

Abstract
The anti-inflammatory effects of the neuronal nicotinic receptor alpha7 (nAChRα7) are proposed to require acetylcholine release from vagal efferents. The necessity for vagal innervation in this anti-inflammatory pathway was tested in the skin, which lacks parasympathetic innervation, using ultraviolet radiation (UVB) to induce a local pro-inflammatory response. Cytokine responses to UV in mice administered chronic oral nicotine, a nAChR agonist, were reduced. Conversely, nAChRα7 knock-out mice exposed to UVB elicit an enhanced pro-inflammatory cytokine response in the skin. Altered pro-inflammatory responses correlated with changes in SOCS3 protein. These results demonstrate that nAChRα7 can participate in modulating a local pro-inflammatory response in the absence of parasympathetic innervation.
Keywords: Neuronal Nicotinic, Inflammation, Ultraviolet Radiation, Cytokine, SOCS3
Introduction
Neuronal nicotinic acetylcholine receptors (nAChR) have been extensively characterized for their function in peripheral and central neurons where they play a role in modulating neurotransmission (Alkondon and Albuquerque, 2004; Hogg et al., 2003). However, nAChRs are also expressed by non-neuronal cells throughout the body where they contribute to diverse physiological processes including immunomodulation (Fujii et al., 2007). Mature nAChRs are pentamers of subunit assemblages from 11 genetically related but distinct subunits (Hogg et al., 2003). In general there are two major nAChRs subtypes that are composed of either homomeric subunits (e.g., nAChRα7) or combinations of alpha (α) and beta (β) subunits, and it is the final subunit configuration that imparts significant functional and pharmacological differences among these receptors. The response of nAChRs to nicotine has been particularly well-examined since it initiates the chemical dependency to tobacco products. However, nicotine also exhibits anti-inflammatory properties in many systems (de Jonge et al., 2005; Metz and Tracey, 2005; Pavlov et al., 2003; Sopori et al., 1998). Among the earliest findings was the observation that nAChRα7 activation altered the capacity of cells to respond to the pro-inflammatory cytokine tumor necrosis factor alpha (TNFα) (Carlson et al., 1998) or inhibited the release of this cytokine from the cell (Metz and Tracey, 2005). These studies have collectively defined an interaction described as the cholinergic anti-inflammatory pathway (Metz and Tracey, 2005; Pavlov et al., 2003). As defined in these reviews, however, the anti-inflammatory properties of acetylcholine (or nicotine) are generally restricted to nAChRα7 function requiring vagal nerve innervation as the source for acetylcholine (Metz and Tracey, 2005).
Recent evidence indicates that nAChRα7 (as well as other subtypes) are also expressed by numerous non-neuronal cell types including distinct populations of astrocytes, epithelial cells, adipocytes, lymphocytes, macrophages and keratinocytes (Conti-Tronconi et al., 1994; Gahring and Rogers, 2005; Kurzen et al., 2007; Misery, 2004). In this context, nAChRα7, in addition to its role in depolarization, exhibits remarkably high calcium permeability that is sufficient to impact on intracellular signaling pathways including those using ERK and CREB (Brunzell et al., 2003). In this report, we have investigated the role of this nAChR in regulating an inflammatory response to ultraviolet radiation (UVR) induced in mouse skin. The skin is not innervated by the vagus nerve (or parasympathetic systems), but there is prominent expression of nAChRs including nAChRα7, and there is local production of acetylcholine and choline (Kurzen et al., 2007; Misery, 2004). Further, UVR exposure is a common and well-characterized method of inducing inflammation in the skin, and we have employed this method for many years to study this effect (Daynes et al., 1986; Gahring et al., 1984). Keratinocyte expression of nAChR has been demonstrated to have important roles in keratinocyte adhesion and motility (Chernyavsky et al., 2004; Grando et al., 1995; Zia et al., 1997), differentiation (Arredondo et al., 2002; Grando et al., 1996), apoptosis (Arredondo et al., 2003; Nguyen et al., 2001), and certain components of inflammation (Kalayciyan et al., 2007; Kurzen et al., 2007).
Our studies examine the influence of nAChRα7 and nicotine on local cytokine production induced by UVR in the UVB range (280 to 320 nm). Results indicate that nicotine administration corresponds with an attenuated inflammatory response by skin to UVB irradiation. Because the pharmacologic effects of nicotine are complex and include both activation of nAChR as well as desensitization of the receptors, including nAChRα7 (Alkondon and Albuquerque, 2004; Gahring and Rogers, 2005), the effects of nAChRα7 on inflammation in the skin were examined directly using knock-out (KO, α7 null mice) mice. When these mice were exposed to UVB the cytokines most altered by the absence of nAChRα7 are IL-1β and IL-6, but not TNFα. Evidence is also presented that the intracellular regulator of cytokine signaling, suppressor of cytokine signaling 3 (SOCS3), is a possible mediator of nAChR effects on inflammation in the skin. Collectively, our results suggest that the impact of nicotine and nAChRs on the inflammatory system may vary considerably among different tissues and the role of α7 in this process is likely to be multifaceted and tissue specific.
Materials and Methods
Animals
All mice were housed in a pathogen free environment with water and standard mouse chow provided ad libitum. Each experiment used groups of 3-7 mice that were age, gender and strain matched. For the ultraviolet radiation dose response and oral nicotine treatment experimental groups, 6-8 week old male or female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, Maine). The nAChRα7KO in the C57BL/6 background were initially purchased from Jackson Laboratories and subsequently maintained as a breeding colony in our animal facility. The α7KO heterozygous mice are bred to produce WT and homozygotes KO mice.
Chronic exposure to oral nicotine
For experiments testing the effects of chronic nicotine exposure, mice were provided water supplemented with nicotine. We and others (Gahring et al., 2004; Matta et al., 2007) have used this method of administration for experiments designed to examine the long-term effects of nicotine exposure via a natural route of drug administration, and the merits of using oral nicotine exposure and standards for using this method have been reviewed extensively (Matta et al., 2007). Mice are initially provided water containing 25 μg/ml of nicotine and 1.5% saccharin. The saccharin is added to mask any adverse flavor of nicotine. The amount of nicotine in the water is increased to 50 μg/ml on day 3, 100 μg/ml on day 5, and to the final concentration of 150 μg/ml on day 7. Mice are maintained on this concentration of nicotine for 6 weeks. Control mice are maintained on 1.5% saccharin in the drinking water for the duration of the experiment.
Exposure to ultraviolet radiation
Ultraviolet (UV) radiation in the UVB range (280-320 nm) was from Westinghouse FS-40 fluorescent sunlamps (Daynes et al., 1986; Gahring et al., 1984; Gahring and Daynes, 1986). The mW/cm2 output of the lamps was measured for each experiment with an IL1350 photometer (International Light Inc., Newburyport MA). All mice (control and UVB exposed) were shaved of back hair with electric clippers one day prior to UVB exposure. On the day of exposure, mice were anesthetized by intraperitoneal injection of 2,2,2, tribromoethanol (Sigma Aldrich, St. Louis MO) and placed in adsorbent paper wraps with all but a 2 × 3 cm square area of back skin exposed (see below and Figure 1A) to produce an equivalent exposure level within a defined region on each animal. The mice were then placed under a bank of three Westinghouse FS-40 bulbs for 30 minutes (4860 J/m2), 20 minutes (3240 J/m2) or 10 minutes (1620 J/m2). Control mice (WT and α7K0) were also anesthetized, and their backs were also shaved of hair, but they were not placed under the sunlamps. At 24, 48, 72, or 96 hrs post UVB exposure, mice were sacrificed and the UVB exposed skin was removed. From the exposed 2 × 3 cm2 patch of skin standardized samples of skin from each exposure site were collected using 6 mm biopsy punches (Miltex Instruments, York, PA). Multiple punch biopsies from each mouse could then be processed for RNA extraction, protein extraction or histology, respectively.
Figure 1.
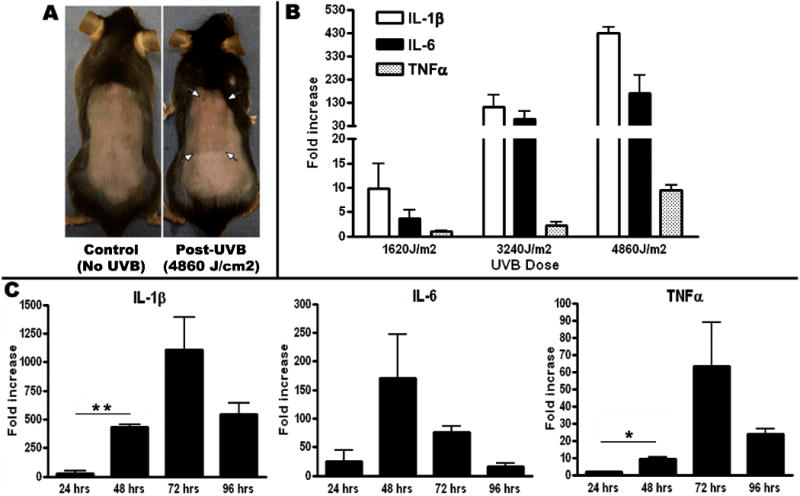
A) For UVB exposure, C57/BL6 mice were shaved and completely covered except for a 2×3 cm2 area on the back. 48 hours following a 30 minute exposure of UVB, the exposed skin showed visible signs of inflammation including thickness and redness. B) Dose response of IL-1β, IL-6 and TNFα mRNA collected from 3 mice per group 48 hrs following 10 minutes (1620J/m2), 20 minutes (3240J/m2) and 30 minutes (4860J/m2) of UVB. Each cytokine shows a dose dependent increase with UVB. IL-1β and TNFα are both significantly (p< 0.001) increased from 1620J/m2 to 4860J/m2. The magnitude of the fold changes in transcripts for IL-1β and IL-6 were much greater than TNFα. C) Time course of IL-1β, IL-6 and TNFα mRNA following 30 minutes (4860J/m2) of UVB exposure. All cytokines had a significant increase or peaked by 48 hrs following UVB. Fold changes represent increases in mRNA over unexposed skin. Error bars represent mean +/- SEM, p<0.01 *; p<0.001 **
Histology
UVB exposed or unexposed skin was placed in formalin overnight at 4°C. The sections were then dehydrated by rocking in increasing concentrations of ethanol and embedded in plastic (Polysciences Immunobed Kit). Skin sections (5 μm) were cut and stained with hematoxylin for 1 hr at 60°C and eosin for 3 minutes at room temperature. Images of the sections were taken using Neuroleucida and a Dage300 digital color camera. Quantification of lymphocytes was done by automated counting of lymphocytes (defined by size and shape) in equivalent areas of 5 sections using ImagePro Plus.
RNA and Real time PCR
To extract RNA, skin biopsies were placed in Trizol (Invitrogen, Carlsbad CA), homogenized with a tissue tearor (Biospec Products, Bartlesville, OK), and processed for RNA according to the manufacturer's instructions. RNA was precipitated in isopropanol and washed in 75% ethanol at room temperature. The concentration and purity of the RNA was determined with a ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington DE). 2 μg of each sample was DNase treated (Promega, Madison WI), and 1μg was used to amplify cDNA (high capacity cDNA Archive Kit, Applied BioSystems, Foster City CA). 20ng of cDNA was loaded into each well and TaqMan probes (Applied BioSystems, Foster City, CA) were used to amplify all genes (IL-1β Mm00434228_m1, IL-6 Mm00446169_m1, TNFα Mm00443258_m1, α2 Mm00460629_m1, α3 Mm00520145_m1, α4 Mm00516561_m1, α5 Mm00616329_m1, α6 Mm00517529_m1, α7 Mm00431639_m1, α9 Mm01221610_m1, α10 (special design), β2 Mm00515323_m1, β3 Mm00532602_m1, β4 Mm00804952_m1 GapD 4352339E). GAPDH was used as an internal control gene in all experiments. All samples were run in duplicate along with a negative RT sample. Fold-changes in RNA expression were calculated relative to the matched control animals, and were determined using the comparative Ct method (see Applied BioSystems for detailed description).
Cytokine Detection by ELISA
A 6mm biopsy punch sample was incubated on ice for 10 minutes in PBS containing the detergent Igepal CA630 (Sigma Aldrich, St. Louis, MO) at 0.1% as described by Matalka et al. (Matalka et al., 2005). Samples were homogenized with a tissue disrupter, incubated again for 10 minutes on ice, and centrifuged at 4°C for 20 minutes at 13000 rpm. The supernatant was then collected and frozen at -80°C or immediately assayed by ELISA for mouse IL-1β, IL-6 and TNFα following the manufacturers' instructions (BioSource/Invitrogen, Carlsbad, CA). All samples were diluted 1:2 to 1:4 for IL-1β and IL-6 or 1:16 for TNFα and run in duplicate. Cytokine concentrations were determined with a standard curve (provided in the ELISA kit) that was run with each ELISA plate and calculated using SOFTmax Pro and Excel.
Western Blots
For Western analysis, 6mm biopsy punches from exposed or unexposed skin were harvested as above for ELISA analysis in 0.1% Igepal detergent in PBS (Matalka et al., 2005). A protein assay was done on the supernatant for each sample and 30 μg of protein was heated in 0.1% Igepal PBS, and SDS-PAGE buffer, and loaded onto SDS-PAGE gels. Gels were transferred to PVDF membrane (Perkin Elmer, Waltham MA) and blocked in 5% nonfat dry milk in PBS plus 0.1% Tween-20. Antibodies for SOCS3 (Abcam, Cambridge, MA) and Actin (Cell Signaling, Danvers, MA) were diluted 1:300 and 1:1000 respectively in 5% nonfat dry milk in PBS- 0.1% Tween-20 and rocked overnight at 4°C. Blots were developed with Chemiluminescence Reagent Plus (Perkin Elmer) and quantitation performed on scans of gel films using ImagePro-Plus quantitation software.
Results
Nicotine Attenuates the Inflammatory Response to UVB
The skin is constantly under assault by environmental as well as infectious agents, and it is a major site of inflammatory cytokine production. We have previously demonstrated that exposure of cultured keratinocytes to ultraviolet radiation (UV) in the UVB (280-320 nm) range results in the production of inflammatory cytokines (Gahring et al., 1984). To establish the parameters capable of inducing inflammatory cytokine expression following in vivo UVB exposure, hair on the dorsal surface of the mice was removed by shaving 24 hrs prior to UVB exposure. Shaving was the preferred method for removing hair since other methods (e.g., depilatory cream products) removed outer epidermal layers and/or produced irritation that interfered with measurements. Just prior to UVB exposure, mice were anesthetized and all but a 2 × 3 cm2 region of their dorsal surface is covered (including head and all extremities). Mice were then placed under the UVB emitting bulbs for the time indicated (10, 20 or 30 minutes corresponding to 1620 J/m2, 3240 J/m2 or 4860 J/m2). Results shown in Figure 1A demonstrate that visible indications of mild inflammation (redness, skin thickening, but no necrosis) were evident at the site of exposure 48 hours post-UVB (Figure 1A). Upon harvesting the irradiated patch of skin, 6 mm biopsies were punched and RNA was extracted from a biopsy. Results in Figure 1B show an increase of RNA for IL-1β, IL-6, and TNFα 48 hours after exposure to as little as 1620 J/m2 of UVB. Exposure to 4860 J/m2 of UVB (30 minutes of exposure), increases cytokine signal to over 400 fold for IL-1β and 100 fold for IL-6. Fold change increases in TNFα transcripts are much less than for IL-1β or IL-6, but protein levels of this cytokine are much higher constitutively. There were no changes in nAChR subunit expression, as measured by transcript analysis, in the skin following UVB exposure at any time (24-96 hrs) in this, or any other experiments (not shown). We next determined the optimal time to measure an increase in cytokine transcripts following the highest dose of UVB (30 minutes, 4860 J/m2). Results shown in Figure 1C demonstrate that each cytokine has a slightly different kinetic response. IL-1β and IL-6 are the prominent cytokines induced by UVB at both 48 and 72 hours. TNFα transcripts are also induced but not to the same extent (10 fold at 48 hours, 50 fold at 72 hours post UVB) as IL-1β or IL-6 (400 fold and 150 fold, respectively, at 48 hours). IL-1β and TNFα both have significant responses at 48 hours which remain elevated at 72 hrs. IL-6 levels were also elevated at 48 and 72 hours post-UVB.
To determine whether systemic nicotine administration affected the local response to UVB-induced inflammation, mice were maintained on nicotine water (containing saccharin, see Methods) or saccharin only water for 6 weeks. This regime of oral nicotine administration has been discussed (Matta et al., 2007), and the effects of this physiologically relevant route of long term administration includes upregulation of high affinity nicotine binding sites and maintenance of neuronal processes during the aging process (Gahring et al., 2004; Rogers et al., 1998). At the end of the 6 week oral nicotine treatment, mice were exposed to 30 minutes of UVB (4860 J/m2, 30 minutes) as described above. Back skin was harvested 24, 48 and 72 hrs later for cytokine RNA and protein. The results of the RNA and protein assays, as determined by ELISA, were essentially the same. Protein results in Figure 2A show that IL-1β production, elicited by UVB exposure, in nicotine treated mice was equivalent to control mice at 24 and 48 hours post-UV irradiation. However, IL-1β protein production in the nicotine treated mice was significantly attenuated at 72 hours post-UVB. In these experiments there was also a trend for the IL-6 response to be attenuated in nicotine treated mice relative to controls, but it failed to achieve statistical significance (not shown). TNFα expression in nicotine treated mice exposed to UVB was equivalent to the response induced in WT mice (not shown).
Figure 2.

Expression of IL-1β protein by ELISA following 30 minutes (4860J/m2) of UVB. A) Protein was collected from skin biopsies from 5 mice at 24, 48, and 72 hours after UVB exposure from mice on oral saccharin or nicotine for 6 weeks. Mice treated with nicotine have a decrease in IL-1β at 72 hrs compared to control mice. B) Expression of nACh receptor subunits in skin determined by real time PCR expressed as a percentage of the internal control gene GAPDH. The backs of WT C57BL/6 mice were shaved and whole back skin was removed for analysis. Transcripts for α2, α6, α9, and β3 subunits were not detected (ND) by real time PCR analysis. Data represents averages of 3-5 mice and was performed on at least 3 separate groups of animals. Subunit expression did not change following exposure to UVB (not shown).
As noted above, nicotine can interact with nAChR expressed on peripheral cells including keratinocytes. To confirm nAChR expression in the skin of mice, real-time (RTi) PCR was performed. For this analysis punch biopsies of back skin (Methods) containing both dermis and epidermis were obtained and RNA extracted. This is shown in Figure 2B. Transcripts for nAChR subunits α3, α4, α5, α7, α10, β2, and β4 were detected before the cut-off level for detection of 37 cycles, but transcripts for α2, α6, α9 or β3 were not detected (ND). Results are expressed as percent of a mouse housekeeping gene, GAPDH (TBP and β-actin have also been used with no difference in results, not shown). These results are in basic agreement with subunit expression in human epidermal and oral keratinocytes and human scalp tissue (Grando, 2006; Grando et al., 1995; Kurzen et al., 2007). We have also examined nAChR subunit expression in separated skin (epidermis separated from dermis using EDTA separation solutions (Sauder et al., 1982)) to determine if the dermis (composed of mostly fibroblasts and collagen) contributes to the nAChR subunit composition. Our results (not shown) suggest that the dermis expresses little if any nAChR subunit transcripts.
We chose to examine the role of nAChRα7 in the response to UVB since this homomeric receptor has been reported to play a key role in regulating inflammatory processes. Since the skin does not receive vagal innervation, examination of the α7KO mice also addresses the question of whether vagal innervation is required for the cholinergic anti-inflammatory pathway. A null mutant mouse lacking α7 was obtained and a small breeding colony established. These mice breed as heterozygotes which allows for generation of both wild-type (WT) and knock-out (KO) mice from the same litters, eliminating any issues of background effects on our results. Groups of 5 WT and age-matched α7KO mice were prepared for UVB exposure as above, and then exposed for 30 minutes to UVB. At various times thereafter, the 2 × 3 cm2 patch of exposed skin was harvested (6 mm skin biopsies were then taken). Cytokine levels in the α7 WT and KO mice were assessed at 24, 48, and 72 hrs post UVB. While differences could be detected at all time points, we determined the 48 hr time point provided the most consistently significant difference between the WT and α7KO mice. As shown in Figure 3A UVB irradiation of the α7KO mouse resulted in significantly more IL-1β and IL-6 transcript production over WT controls as assessed by real-time PCR at 48hrs. Notably, TNFα RNA expression in the α7KO mice, while elevated, was not significantly different from control mice, indicating that modulation of this cytokine following UVB is not related to α7 in this tissue. To determine if the results of RNA expression measurements extended to the protein level, we extracted protein from separate 6 mm biopsies. Cytokine protein was measured by specific ELISA. As shown in Figure 3B, in UV-irradiated α7KO mice, protein levels of IL-1β and IL-6 are also significantly greater than in WT mice at 48 hours post exposure. While message levels of TNFα do increase following UVB exposure (Figure 1C), protein levels are constitutively high and changes in protein not detectable at this 48 hour time point (Figure 3B) in either the WT or the α7KO mouse. The trend for a slight decline (not statistically significant) in TNFα could be attributed to the fact that this cytokine is produced as a precursor protein which is released from the cell upon cell activation. As such, a decline in TNFα protein due to release from the cells at the exposure site is not unexpected.
Figure 3.
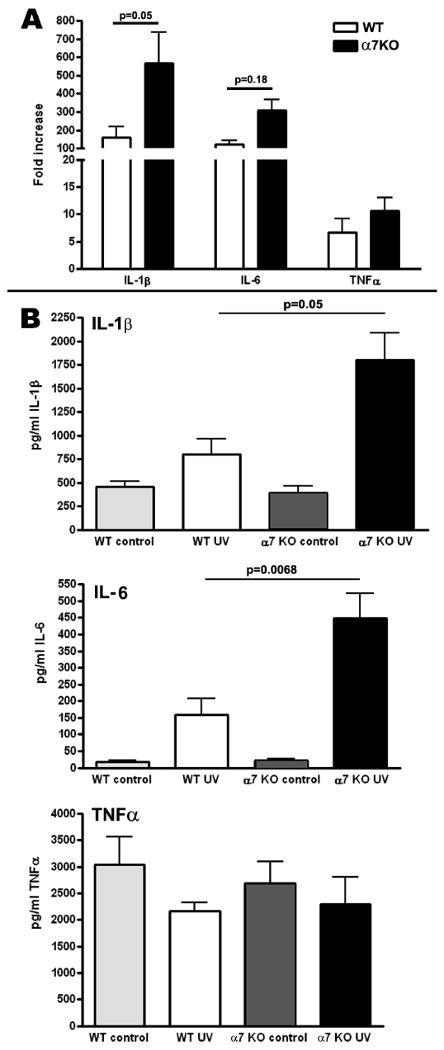
Changes in mRNA determined by real time PCR (A) and protein determined by ELISA (B) in WT and α7 KO mice 48 hrs after 30 minutes of UVB exposure. IL-1β and IL-6 mRNA and proteins levels were both were significantly increased in the α7 KO compared to WT levels. Changes in the already constitutively high levels of TNFα protein were not detectable in the skin of α7 KO mice following UVB. mRNA was collected from 4 mice per group and protein was analyzed from 6-7 mice per group.
Histological Evaluation of the Skin of UVB Exposed Mice
One possible explanation for an increase in cytokines in the UVB exposed skin of the α7KO mouse is that these animals have a greater infiltration by immune cells that in turn produce more cytokines. To assess this possibility, biopsies from UVB exposed (4860 J/m2) C57BL/6 WT and α7KO mice were fixed in formalin, sectioned, and stained with hematoxylin/eosin as described in Methods. Results shown in Figure 4A shows greater thickening/edema in the α7KO mouse compared to the WT mouse. However, when the number of cells per unit area is measured (Figure 4B), the number of cells at 48 hrs post-UVB is not significantly greater in the α7KO mouse.
Figure 4.
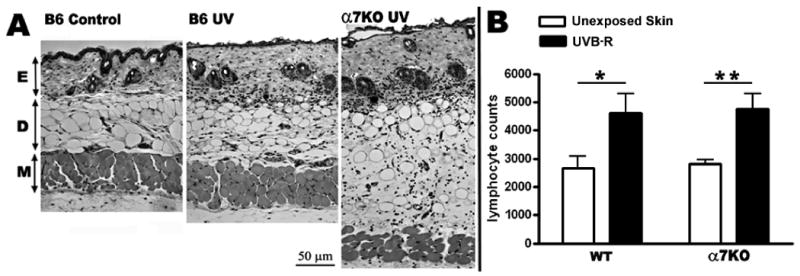
A) H&E stained 5 μm dorsal back skin sections from control (no UV), exposed C57/BL6 WT mice, and exposed α7 KO mice. Mice were exposed to 30 minutes of UVB and harvested 48 hrs later. Sections show epidermis (E), dermis (D) and muscle (M) layers. nAChRα7 KO mice have a visibly thicker dermal layer after UV compared to WT mice. B) Quantification of lymphocytes after 48 hrs in WT and α7 KO mice following 30 minutes of UVB. Lymphocyte numbers were quantitated and averaged from 5 equivalent H&E stained skin sections for each experimental group. The number of lymphocytes quantitiated is significantly increased (p<0.01) in α7 KO following UVB, however the lymphocytes quantitated are approximately equal in both α7 KO and WT mice after UV exposure.
Increased cytokine production by α7KO mice involves SOCS3
The effect of nicotine (attenuated inflammation) versus the effect of the null mutation for α7 (enhanced inflammation) on cytokine production were opposite which indicates that at least part of the attenuated response to nicotine is due to the anti-inflammatory properties of α7. One signaling molecule involved in α7 modulation of inflammatory responsiveness in tissue innervated by the vagal system is the suppressor of cytokine synthesis 3 (SOCS3) protein (Metz and Tracey, 2005; Pavlov et al., 2003). To determine if SOCS3 is involved in the α7KO response in the skin to UVB, we measured SOCS3 protein expression in the skin using Western blot. Results obtained from biopsies pooled from 3 UVB exposed, or unexposed mice, 24 hours following exposure are shown in Figure 5A. SOCS3 expression was readily detected in unexposed skin, and 24 hours following UVB exposure there is little change (perhaps a small decrease, Figure 5A) in this expression. Since SOCS3 expression functions to dampen inflammatory cytokine expression (particularly IL-6 and IL-1β), a decrease in SOCS3 expression would be expected to result in elevated cytokine production. In the α7KO mice, SOCS3 expression was dramatically reduced 24 hours following UVB exposure. Maximal decrease in SOCS3 in the α7KO mouse is observed at 24 hours, as shown, however, decreased expression is also evident at 48 hours post UVB (not shown). We also measured SOCS3 protein in the skin of nicotine treated mice exposed to UVB (Figure 5B). SOCS3 expression in the control mice (saccharin only) is decreased at 24 and 48 hours following UVB and returns to approximately normal levels at 72 hours. The expression of SOCS3 in nicotine treated mice was elevated at 24 hours and decreases at 48 hours followed by increasing levels. These results have been obtained in two completely different sets of nicotine treated (6 weeks) mice. Results suggest that the control of UV induced inflammation in nicotine treated mice and mice lacking α7 may involve SOCS3, resulting in altered inflammation.
Figure 5.
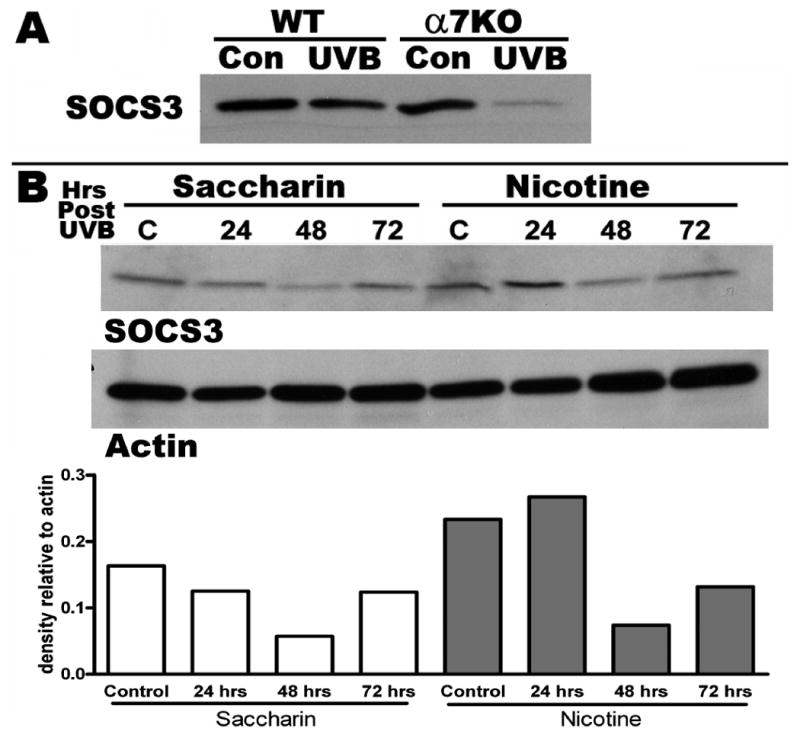
SOCS3 western blots from exposed back skin of 3-5 mice. A) SOCS3 protein expression in WT and α7 KO back skin 24 hrs after a 30 minutes UVB exposure. nAChRα7 KO mice demonstrate a much more dramatic decrease in SOCS3 protein, compared to WT littermates, at 24 hours post-UVB. B) SOCS3 expression in mice treated with oral nicotine or saccharin for 6 weeks. Nicotine treated mice express more SOCS3 at 24 and 48 hrs following UVB as compared to saccharin treated mice. The densitometric scan of the SOCS3 bands are normalized to beta-actin and results are expressed as density relative to actin.
Discussion
It has been reported (Metz and Tracey, 2005; Pavlov et al., 2003) that in tissues innervated by the vagus nerve such as the spleen, efferent processes from this nerve release acetylcholine which can activate nAChR, specifically the homomeric nAChRα7 which is present on cells such as macrophages to inhibit the production/release of inflammatory cytokines. The predominant cytokine reported to be affected in this anti-inflammatory pathway is TNFα. This effect is abrogated following vagotomy suggesting that acetylcholine is provided by the vagus nerve. In this report we questioned the effect of nicotine, as well as well as the α7 knock-out, on an inflammatory response induced in a tissue without vagal efferents. The inflammatory stimulus chosen was ultraviolet radiation since it is well known that exposure of skin to sunlight in the UVB range induces erythema, edema, heat and pain (‘sunburn’). This classical inflammatory response is also characterized by the elevated production of inflammatory cytokines (including IL-1β, IL-6 and TNFα) and chemokines at the site of exposure.
We first determined the effects of nicotine on inflammation induced by UVB and found that chronic oral administration of nicotine attenuates the production of the pro-inflammatory cytokine IL-1β demonstrating that oral administration of nicotine alters inflammation in the skin. IL-6 levels were also decreased in nicotine treated mice, but not to the level of significance, and no effect was observed in TNFα expression at the time points tested. Transcripts for many of the nAChR subunits are present in the skin including α3, α4, α7, β2 and β4. Therefore, nicotine binding receptors composed of α3 or α4 (which can combine with β2 or β4, or both) as well as α7 are all possible in this tissue. However, nAChRα7 has been the nAChR most associated with the anti-inflammatory properties of nicotine (Metz and Tracey, 2005; Pavlov et al., 2003). Our results demonstrate that the lack of α7 results in elevated production of the inflammatory cytokines IL-6 and IL-1β following UVB irradiation of the skin. TNFα protein levels were constitutively high and changes in this cytokine were not observed at this time point. Changes in cytokine levels appear to be initiated by local rather than recruited cells since cytokine expression precedes infiltration by mononuclear cells, and mononuclear cell infiltrate per unit area remains equivalent between WT and α7KO mice following UV exposure. Therefore, the pro-inflammatory response in the skin to UVB, like the spleen in animals treated with LPS (Huston et al., 2006), is increased in the absence of α7, however the influence on this response is not dependent upon vagal innervation and the prominent cytokine affected is IL-1β. It should be noted that experiments which used C57BL/6 mice (time course in Figure 1 and oral nicotine treatment experiment Figure 2) there was a larger IL-1β response compared to experiments using WT α7 mice. This may be attributed to the C57BL/6 129 background that the α7 mice are on (both WT and KO).
Our results raise several issues pertaining to nAChRα7 modulation of the inflammatory response. For example, in the absence of parasympathetic (vagal) innervation, what is the source of the normal ligand for α7 and how is this regulated? Recent studies (Kurzen et al., 2007) have demonstrated that keratinocytes are a significant source of acetylcholine production and release. Further, acetylcholine receptors expressed by keratinocytes are subjected to regulation through autocrine/paracrine mechanisms including those important to normal processes of differentiation and squamatization (Arredondo et al., 2005). That nicotine can influence keratinocytes and other epithelial cells is well documented. In patients with oral ulcerations such as Behcets disease the use of nicotine appears to be anti-inflammatory since patients with this disease have less oral apthae during periods of smoking (Kalayciyan et al., 2007; Soy et al., 2000). Of note is that the skin is not the only tissue where ulcerative disease is associated with the overproduction of inflammatory cytokines such as TNFα, and whose progression is altered by nicotine. Examples include inflammatory bowel diseases of the colon. In particular, ulcerative colitis is sensitive to nicotine which when administered to either humans or to animal models can reduce inflammation and for certain forms of the disease possibly provide therapeutic relief from this autoimmune inflammatory disease (Thomas et al., 2005). While this organ is innervated by the vagal nerve, nicotine acting directly upon the inflammatory process through non-neuronal mechanisms has not been experimentally ruled-out. Further, recent animal models (Orr-Urtreger et al., 2005) and studies of humans (Richardson et al., 2003) suggest a role for non-α7 nAChRs in this process. As such, the possible effect of nicotine or analogs of this compound in certain inflammatory diseases are likely to be diverse, of varied magnitude and not necessarily straight-forward.
One important component of this study is the finding of a corresponding influence of α7 on regulatory mechanisms important to suppressing the production of specific inflammatory cytokines. One pathway indicated as important to this signaling mechanism in peripheral systems has been the NFκB signal transduction pathways, although neurons appear to use alternate pathways to impart nAChR-related signaling (Gahring et al., 2003) including CREB and ERK-mediated signaling pathways (Brunzell et al., 2003). In this general context, we found no alteration in the expression of the NFκB-related molecule, Iκb (phosphorylated or non-phosphorylated) at the time points tested (not shown). While we have not examined the additional adaptor proteins important to this pathway in detail, at present we have not found a major role for the regulation inflammation in the skin by nicotine through the NFκB pathway. Instead, modification of the inflammatory response to UVB appears to correlate with changes in the expression of the inhibitor of cytokine signaling, SOCS3 (Croker et al., 2003). This is also consistent with the reports of others who have identified an influence of nicotine on this pathway (Wang and Campbell, 2002). In this study we find that coincident with UVB exposure, SOCS3 protein expression decreases which is consistent with an increase in the inflammatory cytokines IL-1β and IL-6. While SOCS3 regulation of IL-6 is well-known to require elements of the JAK-STAT pathway, the mechanism through which SOCS3 regulates IL-1β is less clearly resolved (Frobose et al., 2006; Karlsen et al., 2004; Wong et al., 2006). The SOCS3 effect appears to be robust since de Jonge et al. (de Jonge et al., 2005) have reported that vagal anti-inflammatory effects in macrophages through nAChRα7 involve activation of Jak2-STAT3. In addition, murine peritoneal macrophages cultured in vitro that were treated with nicotine showed increased production of STAT3 and SOCS3 upon stimulation with LPS, and the elevated SOCS3 was suggested to attenuate the inflammatory response to LPS in this experimental paradigm (de Jonge et al., 2005). Our results from the skin of UVB exposed mice extend this basic finding and demonstrate several points. SOCS3 protein decreases following exposure of control mice to UVB, with the maximal decrease occurring 48 hours post-exposure and protein levels return to normal thereafter. This decrease in SOCS3 coincides with an increase in inflammatory cytokine production in the skin following UVB exposure. In nicotine treated mice SOCS3 decreases but this decrease occurs 48 hrs post-UVB exposure. This suggests that the nicotine may impact upon the kinetics of the response to UVB and participate in attenuating the UVB response observed at 72 hours. Finally, SOCS3 protein in the α7KO showed a marked decrease at the 24 hour time period following UVB, versus only a small decrease in WT littermates, which would lead to an augmented cytokine response in the α7KO mice, as was observed. The mechanism controlling SOCS3 decline remains to be determined, but other reports suggest degradation following ubiquination (Frobose et al., 2006). Nevertheless, decreases in transcription remain a possible complimentary mechanism to assure SOCS3 regulation.
While reducing the inflammatory response may be of local or short-term benefit, the long-term impact of suppressing an inflammatory response may not be desirable. The need to enhance the immune response to a foreign antigen or to mount a response to opportunistic organisms such as bacteria is critical to survival. Because this is regulated through the inflammatory response, suppression of this effect may impact directly upon the ability to mount an effective immune defense (for discussion see (Gahring and Rogers, 2005)). In the mouth, those exposed to chronic nicotine are more susceptible to diseases such as gingivitis and other oral bacteria. In the skin, nicotine exposure decreases the ability to heal in response to wounding as well as leaves the wound more susceptible to infection. Also, immune suppression may reduce surveillance for abnormal cells, a condition that would favor the growth of transformed cells and possibly cancer. In contrast to the effects of nicotine on the immune response (suppressive), others (Fujii et al., 2007) report that immunization of nAChRα7KO mice results in enhanced production of antigen-specific IgG(1) suggesting that this nicotinic receptor plays a role in cytokine production affecting immunoglobulin synthesis. Further investigations selecting different mouse strains of appropriate genetic susceptibility or experimental design, to define the importance of different nAChRs to this process, the route of inflammatory stimulation and how this system contributes to these processes appears to be of potential clinical relevance and extends the function of the neuronal nicotinic receptors to regulating important components of inflammatory and immune function.
Acknowledgments
The excellent technical abilities of Janelle Lee are greatly appreciated.
Grant Support: These studies were funded by NIH grants DA015148 and DA018930 (LCG), PO1 HL72903 (LCG, SWR) and the Browning Foundation of Utah.
Abbreviations
- nAChR
neuronal nicotinic acetylcholine receptor
- α7
alpha 7
- UVR
ultraviolet radiation
- IL-1β
interleukin 1 beta
- IL-6
interleukin 6
- TNFα
tumor necrosis factor alpha
- SOCS3
suppressor of cytokine signaling 3
- KO
knock-out
Footnotes
Conflict of Interest: The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkondon M, Albuquerque EX. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res. 2004;145:109–120. doi: 10.1016/S0079-6123(03)45007-3. [DOI] [PubMed] [Google Scholar]
- Arredondo J, Chernyavsky AI, Marubio LM, Beaudet AL, Jolkovsky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: regulation of gene expression through alpha3beta2 nicotinic receptor in oral epithelial cells. Am J Pathol. 2005;166:597–613. doi: 10.1016/s0002-9440(10)62281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arredondo J, Nguyen VT, Chernyavsky AI, Bercovich D, Orr-Urtreger A, Kummer W, Lips K, Vetter DE, Grando SA. Central role of alpha7 nicotinic receptor in differentiation of the stratified squamous epithelium. J Cell Biol. 2002;159:325–336. doi: 10.1083/jcb.200206096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arredondo J, Nguyen VT, Chernyavsky AI, Bercovich D, Orr-Urtreger A, Vetter DE, Grando SA. Functional role of alpha7 nicotinic receptor in physiological control of cutaneous homeostasis. Life Sci. 2003;72:2063–2067. doi: 10.1016/s0024-3205(03)00084-5. [DOI] [PubMed] [Google Scholar]
- Brunzell DH, Russell DS, Picciotto MR. In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57Bl/6J mice. J Neurochem. 2003;84:1431–1441. doi: 10.1046/j.1471-4159.2003.01640.x. [DOI] [PubMed] [Google Scholar]
- Carlson NG, Bacchi A, Rogers SW, Gahring LC. Nicotine blocks TNF-alpha-mediated neuroprotection to NMDA by an alpha- bungarotoxin-sensitive pathway. J Neurobiol. 1998;35:29–36. doi: 10.1002/(sici)1097-4695(199804)35:1<29::aid-neu3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Chernyavsky AI, Arredondo J, Marubio LM, Grando SA. Differential regulation of keratinocyte chemokinesis and chemotaxis through distinct nicotinic receptor subtypes. J Cell Sci. 2004;117:5665–5679. doi: 10.1242/jcs.01492. [DOI] [PubMed] [Google Scholar]
- Conti-Tronconi BM, McLane KE, Raftery MA, Grando SA, Protti MP. The nicotinic acetylcholine receptor: structure and autoimmune pathology. Crit Rev Biochem Mol Biol. 1994;29:69–123. doi: 10.3109/10409239409086798. [DOI] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Samlowski WE, Burnham DK, Gahring LC, Roberts LK. Immunobiological consequences of acute and chronic UV exposure. Curr Probl Dermatol. 1986;15:176–194. doi: 10.1159/000412101. [DOI] [PubMed] [Google Scholar]
- de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- Frobose H, Ronn SG, Heding PE, Mendoza H, Cohen P, Mandrup-Poulsen T, Billestrup N. Suppressor of cytokine Signaling-3 inhibits interleukin-1 signaling by targeting the TRAF-6/TAK1 complex. Mol Endocrinol. 2006;20:1587–1596. doi: 10.1210/me.2005-0301. [DOI] [PubMed] [Google Scholar]
- Fujii YX, Fujigaya H, Moriwaki Y, Misawa H, Kasahara T, Grando SA, Kawashima K. Enhanced serum antigen-specific IgG(1) and proinflammatory cytokine production in nicotinic acetylcholine receptor alpha7 subunit gene knockout mice. J Neuroimmunol. 2007;189:69–74. doi: 10.1016/j.jneuroim.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Gahring L, Baltz M, Pepys MB, Daynes R. Effect of ultraviolet radiation on production of epidermal cell thymocyte-activating factor/interleukin 1 in vivo and in vitro. Proc Natl Acad Sci U S A. 1984;81:1198–1202. doi: 10.1073/pnas.81.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahring LC, Daynes RA. Desensitization of animals to the inflammatory effects of ultraviolet radiation is mediated through mechanisms which are distinct from those responsible for endotoxin tolerance. J Immunol. 1986;136:2868–2874. [PubMed] [Google Scholar]
- Gahring LC, Meyer EL, Rogers SW. Nicotine-induced neuroprotection against N-methyl-D-aspartic acid or beta-amyloid peptide occur through independent mechanisms distinguished by pro-inflammatory cytokines. J Neurochem. 2003;87:1125–1136. doi: 10.1046/j.1471-4159.2003.02074.x. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Persiyanov K, Days EL, Rogers SW. Age-related loss of neuronal nicotinic receptor expression in the aging mouse hippocampus corresponds with cyclooxygenase-2 and PPARgamma expression and is altered by long-term NS398 administration. J Neurobiol. 2004 doi: 10.1002/neu.20106. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Rogers SW. Neuronal Nicotinic Acetylcholine Receptor Expression and Function on Non-Neuronal Cells. The AAPS Journal. 2005a;7:E885–E894. doi: 10.1208/aapsj070486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grando SA. Cholinergic control of epidermal cohesion. Exp Dermatol. 2006;15:265–282. doi: 10.1111/j.0906-6705.2006.00410.x. [DOI] [PubMed] [Google Scholar]
- Grando SA, Horton RM, Mauro TM, Kist DA, Lee TX, Dahl MV. Activation of keratinocyte nicotinic cholinergic receptors stimulates calcium influx and enhances cell differentiation. J Invest Dermatol. 1996;107:412–418. doi: 10.1111/1523-1747.ep12363399. [DOI] [PubMed] [Google Scholar]
- Grando SA, Horton RM, Pereira EF, Diethelm-Okita BM, George PM, Albuquerque EX, Conti-Fine BM. A nicotinic acetylcholine receptor regulating cell adhesion and motility is expressed in human keratinocytes. J Invest Dermatol. 1995;105:774–781. doi: 10.1111/1523-1747.ep12325606. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol. 2003;147:1–46. doi: 10.1007/s10254-003-0005-1. [DOI] [PubMed] [Google Scholar]
- Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, Tracey KJ, Ulloa L. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203:1623–1628. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalayciyan A, Orawa H, Fimmel S, Perschel FH, Gonzalez JB, Fitzner RG, Orfanos CE, Zouboulis CC. Nicotine and biochanin A, but not cigarette smoke, induce anti-inflammatory effects on keratinocytes and endothelial cells in patients with Behcet's disease. J Invest Dermatol. 2007;127:81–89. doi: 10.1038/sj.jid.5700492. [DOI] [PubMed] [Google Scholar]
- lsen AE, Heding PE, Frobose H, Ronn SG, Kruhoffer M, Orntoft TF, Darville M, Eizirik DL, Pociot F, Nerup J, Mandrup-Poulsen T, Billestrup N. Suppressor of cytokine signalling (SOCS)-3 protects beta cells against IL-1beta-mediated toxicity through inhibition of multiple nuclear factor-kappaB-regulated proapoptotic pathways. Diabetologia. 2004;47:1998–2011. doi: 10.1007/s00125-004-1568-3. [DOI] [PubMed] [Google Scholar]
- Kurzen H, Wessler I, Kirkpatrick CJ, Kawashima K, Grando SA. The non-neuronal cholinergic system of human skin. Horm Metab Res. 2007;39:125–135. doi: 10.1055/s-2007-961816. [DOI] [PubMed] [Google Scholar]
- Matalka KZ, Tutunji MF, Abu-Baker M, Abu Baker Y. Measurement of protein cytokines in tissue extracts by enzyme-linked immunosorbent assays: application to lipopolysaccharide-induced differential milieu of cytokines. Neuro Endocrinol Lett. 2005;26:231–236. [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- Metz CN, Tracey KJ. It takes nerve to dampen inflammation. Nat Immunol. 2005;6:756–757. doi: 10.1038/ni0805-756. [DOI] [PubMed] [Google Scholar]
- Misery L. Nicotine effects on skin: are they positive or negative? Exp Dermatol. 2004;13:665–670. doi: 10.1111/j.0906-6705.2004.00274.x. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Ndoye A, Hall LL, Zia S, Arredondo J, Chernyavsky AI, Kist DA, Zelickson BD, Lawry MA, Grando SA. Programmed cell death of keratinocytes culminates in apoptotic secretion of a humectant upon secretagogue action of acetylcholine. J Cell Sci. 2001;114:1189–1204. doi: 10.1242/jcs.114.6.1189. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Kedmi M, Rosner S, Karmeli F, Rachmilewitz D. Increased severity of experimental colitis in alpha5 nicotinic acetylcholine receptor subunit-deficient mice. Neuroreport. 2005;16:1123–1127. doi: 10.1097/00001756-200507130-00018. [DOI] [PubMed] [Google Scholar]
- Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med. 2003;9:125–134. [PMC free article] [PubMed] [Google Scholar]
- Richardson CE, Morgan JM, Jasani B, Green JT, Rhodes J, Williams GT, Lindstrom J, Wonnacott S, Peel S, Thomas GA. Effect of smoking and transdermal nicotine on colonic nicotinic acetylcholine receptors in ulcerative colitis. Qjm. 2003;96:57–65. doi: 10.1093/qjmed/hcg007. [DOI] [PubMed] [Google Scholar]
- Rogers SW, Gahring LC, Collins AC, Marks M. Age-related changes in neuronal nicotinic acetylcholine receptor subunit alpha4 expression are modified by long-term nicotine administration. J Neurosci. 1998;18:4825–4832. doi: 10.1523/JNEUROSCI.18-13-04825.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauder DN, Carter CS, Katz SI, Oppenheim JJ. Epidermal cell production of thymocyte activating factor (ETAF) J Invest Dermatol. 1982;79:34–39. doi: 10.1111/1523-1747.ep12510569. [DOI] [PubMed] [Google Scholar]
- Sopori ML, Kozak W, Savage SM, Geng Y, Kluger MJ. Nicotine-induced modulation of T Cell function. Implications for inflammation and infection. Adv Exp Med Biol. 1998;437:279–289. doi: 10.1007/978-1-4615-5347-2_31. [DOI] [PubMed] [Google Scholar]
- Soy M, Erken E, Konca K, Ozbek S. Smoking and Behçet's Disease. Clinical Rheumatology. 2000;19:508–509. doi: 10.1007/s100670070020. [DOI] [PubMed] [Google Scholar]
- Thomas GA, Rhodes J, Ingram JR. Mechanisms of disease: nicotine--a review of its actions in the context of gastrointestinal disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:536–544. doi: 10.1038/ncpgasthep0316. [DOI] [PubMed] [Google Scholar]
- Wang J, Campbell IL. Cytokine signaling in the brain: putting a SOCS in it? J Neurosci Res. 2002;67:423–427. doi: 10.1002/jnr.10145. [DOI] [PubMed] [Google Scholar]
- Wong PK, Egan PJ, Croker BA, O'Donnell K, Sims NA, Drake S, Kiu H, McManus EJ, Alexander WS, Roberts AW, Wicks IP. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest. 2006;116:1571–1581. doi: 10.1172/JCI25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zia S, Ndoye A, Nguyen VT, Grando SA. Nicotine enhances expression of the alpha 3, alpha 4, alpha 5, and alpha 7 nicotinic receptors modulating calcium metabolism and regulating adhesion and motility of respiratory epithelial cells. Res Commun Mol Pathol Pharmacol. 1997;97:243–262. [PubMed] [Google Scholar]