

Abstract
A rare subpopulation of cells within malignant gliomas, which shares canonical properties with neural stem cells (NSCs), may be integral to glial tumor development and perpetuation. These cells, also known as tumor initiating cells (TICs), have the ability to self-renew, develop into any cell in the overall tumor population (multipotency), and proliferate. A defining property of TICs is their ability to initiate new tumors in immunocompromised mice with high efficiency. Mounting evidence suggests that TICs originate from the transformation of NSCs and their progenitors. New findings show that TICs may be more resistant to chemotherapy and radiation than the bulk of tumor cells, thereby permitting recurrent tumor formation and accounting for the failure of conventional therapies. The development of new therapeutic strategies selectively targeting TICs while sparing NSCs may provide for more effective treatment of malignant gliomas.
Keywords: Central nervous system, Glioblastoma, Cancer stem cells, Brain cancer
Introduction
The hypothesis that tumor growth may be sustained by a rare subpopulation of cells, termed tumor initiating cells (TICs), is currently being explored in different types of cancer [1-6]. These cells have also been referred to as cancer stem cells (CSCs), although this nomenclature has been questioned due to the controversial nature of their “stemness” [7, 8]. The terms CSCs and TICs are often used interchangeably. Recently, an attempt at defining TICs from CSCs has been made based on the number of mutations present in these cells and their ability to support tumor growth [9]. TICs are identified in the early stages of tumor development and may not yet have acquired the full tumorigenic potential of CSCs, therefore may carry less mutations and may not grow as well in animal models for tumorigenicity. In this review, we will use the terminology of TICs.
The most common and malignant cancer of the adult central nervous system, known as astrocytoma WHO grade IV or glioblastoma multiforme (GBM), has been shown to contain TICs [10, 11]. Cells with the ability to regenerate a GBM have been isolated from fresh specimens based on their expression of a 120-kDa cell-surface protein known as CD133 (Prominin-1) [10]. CD133 was initially identified as a marker of normal human neural precursors and is also a marker for hematopoietic stem cells [12-14]. CD133+ GBM-initiating cells possess in vitro neural stem-cell like characteristics of extensive self-renewal, multipotency (capability to develop into any cell in the overall tumor population) and the proliferative ability for generation of many progeny. They are able to initiate new tumors in vivo when transplanted into immunocompromised mice at low cell numbers [11].
Glioblastoma continues to be one of the most lethal cancers despite all conventional therapies [15]. These highly vascularized and diffusely infiltrative tumors are radioresistant, chemoresistant, and tend to recur in a local fashion despite surgical resection [16-18]. Complete surgical excision of GBM tumors is impossible, as individual tumor cells can deeply infiltrate adjacent normal brain. Initial findings on GBM-initiating cells provide new explanations for patient relapse. Recent evidence has shown that a subpopulation of TICs resides in a vascular niche and that TICs promote radioresistance [19-21] and chemoresistance [22-24]. Targeted therapy against GBM-initiating cells or their vascular niche may form the basis of new treatments.
Definition and source of neural stem cells in the brain
Neural stem cells and GBM-initiating cells share both phenotypic and functional similarities. The have both been shown to express the CD133 cell surface marker and are self-sustaining and able to self-renew. Reynolds and Weiss were the first to isolate adult neural stem cells from the adult striatum that could proliferate and generate multipotent clones of cells in vitro termed neurospheres [25, 26]. Neurosphere culture relies on a serum-free, selective growth factor [epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF-2)] system in which most differentiating or differentiated cells rapidly die [27-30]. Growth-factor-responsive neural stem cells can be passaged and expanded indefinitely with little change in their growth or differentiation characteristics. Removal of growth factors induces the differentiation of the progeny of neural stem cells into neurons, astrocytes, and oligodendrocytes. As a result, adult neural stem cells possess the stem-cell criteria of self-renewal, multipotency, and the generation of many progeny.
Neural stem cells have been isolated from the subventricular zone lining of the lateral ventricles, the dentate gyrus within the hippocampus, and the subcortical white matter [31-34].
The largest of these germinal regions in humans is the subventricular zone, located between the ependymal layer of the lateral ventricles and the parenchyma of the striatum (Fig. 1). The subventricular zone is thought to be the likely source of GBM-initiating cells [35]. A subset of cells in this region display astrocyte features; yet, they can function as full-fledged neural stem cells [32, 36]. These astrocytic stem cells express the astroglial marker glial fibrillary acidic protein as do mature astrocytes of the brain parenchyma, which do not possess stem-cell properties. They are able to give rise to neurons, astrocytes, and oligodendrocytes through the generation of uncommitted progenitor cells known as transit-amplifying progenitor cells (TACs) [37-40]. Fast-cycling TACs give rise to committed progenitor cells (neuronal or glial), which retain the capacity of producing progeny along either neuronal or glial lineages (oligodendrocytes and/or astrocytes), but not both (Fig. 2). Committed progenitor cells (PC) can possess replicative ability, but they do not usually have the self-renewal capacity of stem cells. Neural stem cells, committed progenitor cells, and differentiated adult glial cells are all thought to be substrates for neoplastic transformation and ultimate formation of GBM tumors [1].
Fig. 1.

Source of neural stem cells and GBM tumors. The subventricular zone (SVZ) is found beneath the ependymal (E) layer that lines the lateral ventricles (LV) of the brain and is believed to be the origin of GBM tumors (upper section). Neural stem cells (NSCs) originate from this area of the brain and are characterized by the cell surface markers CD133 and nestin. Immunohistochemistry staining of the SVZ shows the presence of nestin-expressing cells, some of which represent NSCs (lower section). The structures on the right side of the upper section are choroid plexus protrusions from which the CSF filling the ventricles originates
Fig. 2.

Possible lineage relationships for the ontogeny and production of tumor-initiating cells (TICs) and generation of GBM tumors. During normal CNS differentiation neural stem cells (NSCs) undergo an amplification step to produce transit-amplifying progenitor cells (TACs), which then differentiate into committed progenitor cells (PCs*). PC* (neuronal or glial) retain the capacity of producing progeny along either neuronal or glial lineages (oligodendrocytes and/or astrocytes), but not both. Mutations generating gliomas can occur at all levels within this lineage and produce TICs or differentiated tumor cells (DTCs). TICs are thought to be stem-like in behavior by their ability to self-renew, proliferate, and generate DTCs or cancer progenitor cells (CPG) within the tumor mass. While this model suggests unilateral progression toward mutated cell populations that become terminally differentiated cancer cells, in reality, there is evidence in the literature for more plasticity, and de-differentiation events may also take place to generate self-propagating cancer cells from astrocytes and oligodendrocytes. It is unknown whether neurons can generate cancer cells
GBM-initiating cells
Genetic evidence for the existence of a precursor cell population in glioblastoma from which the bulk of the tumor was derived has existed for some time [41]. However, it was only recently that the first experimental evidence of cells with stem-like characteristics was reported in human GBM [42]. Clonogenic, neurosphere-forming precursor cells were isolated from GBM specimens that expressed neuronal and astroglial markers upon differentiation. By applying the same conditions used for the isolation of human neural stem cells, others were later able to isolate clonogenic neurospheres from adult human GBM [10, 11, 43, 44]. Purification of CD133-positive cells from human gliomas by flow cytometry can also allow for the isolation and growth of TICs [45]. These cells are capable of self-renewal, multipotency, and generation of many progeny in vitro (Fig. 2). CD133+ tumor neurospheres under neural stem cell culture conditions express the stem cell marker, nestin, and upon exposure to serum, differentiate into a mixed population of neurons, astrocytes, and oligodendrocytes. A small fraction of CD133+GBM-initiating cells (as few as 100 implanted cells) can initiate tumors in vivo when transplanted into immunocompromised mice. These tumors exhibit many of the key malignant features of GBM, including infiltration and microvascular proliferation. CD133- cells derived from the tumors studied by Singh et al. failed to form orthotopic tumors in mice even following injection of a larger cell inoculum (105 per injection) [45]. More recently, several reports suggest a less clear distinction between the ability of CD133+ and CD133- cells to form orthotopic tumors [20, 46-48]. CD133- cells isolated from primary GBM tumors have been reported to equally form orthotopic tumors as the CD133+ subpopulation. Implanted CD133- cells have also been shown to form CD133+ cells in orthotopic tumors [49]. The reasons for these discrepancies are currently unclear. This may reflect in part the presence of other types of TICs in GBM, which are devoid of CD133.
Purification of TICs from malignant gliomas was also achieved through the side population technique [50-52]. This technique was originally used to identify adult stem cells in bone-marrow-derived cells. Those cells that did not accumulate an appreciable amount of Hoechst dye 33342 were classified as a Hoechst side population (SP) that remarkably was highly enriched for hematopoietic stem cells. The SP approach represents a valid marker-independent method to identify TICs in various cancers, including brain [51]. SP cells from the rat C6 glioma cell line can be passaged and maintained in vitro by serum-free medium containing platelet-derived growth factor (PDGF) and FGF-2. SP cells can also form tumors in immunocompromised mice. The current lack of a single marker to identify all TICs in malignant gliomas confirms the probable molecular heterogeneity among these cells. Enrichment of TICs relies on either neurosphere culture, isolation of CD133+ cells, or use of the SP approach on glioblastoma tumor cells [53].
GBM-initiating cell niche
An important characteristic of at least one subset of normal neural stem cells is that they are concentrated in regions of the brain that are rich in blood vessels, called “vascular niches” [54-57]. Within these niches, neural stem cells are thought to be sheltered from apoptotic stimuli to maintain a proper balance between self-renewal and differentiation. Endothelial cells are a key component of the vascular niche. They secrete paracrine factors that promote stem cell survival and self-renewal. The high vascularity of glioblastoma tumors is well known. Microvascular proliferation is a pathologic hallmark of glioblastoma [58]; therefore, it was conceivable that the tumor might be an alternative harbor for cells requiring such paracrine factors for their maintenance and survival. Stem cells are also known to thrive in hypoxic regions [59], and the tumor may provide a hypoxic niche [18, 60]. Upon examination, GBM-initiating cells, like neural stem cells, were found intimately associated with the vascular niche in the tumor [19]. CD133+/nestin+ GBM stem cells were frequently found close to capillaries within GBM. When the CD133+ tumor cells were cultured with primary human endothelial cells, they rapidly and selectively associated with endothelial cells, while the majority of tumor cells did not. The endothelial cells were also found to enhance the self-renewal capacity of the CD133+GBM-initiating cells. In vivo, CD133+ tumor cells transplanted with endothelial cells grew more rapidly than cells transplanted alone. Self-renewing rat C6 GBM stem cells have also been maintained in vitro by factors secreted from endothelial cells [61]. This was not due to increased vascular density but rather a reduction in time needed to establish the level of vasculature needed for tumor growth. In addition, tumors established in the presence of endothelial cells contained up to 25 times more CD133+ CSCs. These data suggest that endothelial cells enhance the self-renewal of the TICs in vitro and promote growth of brain tumors in vivo.
Further evidence of a functional relationship between the tumor vasculature and GBM stem cells shows that cancer-initiating cells also sustain vascular development [62]. CD133+GBM stem cells show higher level production of vascular endothelial growth factor (VEGF) than CD133-cells. This likely explains why CD133+ and not CD133-GBM cells readily formed highly vascular and hemorrhagic tumors in the brains of immunocompromised mice.
Genesis of glioblastoma
All forms of cancer are believed to originate from a series of mutations in single cells (clones), which endow them with biological properties that permit their growth into tumors, including disturbances in proliferation, apoptosis, and tissue invasion [63]. A number of genetic aberrations involved in the genesis of glioblastomas have been well described for both primary (de novo) and secondary (progressive) tumors [64, 65]. The “cancer-stem cell hypothesis” [2] suggests that a rare subset of cells within GBM tumors may have significant expansion capacity and the ability to generate new tumors. The remainder of tumor cells, which predominantly make up GBM tumors, may represent partially differentiated cells with limited progenitor capacity or terminally differentiated cells that cannot form new tumors.
Mounting evidence supports the observation that transformation of neural stem cells and their progenitors may lead to the formation of GBM-initiating cells and tumors [66-68] rather than from a differentiated cell type. Micro-array studies have revealed considerable overlap between the gene-expression signatures of glioblastomas and progenitor cells of the developing forebrain [69]. Gene expression similarities suggest common signaling pathways present in neural stem cells and GBM-initiating cells [43]. Genetic studies in mice have brought some insight into the genetic alterations (deletion of tumor suppressors and/or activation of oncogenes) that can lead to neural stem cell and progenitor cell transformation [70]. Retroviral transfer of signal transduction proteins, Akt and Ras, in the brain leads to the transformation undifferentiated nestin-expressing progenitor cells into tumors with aggressive features reminiscent of GBM histology. Expression of ras and myc oncogenes in oligodendrocyte progenitors yields cells that readily form high-grade gliomas when transplanted in vivo [71]. Others have shown transgenic Ink4a-Arf -/- mouse neural stem cells form aggressive GBM-like tumors in vivo when transfected with a constitutively active EGF receptor (EGFR) [72]. In contrast, mature, differentiated mouse astrocytes appear less prone to transformation, although this is still a subject of controversy. In vitro, human astrocytes need three defined transformation events, expression of human telomerase reverse transcriptase, and activation of two oncogenic pathways, for example, ras and Akt [73, 74]. Evidence that both astrocytes and neural stem cells can serve as the cell-of-origin for malignant gliomas exists [72]. The combined loss of p16INK4a and p19ARF enables astrocyte dedifferentiation in response to EGFR activation, while transduction of Ink4a/Arf-/- neural stem cells with constitutively active EGFR induces high-grade gliomas.
Genes involved in cell cycle and apoptosis have been implicated in the proliferation and tumorigenicity of GBM-initiating cells. The HEDGEHOG (HH)-GLI signaling pathway has been shown to control the behavior of human GBM TICs by regulating their self-renewal and tumorigenicity [75]. Another transcription factor, OLIG2, has been shown to promote proliferation of both neural progenitors and GBM-initiating cells, possibly by repressing the p21 tumor suppressor [67]. Jackson and colleagues [76] have shown that amplification of the PDGF signaling in neural stem cells induces the formation of large GBM tumors. Deletion of Nf1 and Trp53 from neural stem cells in mice initiates gliomagenesis in the subventricular zone where neural stem cells reside [77].
Impact of TICs on GBM therapy
Conventional therapies for GBM involving surgery, radiation, and chemotherapy (temozolomide) offer modest benefits to patients with GBM [78]. Virtually, all patients with GBM have a local relapse of their disease at the site of their initial treatment. Current evidence proposes that despite making up a small fraction of tumor cells, GBM-initiating cells are necessary for the initiation and maintenance of tumors (Fig. 3). The presence of a rare subpopulation of cells within GBM possibly responsible for generating the entire mass of tumor cells has important implications for the understanding of the tumor biology and the efficacy of existing therapies. A re-evaluation of our current knowledge needs to be made in light of this new hypothesis.
Fig. 3.

TICs as the origin of GBM tumor resistance to therapy. Recurrence of malignant brain tumors in 3-6 months may be related to the resistance of TICs to standard therapies, such as chemoradiation, and their ability to generate new tumors. Note that the bulk of the tumor (shown by empty circles marked by X) responds to therapy, but the TICs are unaffected and regenerate the tumor. It is currently unknown whether CPGs may possess the capacity to regenerate the tumor as well. Clearly, this is a simplified model, and different cell populations in the tumor will respond differentially to individual therapies, depending on their intrinsic resistance mechanisms; these may include expression of drug transporters, genetic composition, dependence upon specific signaling pathways, aerobic and anaerobic metabolism, and ability to cope with reactive oxygen species (ROS) stress. TIC tumor-initiating cell, CPG cancer progenitor cell, DTC differentiated tumor cell
The last few decades of progress made on the individual parameters that characterize a tumor (proliferation, apoptosis, invasion, angiogenesis, immune resistance, etc.) have all been obtained through analysis of the bulk of the tumor. These studies have been the “guiding principle” for identifying oncogenic pathways for therapeutic targeting. Therapeutic clinical trials that have been performed based on these premises for glioblastoma have largely been a failure. As a result, the question remains as to whether the proper cellular target is missed and whether the signaling pathways driving their transformed state are radically different.
Based on the current TIC hypothesis, the bulk of the tumor is comprised of the differentiated progeny from TICs. Glioblastomas may consist of three broad categories of cells: TICs, cancer progenitor cells (CPGs), and differentiated tumor cells (DTCs) [79, 80]. CPGs, also known as progenitor-like glioma cells, may originate from TICs and lead to DTCs by mutational events or differentiation. These are purely speculative observations. The subdivision of the tumor into different cell populations is an attractive hypothesis but needs further substantiation. A hierarchical system of TICs and CPGs may both lead to the development of DTCs. Treatments that target the TIC may be effective at treating the entire tumor mass (Fig. 4). In addition, if the majority of cells contain the genetic alterations that initiated transformation in the neural stem cell population, targeting of these pathways is warranted. Ultimately, targeting GBM-initiating cells may offer better therapeutic strategies that could translate to much better patient survival and quality of life. Several strategies have been shown already to be effective at targeting GBM-initiating cells and causing their demise.
Fig. 4.
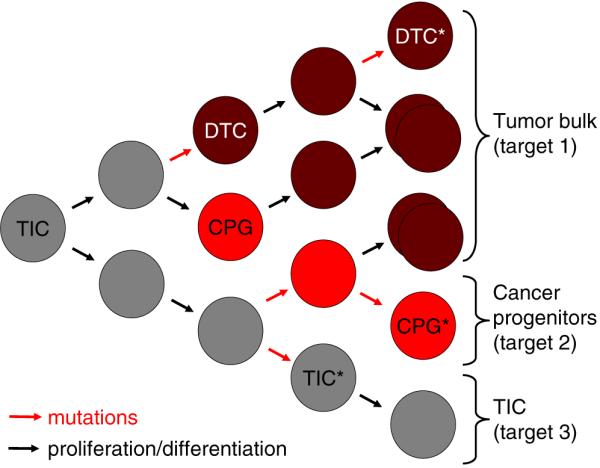
Therapeutic intervention for CNS tumors requires targeting of multiple cell types. Treatments for CNS tumors may require targeting of differentiated tumor cells (DTC), which likely make up the bulk of the tumor (target 1), cancer progenitor cells (CPG; target 2), and TICs (target 3). TICs can give rise to DTCs through mutational events, while TICs may develop CPGs by mutations or differentiation events. CPGs are likely committed to the development of DTCs after proliferation and differentiation. Each of these cell populations may require distinct therapies, either delivered simultaneously or sequentially. Clonal evolution of these cell populations in the tumor is linked to the sequential accumulation of genetic defects (mutations), which will drive cell expansion. Activation of differentiation programs will limit the pluripotency of certain cells but not necessarily their replicative potential. It is currently unknown whether DTC can undergo multiple rounds of cell division or are rapidly terminally differentiated and cease proliferation. This simplified schematic does not exclude the existence of a whole hierarchy of TIC, CPG, and DTC with multiple genetic defects (asterisk). It also allows for the coexistence of different cell populations, which may share initial genetic defects but have distinct downstream mutations. Conceivably, such cell populations may even gain a mutual benefit by providing reciprocal cell survival signals. TIC tumor-initiating cell, CPG cancer progenitor cell, DTC differentiated tumor cell
New therapeutic strategies targeting GBM-initiating cells
Recent evidence supports the notion that GBM-initiating cells may be implicated in the resistance of tumors to radiation and chemotherapy [21] (Table 1). Bao et al. suggest that CD133+GBM-initiating cells are resistant to ionizing radiation (IR) because they are more efficient at inducing repair of damaged DNA than the bulk of tumor cells [20]. They have shown that CD133+GBM-initiating cells escape the lethal DNA-damaging effects of IR by preferential activation of DNA repair checkpoints through phosphorylation of the checkpoint proteins Chk1 and Chk2. In both cell culture and the brains of immunocompromised mice, CD133-expressing glioma cells survive IR in increased proportions relative to most tumor cells, which lack CD133. After irradiation of tumors, CD133+ cells become enriched up to fivefold compared to CD133- cells as a result of apoptosis reduction. The authors further showed that inhibition of the checkpoint kinases, Chk1 and Chk2, with small molecules can reverse the radioresistance of CD133+GBM-initiating cells, suggesting new options for combination radiotherapy using Chk1/2 inhibitors as radio-sensitizers.
Table 1.
Summary of various therapeutic strategies against TICs in the brain
| Technology | Mechanism |
|---|---|
| BMPs (bone morphogenic proteins) | Promote differentiation of TICs |
| Anti-angiogenesis molecules (bevacizumab) | Disruption of vascular niche |
| DNA repair inhibitors (Chk1 and Chk2 inhibitors) | Radiosensitization of TICs |
| Nanotechnology (magnetic nanoparticles) | Imaging and tracking of TICs; Potential targeted therapy (drug binding or local hyperthermia) |
| Immunotherapy | Elicit a natural immune response to TICs |
| Oncolytic therapy (viruses) | Viral constructs specific to TICs may conditionally replicate and lyse cells |
Chemoresistance of GBM-initiating cells may also account for glioblastoma tumor progression. Recently, it was shown that GBM-initiating cells are resistant to the currently used adjuvant agent, temozolomide [75]. Several mechanisms of CSC drug resistance have been described in various cancers. Elevated expression of transporters that pump out chemotherapeutic agents may be one important mechanism [24]. In hematopoietic cancers, ATP-binding cassette (ABC) drug transporters have been shown to protect cancer stem cells from chemotherapeutic agents [22]. As mentioned above a distinct SP of cells can be isolated from human tumors and represent a class of CSCs with inherently high resistance to chemotherapeutic agents due to their high drug efflux capacity [23]. SP cells were initially described as a primitive CD34low/neg stem cell population in the normal bone marrow and muscle. This subset of cells was shown to have the unique capacity to efflux lipophilic dyes such as Hoechst 33342 and also to function as stem cells in the tissues from which they were isolated [81, 82]. SP cells have been shown to express high levels of the ABC drug transporter genes, ABCG2 and ABCA3, and have been found in glioblastoma cell lines [23].
While these studies highlight the need to develop therapeutic regimens tailored to GBM TIC, such approaches may have unwanted side effects on normal central nervous system (CNS) stem cells. The vulnerability of CNS progenitor cells to chemotherapeutic agents has recently been shown [83]. Clinically relevant concentrations of BCNU, cisplatin, or cytarabine were shown to be more toxic for progenitor cells than glioma cell lines. When administered systemically in mice, these chemotherapeutic agents were associated with increased cell death and decreased cell division in the subventricular zone of the CNS, in the dentate gyrus of the hippocampus, and in the corpus callosum. The vulnerability of normal neural stem cells and their progenitors to chemotherapeutic agents raises the concern of normal neurologic damage when targeting TICs. Novel therapies need to be developed that can penetrate the CNS effectively so as to be able to reach tumor cells deeply infiltrated into the CNS, yet that also show some selectivity in the killing of TIC versus normal CNS stem cell populations so as to avoid induction of neurological deficits.
Differentiation of GBM-initiating cells
Bone morphogenetic proteins (BMPs) are a family of cytokines that can regulate differentiation of normal neural stem cells [44, 84, 85]. In early embryonic neural stem cells, BMPs can promote both proliferation as well as neuronal differentiation. In contrast, neural stem cells derived from older animals undergo astrocytic differentiation in response to BMPs [86]. The difference in response of neural stem cells appears to be the result of acquisition of a new signaling pathway switch by the older stem cells. Older stem cells express the receptor, BMPR1B, which can activate STAT 3 and promote astrocytic differentiation. Early neural stem cells express BMPR1A and not BMPR1B and therefore are unable to induce activation of STAT3 [44].
Promoting differentiation of GBM-initiating cells by BMPs, specifically BMP4, has been shown to trigger a significant reduction in GBM stem cells [87]. Most CD133+GBM-initiating cells have a functional BMP receptor pathway and exposure to BMP4 resulted in a depletion of these cells in vitro as well as a reduction in their proliferation. Transient BMP4 exposure of engrafted CD133+ tumors in mice reduced tumor growth rate and extended mice survival. The BMP-treated tumor cells engrafted into mice were more mature and less invasive. CD133+ cells could not be recovered from these small tumors, and serial engraftment of mice was unable to be performed with treated tumors. A small number of animals with tumors did not respond to BMP and died 3 months after treatment. More recently, Lee and colleagues [44] have confirmed that BMPs promote glial differentiation of GBM stem cells. However, they were also able to show in one GBM cell line that BMPs failed to induce glial differentiation of GBM-initiating cells, causing instead their proliferation and tumorigenesis. GBM-initiating cells did not induce STAT3-dependent glial differentiation in that cell line due to the epigenetic silencing of BMP1RB by an EZH2-dependent mechanism. In their study, they found that a significant number (20%) of GBM tumor samples had low levels of BMP1RB, and the majority of these samples had hypermethylation of the BMP1RB gene promoter. These results provide evidence for epigenetic resistance mechanisms to differentiation therapy and suggest that BMPs in combination therapy with epigenetic modulators may have to be tailored to patients with BMP1RB silencing.
GBM-initiating cell niche
Disruption of GBM-initiating cell maintenance by targeting their vascular niche has shown promise in targeted therapy of these cells [56, 57]. Clinical trials of the anti-VEGF antibody, bevacizumab, and the pan-VEGF receptor tyrosine kinase inhibitor, cediranib (AZD2171), have demonstrated efficacy in GBM patients [88, 89]. The treatment of mice with bevacizumab, after GBM implantation, resulted in a large reduction in the number of GBM-initiating cells and in the growth rate of the tumors [19]. Treatment with bevacizumab did not have much of an effect on the proliferation or survival of most of the cells in the tumors, suggesting that the GBM-initiating cells were targeted. GBM-initiating cells secrete elevated levels of VEGF, which increases endothelial cell migration and tube formation [62]. Treatment of CD133+GBM-initiating cells with bevacizumab blocked their ability to induce endothelial cell migration and tube formation in culture and initiate tumors in vivo. The benefit of anti-angiogenic strategies in combination with conventional cytotoxic drugs has also been shown with rat C6 GBM-initiating cells [61]. Combined, these results suggest that inhibition of blood vessel growth may be an effective method for eliminating GBM-initiating cells.
Perspectives
The discovery of GBM-initiating cells has provided the impetus to rethink our understanding of the growth of glioblastoma and generated interest in developing targeted therapeutics against these cells [19, 20, 23]. Controversy still exists as to whether the relentless growth of GBM is exclusively driven by the TICs [90] or whether all the cells of a tumor contribute to its growth and regeneration following therapy, including the DTCs [91]. There may well be co-evolution of different cell populations in the tumors, which diverge in their genetic alterations and may co-exist and even be co-dependent (Figs. 4, 5, and 6). The picture is further complicated by the recruitment of stromal cells to the tumor, which are known to include vascular cells, immune cells, and even normal neural stem cells [92, 93]. Vascular stroma and TIC interactions have been reported, and other TIC stromal interactions remain to be investigated [19]. Such stroma can contribute growth factors and potentially even be modified by adjacent cancer cells through the transfer of oncogenic proteins through microvesicles or exosomes [94-97]. In sum, we are dealing with a very complex multi-headed target and the heterotypic cell-cell communications between all the tumor elements need to be better understood (Fig. 5). Clearly, the CD133 marker used to define GBM-initiating cells is likely only one of a variety of cell specific markers, and we may have just characterized the tip of the iceberg. If the knowledge acquired with lineage differentiation in blood cancers can serve as a guide, current research needs to develop a nomenclature for GBM-initiating cells and possibly a whole hierarchy of offspring “cancer progenitor” cells (CPG) that is reproducible and consistent. Until a more comprehensive understanding of markers characterizing the different cell populations comprising GBM is acquired, isolation of TICs for study and therapeutic targeting must be performed by harvesting and culturing clonogenic neurospheres with a serum-free culture method utilizing growth factors [43].
Fig. 5.
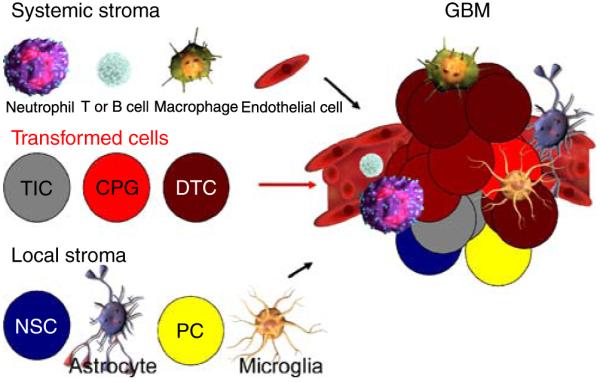
Heterogeneous composition of GBM including vascular niche and other cells. The cellular complexity of the tumor is shown, highlighting the fact that any therapy will have to take into consideration all cellular elements of this complex tissue, which likely functions as a self-sustaining and expanding cancer organ growing as a “parasite” of the normal host. Systemic stromal cells (neutrophils, T or B cells, macrophages, and endothelial cells) and local stromal cells (NSCs, astrocytes, PCs, and microglia) are shown to potentially interact with the transformed cells (TICs, CPGs, and DTCs) to form the GBM tumor. The vascular niche provided by endothelial cells is a well-established component to the maintenance and survival of TICs and GBM tumors. It will be the key to determine which stromal cell populations (systemic or local) contribute to tumor initiation and progression and what extracellular signals regulate their recruitment to the tumor. Antagonizing such signals, possibly in the systemic blood circulation, may be a novel approach to therapy. NSC neural stem cell, PC progenitor cell, TIC tumor-initiating cell, CPG cancer progenitor cell, DTC differentiated tumor cell
Fig. 6.
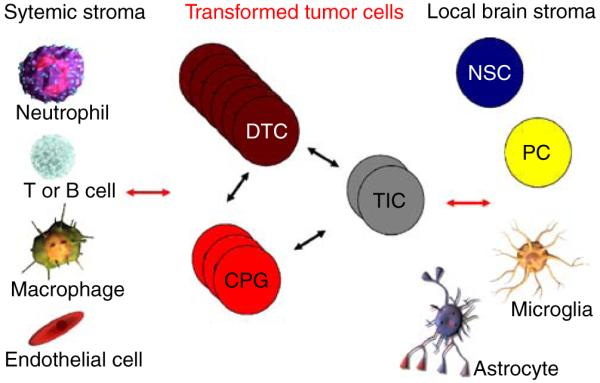
Hypothetical interactions between tumor cells and stromal cells. The complexity of heterotypic cell-cell communication in GBM needs extensive study. Are critical signals present which sustain and help the growth of the different tumor cell populations (TIC, CPG, and DTC)? How do these interact with stromal cells, either from the local brain microenvironment or recruited systemically, and are such signals essential for tumor maintenance, progression, and recurrence? Is there transfer of oncogenic proteins from brain cancer cells to normal stroma through secreted vesicles, e.g., oncosomes? Are there tumor suppressor effects from normal stroma onto cancer cells that may be amplified for therapy? TIC tumor-initiating cell, CPG cancer progenitor cell, DTC differentiated tumor cell, NSC neural stem cell, PC progenitor cell
The clonal evolution model for tumorigenesis predicts that the genetic alterations in GBM-initiating cells will be all present in the more differentiated tumor cell populations that comprise the bulk of tumors [41]. It also predicts that the cancer cells undergoing terminal differentiation may have acquired more alterations than their precursors (Fig. 4). As discussed above, the initial investigations comparing the biological properties of TICs with the cells derived from the bulk of the tumor have already identified clear differences [20, 21, 43, 44]. These findings highlight the need to reexamine all the biological tenets that we use as current foundation upon which to guide new therapy development. It also brings forth the pressing need to develop new technology to track these cells in patients, as their fate in response to therapy might become a new guiding principle for judging the efficacy of clinical trials (Fig. 4).
Imaging of TICs will be very important in the targeted therapy of these cells in glioblastomas. The ability to monitor the eradication of these cells by magnetic resonance imaging or positron emission tomography imaging will permit a better understanding of the relationship between TICs and tumor response to therapy. Emerging nanotechnologies may permit simultaneous imaging of TICs as well as targeted therapy with conjugated antitumor agents and/or the ability to generate local hyperthermia due to their magnetic properties [98-101].
Immunotherapy may be possible with vaccine peptides that can elicit a natural immune response to TICs [102]. Surface characterization of GBM-initiating cells needs to be further determined to permit the development of peptides derived from TICs that are specific and able to generate an immune response. Use of dendritic cells pulsed with GBM-initiating cells may form the basis of selective immunotherapy against these cells [103]. Such novel approaches will need to carefully examine the cost-benefit ratio of eliminating TIC and possibly normal stem cells in the brain as they are likely to overlap in marker expression.
As a further example, oncolytic therapy agents in the form of viruses [herpes simplex virus 1 (HSV-1), adenoviruses, etc.] may be designed to selectively infect and replicate within GBM-initiating cells [104, 105]. Selective uptake of these viruses by TICs will need to be based on viral constructs that can express a ligand specific to TICs.
In summary, the “cancer stem cell” hypothesis has invigorated the neuro-oncology field with a breath of fresh thinking that may end up shaking the foundation of old dogmas, such as the widely held belief that GBM tumors are incurable because of infiltrative disease. If the infiltrated cells are in fact DTCs, their dissemination beyond the surgical boundary may not be the primary cause of tumor recurrence. It is also important to keep in mind that these emerging concepts are still rapidly evolving and questioned. Recent findings ponder the rarity of CSC and suggest that the so-called CSC may simply represent a fraction of tumor cells that can evade the immune system in mice models that are only partially immunocompromised such as the nu/nu and NOD/SCID strains [91]. Further work on all biological fronts will consolidate or deflate the pillars on which we build our current therapies. This is an exciting time for new discoveries and provides new hope for patients who rely on all of us to find a cure for this devastating disease.
Acknowledgments
We wish to thank Dr. Daniel Brat who kindly provided the microscopic sections of the brain and the immunohistochemistry staining present in Fig. 1.
Financial support This work was supported in part by grants from the NIH (CA86335, CA116804 to EGVM, NS053454 to CGH), American Brain Tumor Association (to CGH), Brain Tumor Foundation for Children (to EGVM), Southeastern Brain Tumor Foundation (to CGN and EGVM), Goldhirsh Foundation (to EGVM), and the Georgia Cancer Coalition, Distinguished Cancer Clinicians and Scientists Program (to CGH).
Contributor Information
Costas G. Hadjipanayis, Laboratory of Molecular Neuro-Oncology, Departments of Neurosurgery, Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA 33022, USA, e-mail: chadjip@emory.edu
Erwin G. Van Meir, Laboratory of Molecular Neuro-Oncology, Departments of Neurosurgery, Hematology and Oncology, Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA 30322, USA
References
- 1.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 2.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 3.Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14:43–47. doi: 10.1016/j.gde.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. discussion 1895-1886. [DOI] [PubMed] [Google Scholar]
- 5.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 6.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. ProcNatl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy JA, Barabe F, Poeppl AG, Wang JC, Dick JE. Comment on “Tumor growth need not be driven by rare cancer stem cells”. Science. 2007;318:1722. doi: 10.1126/science.1149590. author reply 1722. [DOI] [PubMed] [Google Scholar]
- 9.Reilly KM, Rubin JB, Gilbertson RJ, Garbow JR, Roussel MF, Gutmann DH. Rethinking brain tumors: The fourth mouse models of human cancers consortium nervous system tumors workshop. Cancer Res. 2008;68:5508–5511. doi: 10.1158/0008-5472.CAN-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 11.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, DeVitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 12.Shmelkov SV, St Clair R, Lyden D, Rafii S. AC133/CD133/Prominin-1. Int J Biochem Cell Biol. 2005;37:715–719. doi: 10.1016/j.biocel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamaki S, Eckert K, He D, Sutton R, Doshe M, Jain G, Tushinski R, Reitsma M, Harris B, Tsukamoto A, Gage F, Weissman I, Uchida N. Engraftment of sorted/expanded human central nervous system stem cells from fetal brain. J Neurosci Res. 2002;69:976–986. doi: 10.1002/jnr.10412. [DOI] [PubMed] [Google Scholar]
- 15.Legler JM, Ries LA, Smith MA, Warren JL, Heineman EF, Kaplan RS, Linet MS. Cancer surveillance series [corrected]: brain and other central nervous system cancers: recent trends in incidence and mortality. J Natl Cancer Inst. 1999;91:1382–1390. doi: 10.1093/jnci/91.16.1382. [DOI] [PubMed] [Google Scholar]
- 16.Van Meir EG, Polverini PJ, Chazin VR, Su Huang HJ, de Tribolet N, Cavenee WK. Release of an inhibitor of angiogenesis upon induction of wild type p53 expression in glioblastoma cells. Nat Genet. 1994;8:171–176. doi: 10.1038/ng1094-171. [DOI] [PubMed] [Google Scholar]
- 17.Van Meir EG, Bellail A, Phuphanich S. Emerging molecular therapies for brain tumors. Semin Oncol. 2004;31:38–46. doi: 10.1053/j.seminoncol.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7:134–153. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 20.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 21.Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980–8984. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
- 22.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 23.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin harmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 27.Gritti A, Frolichsthal-Schoeller P, Galli R, Parati EA, Cova L, Pagano SF, Bjornson CR, Vescovi AL. Epidermal and fibroblast growth factors behave as mitogenic regulators for a single multipotent stem cell-like population from the subventricular region of the adult mouse forebrain. J Neurosci. 1999;19:3287–3297. doi: 10.1523/JNEUROSCI.19-09-03287.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vescovi AL, Parati EA, Gritti A, Poulin P, Ferrario M, Wanke E, Frolichsthal-Schoeller P, Cova L, Arcellana-Panlilio M, Colombo A, Galli R. Isolation and cloning of multipotential stem cells from the embryonic human CNS and establishment of transplantable human neural stem cell lines by epigenetic stimulation. Exp Neurol. 1999;156:71–83. doi: 10.1006/exnr.1998.6998. [DOI] [PubMed] [Google Scholar]
- 29.Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- 30.Gritti A, Parati EA, Cova L, Frolichsthal P, Galli R, Wanke E, Faravelli L, Morassutti DJ, Roisen F, Nickel DD, Vescovi AL. Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. J Neurosci. 1996;16:1091–1100. doi: 10.1523/JNEUROSCI.16-03-01091.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 32.Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, Lawton MT, McDermott MW, Parsa AT, Manuel-Garcia Verdugo J, Berger MS, Alvarez-Buylla A. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. 2004;427:740–744. doi: 10.1038/nature02301. [DOI] [PubMed] [Google Scholar]
- 33.Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G, 2nd, Jiang L, Kang J, Nedergaard M, Goldman SA. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- 34.Ayuso-Sacido A, Roy NS, Schwartz TH, Greenfield JP, Boockvar JA. Long-term expansion of adult human brain subventricular zone precursors. Neurosurgery. 2008;62:223–229. doi: 10.1227/01.NEU.0000311081.50648.4C. discussion 229-231. [DOI] [PubMed] [Google Scholar]
- 35.Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- 36.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 37.Rao MS, Noble M, Mayer-Proschel M. A tripotential glial precursor cell is present in the developing spinal cord. Proc Natl Acad Sci USA. 1998;95:3996–4001. doi: 10.1073/pnas.95.7.3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayer-Proschel M, Kalyani AJ, Mujtaba T, Rao MS. Isolation of lineage-restricted neuronal precursors from multipotent neuroepithelial stem cells. Neuron. 1997;19:773–785. doi: 10.1016/s0896-6273(00)80960-5. [DOI] [PubMed] [Google Scholar]
- 39.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 40.Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- 41.Fulci G, Ishii N, Maurici D, Gernert KM, Hainaut P, Kaur B, Van Meir EG. Initiation of human astrocytoma by clonal evolution of cells with progressive loss of p53 functions in a patient with a 283H TP53 germ-line mutation: evidence for a precursor lesion. Cancer Res. 2002;62:2897–2905. [PubMed] [Google Scholar]
- 42.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 43.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 44.Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, Kim M, Totonchy M, Cusack T, Ene C, Ma H, Su Q, Zenklusen JC, Zhang W, Maric D, Fine HA. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 46.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 47.Sakariassen PO, Prestegarden L, Wang J, Skaftnesmo KO, Mahesparan R, Molthoff C, Sminia P, Sundlisaeter E, Misra A, Tysnes BB, Chekenya M, Peters H, Lende G, Kalland KH, Oyan AM, Petersen K, Jonassen I, van der Kogel A, Feuerstein BG, Terzis AJ, Bjerkvig R, Enger PO. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc Natl Acad Sci USA. 2006;103:16466–16471. doi: 10.1073/pnas.0607668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng X, Shen G, Yang X, Liu W. Most C6 cells are cancer stem cells: evidence from clonal and population analyses. Cancer Res. 2007;67:3691–3697. doi: 10.1158/0008-5472.CAN-06-3912. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Sakariassen PO, Tsinkalovsky O, Immervoll H, Boe SO, Svendsen A, Prestegarden L, Rosland G, Thorsen F, Stuhr L, Molven A, Bjerkvig R, Enger PO. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer. 2008;122:761–768. doi: 10.1002/ijc.23130. [DOI] [PubMed] [Google Scholar]
- 50.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2-cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–6219. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 51.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci USA. 2004;101:781–786. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3:1337–1345. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- 53.Harris MA, Yang H, Low BE, Mukherje J, Guha A, Bronson RT, Shultz LD, Israel MA, Yun K. Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res. 2008;68:10051–10059. doi: 10.1158/0008-5472.CAN-08-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramirez-Castillejo C, Sanchez-Sanchez F, Andreu-Agullo C, Ferron SR, Aroca-Aguilar JD, Sanchez P, Mira H, Escribano J, Farinas I. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat Neurosci. 2006;9:331–339. doi: 10.1038/nn1657. [DOI] [PubMed] [Google Scholar]
- 55.Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 56.Yang ZJ, Wechsler-Reya RJ. Hit ’em where they live: targeting the cancer stem cell niche. Cancer Cell. 2007;11:3–5. doi: 10.1016/j.ccr.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 57.Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 58.Brat DJ, Van Meir EG. Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: a new world of angiogenesis research. Am J Pathol. 2001;158:789–796. doi: 10.1016/S0002-9440(10)64025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20:7377–7383. doi: 10.1523/JNEUROSCI.20-19-07377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67:3560–3564. doi: 10.1158/0008-5472.CAN-06-4238. [DOI] [PubMed] [Google Scholar]
- 62.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 63.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 64.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 65.Ishii N, Tada M, Hamou MF, Janzer RC, Meagher-Villemure K, Wiestler OD, Tribolet N, Van Meir EG. Cells with TP53 mutations in low grade astrocytic tumors evolve clonally to malignancy and are an unfavorable prognostic factor. Oncogene. 1999;8:5870–5878. doi: 10.1038/sj.onc.1203241. [DOI] [PubMed] [Google Scholar]
- 66.Dietrich J, Imitola J, Kesari S. Mechanisms of Disease: the role of stem cells in the biology and treatment of gliomas. Nat Clin Pract Oncol. 2008;5:393–404. doi: 10.1038/ncponc1132. [DOI] [PubMed] [Google Scholar]
- 67.Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, Anderson DJ, Stiles CD, Rowitch DH. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 69.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 70.Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 71.Barnett SC, Robertson L, Graham D, Allan D, Rampling R. Oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells transformed with c-myc and H-ras form high-grade glioma after stereotactic injection into the rat brain. Carcinogenesis. 1998;19:1529–1537. doi: 10.1093/carcin/19.9.1529. [DOI] [PubMed] [Google Scholar]
- 72.Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, Rowitch DH, Louis DN, DePinho RA. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 73.Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, Pieper RO. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61:4956–4960. [PubMed] [Google Scholar]
- 74.Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS, Pieper RO. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of lioma. Cancer Res. 2001;61:6674–6678. [PubMed] [Google Scholar]
- 75.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicty. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, Alvarez-Buylla A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–199. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 77.Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozoloide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 79.Ehtesham M, Mapara KY, Stevenson CB, Thompson RC. CXCR4 mediates the proliferation of glioblastoma progenitor cells. Cancer Lett. 2009;274:305–312. doi: 10.1016/j.canlet.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Canoll P, Goldman JE. The interface between glial progenitors and gliomas. Acta Neuropathol. 2008;116:465–477. doi: 10.1007/s00401-008-0432-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gussoni E, Soneoka Y, Strickland CD, Buzney EA, Khan MK, Flint AF, Kunkel LM, Mulligan RC. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 1999;401:390–394. doi: 10.1038/43919. [DOI] [PubMed] [Google Scholar]
- 82.Jackson KA, Mi T, Goodell MA. Hematopoietic potential of stem cells isolated from murine skeletal muscle. Proc Natl Acad Sci USA. 1999;96:14482–14486. doi: 10.1073/pnas.96.25.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dietrich J, Han R, Yang Y, Mayer-Proschel M, Noble M. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J Biol. 2006;5:22. doi: 10.1186/jbiol50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Panchision DM, McKay RD. The control of neural stem cells by morphogenic signals. Curr Opin Genet Dev. 2002;12:478–487. doi: 10.1016/s0959-437x(02)00329-5. [DOI] [PubMed] [Google Scholar]
- 85.Nakano I, Saigusa K, Kornblum HI. BMPing off glioma stem cells. Cancer Cell. 2008;13:3–4. doi: 10.1016/j.ccr.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 86.Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713–726. doi: 10.1016/s0896-6273(00)00148-3. [DOI] [PubMed] [Google Scholar]
- 87.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 88.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Wagner M, Bigner DD, Friedman AH, Friedman HS. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 89.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY, Jain RK. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblasoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Adams JM, Strasser A. Is Tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 2008;68:4018–4021. doi: 10.1158/0008-5472.CAN-07-6334. [DOI] [PubMed] [Google Scholar]
- 91.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jurvansuu J, Zhao Y, Leung DSY, Boulaire J, Yu YH, Ahmed S, Wang S. Transmembrane protein 18 enhances the tropism of neural stem cells for glioma cells. Cancer Res. 2008;68:4614–4622. doi: 10.1158/0008-5472.CAN-07-5291. [DOI] [PubMed] [Google Scholar]
- 94.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr., Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, Breakefield XO, Snyder EY. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D, Galli R, Selleri S, Di Meco F, De Fraja C, Vescovi A, Cattaneo E, Finocchiaro G. Gene therapy of experimental brain tumors using neural progenitor cells. Nat Med. 2000;6:447–450. doi: 10.1038/74710. [DOI] [PubMed] [Google Scholar]
- 97.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell iol. 2008;10:619–624. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 98.Hadjipanayis CG, Bonder MJ, Balakrishnan S, Wang X, Mao H, Hadjipanayis GC. Metallic iron nanoparticles for MRI contrast enhancement and local hyperthermia. Small. 2008;4:1925–1929. doi: 10.1002/smll.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Veiseh O, Sun C, Gunn J, Kohler N, Gabikian P, Lee D, Bhattarai N, Ellenbogen R, Sze R, Hallahan A, Olson J, Zhang M. Optical and MRI multifunctional nanoprobe for targeting gliomas. Nano Lett. 2005;5:1003–1008. doi: 10.1021/nl0502569. [DOI] [PubMed] [Google Scholar]
- 100.Maier-Hauff K, Rothe R, Scholz R, Gneveckow U, Wust P, Thiesen B, Feussner A, von Deimling A, Waldoefner N, Felix R, Jordan A. Intracranial thermotherapy using magnetic nanoparticles combined with external beam radiotherapy: results of a feasibility study on patients with glioblastoma multiforme. J Neurooncol. 2007;81:53–60. doi: 10.1007/s11060-006-9195-0. [DOI] [PubMed] [Google Scholar]
- 101.Jordan A, Scholz R, Maier-Hauff K, van Landeghem FK, Waldoefner N, Teichgraeber U, Pinkernelle J, Bruhn H, Neumann F, Thiesen B, von Deimling A, Felix R. The effect of thermotherapy using magnetic nanoparticles on rat malignant glioma. J Neurooncol. 2006;78:7–14. doi: 10.1007/s11060-005-9059-z. [DOI] [PubMed] [Google Scholar]
- 102.Wu A, Wiesner S, Xiao J, Ericson K, Chen W, Hall WA, Low WC, Ohlfest JR. Expression of MHC I and NK ligands on human CD133+glioma cells: possible targets of immunotherapy. J Neurooncol. 2007;83:121–131. doi: 10.1007/s11060-006-9265-3. [DOI] [PubMed] [Google Scholar]
- 103.Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, Caldera V, Nava S, Ravanini M, Facchetti F, Bruzzone MG, Finocchiaro G. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247–10252. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 104.Skog J, Edlund K, Bergenheim AT, Wadell G. Adenoviruses 16 and CV23 efficiently transduce human low-passage brain tumor and cancer stem cells. Mol Ther. 2007;15:2140–2145. doi: 10.1038/sj.mt.6300315. [DOI] [PubMed] [Google Scholar]
- 105.Nandi S, Ulasov IV, Tyler MA, Sugihara AQ, Molinero LYH, Zhu ZB, Lesniak MS. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008;68:5778–5784. doi: 10.1158/0008-5472.CAN-07-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]