethanol. 2007 Mar 14; doi: 10.1093/carcin/bgm057. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 44.Oyama T, Isse T, Ogawa M, Muto M, Uchiyama I, Kawamoto T. Susceptibility to inhalation toxicity of acetaldehyde in Aldh2 knockout mice. 2007;12:1927–34. doi: 10.2741/2198. [DOI] [PubMed] [Google Scholar]