

Summary
Disease-causing mutations occur in genes for aminoacyl tRNA synthetases. That some mutations are dominant suggests a gain-of-function. Native tRNA synthetases, like TyrRS and TrpRS, catalyze aminoacylation and are also procytokines that are activated by natural fragmentation. In principle, however, gain-of-function phenotypes could arise from mutational activation of synthetase procytokines. From crystal structure analysis we hypothesized that a steric block of a critical ELR motif in full-length TyrRS suppresses the cytokine activity of a natural fragment. To test this hypothesis, we attempted to uncover ELR in the procytokine by mutating a conserved tyrosine (Y341) that tethers ELR. Site-specific proteolytic cleavage and small angle X-ray scattering established subtle opening of the structure by the mutation. Strikingly, four different assays demonstrated mutational activation of cytokine functions. The results prove the possibilities for constitutive gain-of-function mutations in tRNA synthetases.
Keywords: Aminoacyl-tRNA synthetase, Cytokine function, Tyrosyl-tRNA synthetase, Structure
Introduction
Aminoacyl tRNA synthetases (AARS) are a group of 20 enzymes, one for each amino acid, which catalyze the first step of protein synthesis by aminoacylation of tRNAs. Interestingly, both recessive [1, 2] and dominant [3–10] mutations in genes for tRNA synthetases have causal associations with diseases in humans and the mouse. These causal associations of disease with mutations in synthetases have, in some instances, been shown to not be associated with defects in aminoacylation activity [3, 10, 11]. Alternatively, the connections with human diseases may reflect a long evolution, during which the enzymes expanded their functions to act in translational and transcriptional regulation, RNA splicing and, in higher eukaryotes, signal transduction pathways [12, 13].
After the discovery of the synthetase-interacting factor AIMP1 (also known as p43 and EMAP II), which has broad cell-signaling activities [14, 15], human tyrosyl-tRNA synthetase (TyrRS) was the first example of a tRNA synthetase having activities in cell-signaling and angiogenesis [16, 17]. These activities are manifested when the enzyme is secreted, during an inflammatory response, as a procytokine subsequently activated by natural proteolysis. Later, more examples further supported the idea of functional expansion of AARSs in higher eukaryote systems [13, 18]. Human tryptophanyl-tRNA synthetase (TrpRS) is another example, where natural fragmentation by either alternative splicing or proteolysis leads to production of a potent angiostatic cytokine [19, 20]. The expanded functions of Trp-, Tyr- and other tRNA synthetases, and of synthetase-interacting proteins, suggest a broad connection of translation to signal transduction pathways in angiogenesis, inflammation, and neuronal development. Not fully understood, however, is how the cytokine functions of a tRNA synthetase procytokine are activated so that connections are made to pathways beyond translation. It is this question—the mechanism of activation of a synthetase procytokine—that is addressed here. We believe that this question is closely related to that of understanding how dominant, presumptive gain-of-function mutations might occur in genes for synthetases in the human population. For this purpose, we focused on TyrRS as an example where detailed structural information could guide our experimental approach.
Results
Background on structure and cytokine activity of human TyrRS
TyrRS is a member of the class I tRNA synthetases, which are characterized in part by a Rossmann fold catalytic domain with an 11 amino acid signature sequence that ends in the tetrapeptide HIGH [21]. In prokaryotes and lower eukaryotes, the catalytic domain is fused on the C-terminal side with an anticodon binding domain. This format—Rossmann fold followed by anticodon binding domain—is conserved amongst all TyrRSs in evolution. In higher eukaryotes including mammals, an additional domain, known as the ‘C-domain’, is appended to the C-terminal side of the anticodon binding domain (Figure 1A). The C-domain, which is homologous to EMAP II (endothelial monocyte activating protein II) [22], regulates the cytokine activity of TyrRS in a way that is not understood and is investigated here.
Figure 1.
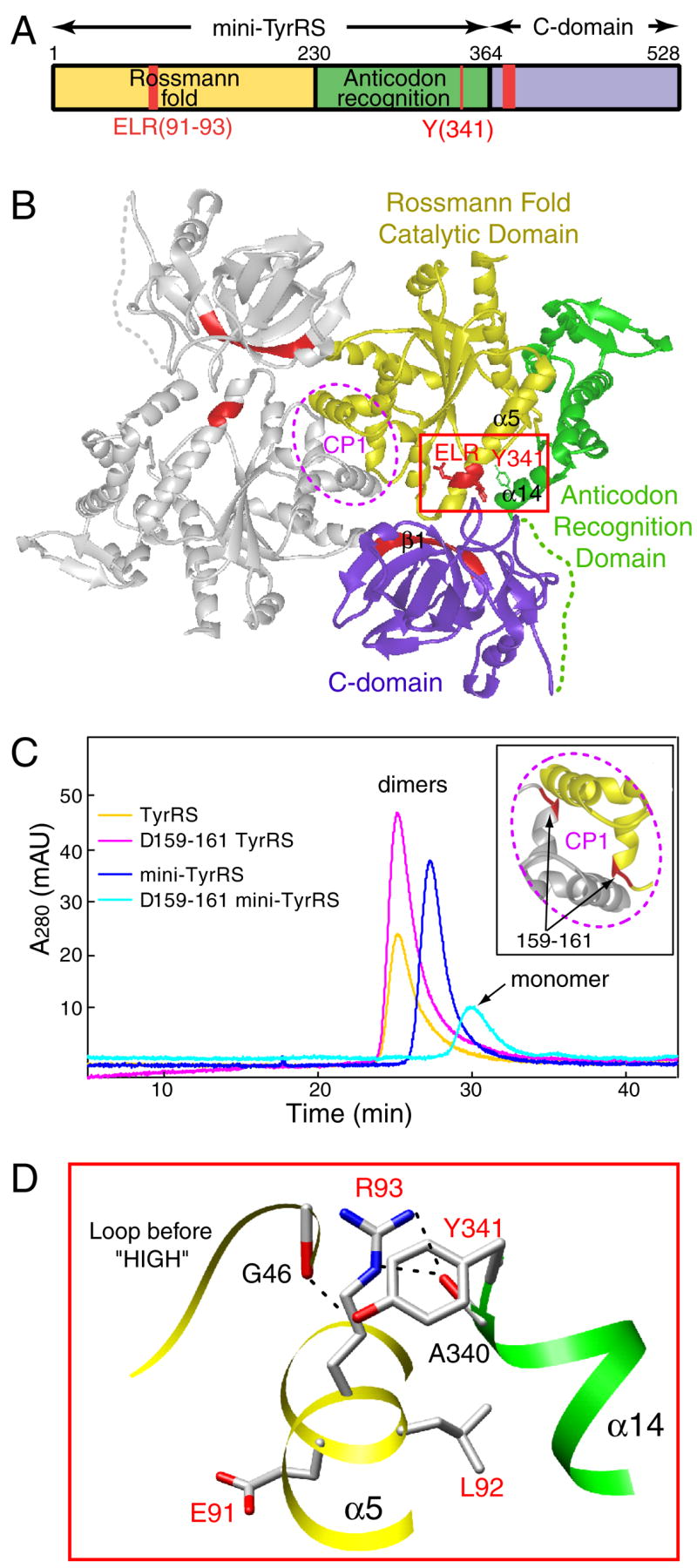
Organization of domains and the ELR motif of human TyrRS. (A) Human TyrRS contains three domains: Rossmann fold catalytic domain, anticodon recognition domain and C-domain. The first two domains constitute mini-TyrRS and are essential for the aminoacylation of tRNATyr. The C-domain is dispensable for aminoacylation. Mini-TyrRS also has cytokine activity, and an ELR (red) motif in the Rossmann fold catalytic domain is critical for that activity. C-domain alone also has cytokine activity, and a heptapeptide (red) sequence close to the N-terminus of the C-domain was shown to be critical for its PMN migration activity. (B) Structure model of full-length TyrRS. The ELR motif (red) on mini-TyrRS and the heptapeptide (red) on the C-domain are likely to be masked in the full-length protein, where the C-domain is linked to mini-TyrRS. (C) Analytical gel filtration experiment suggested that the C-domain contributed to the dimer interactions in a way consistent with the structural model. (D) The interactions around the ELR motif. Y341 is of special significance, as it plays a key role in tethering helix α14 to the ELR.
Full-length human TyrRS has no known cytokine activities. However, when the appended C-domain is removed by natural proteolysis, the remaining mini-TyrRS can induce migration of PMN and endothelial cells, and is pro-angiogenic [16, 17]. (The EMAP II-like C-domain of TyrRS is also a procytokine that is activated when separated from the rest of TyrRS [23].) Removal of the C-domain does not affect aminoacylation activity—mini-TyrRS charges its cognate tRNA with the same efficiency as the full-length enzyme [16].
An ELR motif embedded in the sequence of mini-TyrRS was shown by mutational analysis to be critical for its cytokine activity [16, 23]. An ELR motif is also present in, and required for, activity of some CXC chemokines, such as IL-8 [24]. The importance of the ELR motif for mini-TyrRS is consistent with the observation that mini-TyrRS binds with high affinity to the IL-8 receptor CXCR1 [16]. In earlier work we separately determined high resolution crystal structures of human mini-TyrRS [25] and of the C-domain [26] (embedded in Figure 1B). The ELR motif was mostly exposed in mini-TyrRS and was able, in principle, to interact with a receptor. In contrast, in the native, full-length enzyme, we imagine this motif would be sterically shielded by the C-domain.
As for the EMAP-II like C-domain, a heptapeptide sequence close to the N-terminus of the C-domain was shown to be critical for its PMN migration activity [23]. In our crystal structure of the C-domain (embedded in Figure 1B), this heptapeptide is located on strand β1 (among the 7 β-strands that form a β-barrel structure) and is essentially exposed on the surface of the C-domain when that domain is a stand-alone protein. Presumably, this heptapeptide is also masked in the full-length TyrRS by mini-TyrRS.
Structural model of human TyrRS
While no structure of human TyrRS has been reported, from the structures of mini-TyrRS and C-domain, we were able to build a working model of the full-length protein (Figure 1B). Mini-TyrRS contains the Rossmann fold catalytic domain and anticodon recognition domain. It forms a dimer with the CP1 (connective peptide 1) insertion (in the Rossmann fold domain) forming the dimerization interface. In the model, the C-domain is linked to the C-terminus of mini-TyrRS via a flexible loop identified in the crystal structure of mini-TyrRS [25]. As shown previously, the surface electrostatic potential of the ELR region in mini-TyrRS is positively charged, while that of the heptapeptide region in C-domain is negatively charged [26]. Therefore, the two critical motifs have complementarily charged surfaces that, in principle, can be brought together for mutual shielding in the full-length TyrRS. If this is the case, because of the proximity of the ELR motif to the dimer interface (Figure 1B), the C-domain would also be close to the dimer interface and could contribute additional dimerization interactions. Experiments described below were designed to test whether the C-domain strengthens the dimer interactions.
The dimer interface of mini-TyrRS mainly involves backbone H-bonding interactions between residues P159, L161 and S162 from one subunit, with S137, V140 and Q142 from the other subunit, and vice versa. We constructed a 3-residue (P159L160L161) deletion mutant, designated as Δ159-161 TyrRS, to disrupt the dimer interaction. In contrast to wild-type mini TyrRS, using analytical gel filtration chromatography, Δ159-161 mini-TyrRS was a monomer (Figure 1C). Thus, the Δ159-161 deletion successfully disrupted dimer formation of mini-TyrRS. We also createdΔ159-161 TyrRS, that is, the full-length protein with the same deletion. Significantly, this deletion did not disrupt dimer formation of full-length TyrRS. (Both Δ159-161 and wild-type TyrRS eluted as dimers from the gel filtration column (Figure 1C).) These results are consistent with the C-domain being important for providing dimer stabilization beyond that which comes from contacts made between partner subunits through the CP1 insertion. They support the structural model of dimeric TyrRS having two C-domains located near (with each on one side of) the dimer interface of mini-TyrRS (Figure 1B).
This model for full-length TyrRS provided the structural basis to understand the mutual shielding of the cytokine activities of mini-TyrRS and C-domain in the native enzyme. We reasoned that, if we could specifically remove the shield to unmask either of the two critical motifs, then this AARS procytokine would be converted to a cytokine. To test this hypothesis, we examined the structure to find a way to ‘open’ that structure, so as to expose the ELR tripeptide without breaking the covalent polypeptide backbone. This analysis led to the idea that a specific point mutation could unhitch a non-covalent tether, and thereby provide access to ELR and yield a mutant full-length enzyme that would have the cytokine activities of mini-TyrRS.
Identification of a tether that enables the C-domain to shield the ELR motif
The partially exposed ELR motif of mini-TyrRS is located within a long α helix (α5) of the Rossmann fold catalytic domain (Figure 1B). Interestingly, the C-terminal end of mini-TyrRS, which forms helix α14, is close to the ELR motif. In fact, α14 is tethered to the ELR motif through H-bonding and stacking interactions that deploy a conserved tyrosine Y341 on α14 (Figure 1D). Therefore, when the C-domain is attached to the C-terminal end of mini-TyrRS, the ELR motif can be easily masked by the C-domain (Figure 1B). We hypothesized that, if we mutationally disrupted the interactions between the ELR motif and Y341, then the tether that brings the C-domain into the proximity of the ELR motif would be broken. As a result, the ELR motif would be exposed, and mini-TyrRS-like activity would be activated in the full-length protein.
For this purpose, we constructed a Y341A substitution to give Y341A TyrRS. The resulting recombinant protein was stable so that quantities sufficient for the studies reported below could be prepared. To use as a control, we also constructed Y341A mini-TyrRS and found it to be stable. As a further investigation of the conformational integrity of the two proteins, their aminoacylation activities were determined. Both Y341A mutants had about 50% of the aminoacylation activity of the respective wild-type counterpart (data not shown). This reduction was expected because, in the crystal structure of mini-TyrRS, Y341 makes an H-bond to G46, which is an absolutely conserved residue in the middle of the aforementioned 11-amino acid signature sequence. That sequence motif is important for synthesis and binding of tyrosyl-AMP. As expected, the tyrosine-dependent ATP-PPi exchange activity was significantly reduced for both Y341A mutant proteins (data not shown). However, possibly because the rate limiting step in aminoacylation for many tRNA synthetases is after adenylate synthesis (transfer of the aminoacyl-moiety from AMP to tRNA [27]), the effect of the Y341A mutation on aminoacylation activity is not pronounced.
Demonstration of a broken ‘tether’ by protease digestion
The Y341A TyrRS mutant proteins was expressed and purified side-by-side with the previously cloned wild-type TyrRS. To evaluate whether the Y341A substitution disrupted the interaction between the ELR motif and helix α14, proteases were used as probes to detect any subtle conformational change caused by the mutation. We imagined that, with a more opened-up structure, the Y341A mutant proteins could have additional cleavages site(s) relative to the wild-type enzyme.
First, we explored protease digestion of wild-type TyrRS, using plasmin and leukocyte elastase, both of which are extracellular enzymes important for angiogenesis signaling pathways [28, 29]. Plasmin cleaves after accessible lysines or, occasionally, arginines. Four major fragments of TyrRS were generated by limited plasmin digestion, and the identity of each fragment was determined by mass spectrometry and N-terminal sequencing (Figure 2A left). This analysis established that the four fragments arose from cleavages at three sites, located at K154, K352 and K356. K154 is a minor cleavage site located on a long internal loop of the CP1 insertion that splits the Rossmann nucleotide binding fold (Figure 2B). K352 and K356 suggest a major cleavage site located on a different loop-the linker that joins the C-domain to mini-TyrRS (Figure 2B). Therefore, because the major cleavage at K352 and K356 is within the linker loop, plasmin can liberate mini-TyrRS from the C-domain, and thereby activate the cytokine activities imbedded in TyrRS.
Figure 2.

Limited proteolysis of wild-type and Y341A TyrRS to probe for structural changes. (A) Additional bands (boxed) were observed with Y341A TyrRS using either plasmin (top) or elastase (bottom) for digestion. (B) The cleavage sites generated by plasmin (magenta) and elastase (cyan) are marked on the structure. Additional cleavage sites were generated for Y341A relative to wild-type TyrRS, and those additional sites are located on or near helix α14.
Limited elastase cleavage also generated four major fragments, arising from three cleavages located at V153, A355 and S366 (Figure 2A left and B). Although the sequence specificity of elastase is different than that of plasmin, the three elastase cleavage sites are essentially juxtaposed with those of plasmin. This observation-of the same spatial locations for three distinct cleavage sites with two different proteases-gave strong confidence that the protease probes were primarily revealing structural information and not information that was idiosyncratic to the protease.
When Y341A TyrRS was digested with plasmin, three fragments in addition to those seen with wild-type TyrRS were observed. Consistently, elastase also gave additional cleavage fragments for the Y341A mutant compared to wild-type TyrRS (Figure 2A right). Those additional fragments corresponded to Y341A-specific cleavage sites at K319, K327 and K334 by plasmin, and L333 and A337 by elastase, respectively. These 5 additional cleavage sites are either on helix α14 or at the end of α13 that connects to α14. This suggests that helix α14, and part of α13, becomes unwound by virtue of the Y341A substitution. Most likely, this part of the structure of the mutant protein is present as a flexible loop, presumably because the tether between α14 and the ELR motif was broken.
Demonstration of subtle structure-opening by small angle X-ray scattering
With the broken tether that holds the C-domain close to the ELR motif, we imagined that the Y341A mutation would generate a more extended overall conformation. We thought that the conformational difference between Y341A and wild-type TyrRS might be sufficient to be detected by small angle X-ray scattering. Pursuant to this objective, scattering curves were measured for both wild-type and Y341A human TyrRS at Beam Line 4-2 at the Stanford Synchrotron Radiation Laboratory. The radius of gyration (Rg) evaluated by Guinier plots of the individual scattering curves, recorded at concentrations of 2, 4, 6, 9 and 20 mg/ml, showed no obvious concentration dependence, for both the wild-type and Y341A enzyme. The Rg values were then extrapolated to zero concentration, yielding Rg = 46.5 Å (±1.0) for the wild-type and Rg = 49.7 (±1.0) Å for the Y341A mutant protein. These values support the hypothesis that Y341A is more open, and therefore more extended than, the wild-type enzyme.
Electron pair distance distribution P(r) function was computed using the composite data covering the momentum transfer Q = 0.01 to 0.25 Å−1, and consisting of the very small angle portion from the lowest concentration point (2 mg/ml) and the intermediary angle from either the 9 or 20 mg/ml sample (Figure 3). The maximum dimension of the molecule Dmax = 152 (±3) Å was obtained for the wild-type enzyme, while the Y341A mutant enzyme gave Dmax = 155 (±5) Å. The maximum dimension of our TyrRS model shown in Figure 1B is about 20 Å smaller than the obtained Dmax, suggesting that, as the C-domain is linked to mini-TyrRS via the flexible loop, multiple conformations of TyrRS, including those that are more extended than our TyrRS model, may be present in solution. Nevertheless, a slightly larger Dmax of Y341A TyrRS relative to the wild-type enzyme is consistent with Y341A TyrRS having a more opened conformation.
Figure 3.
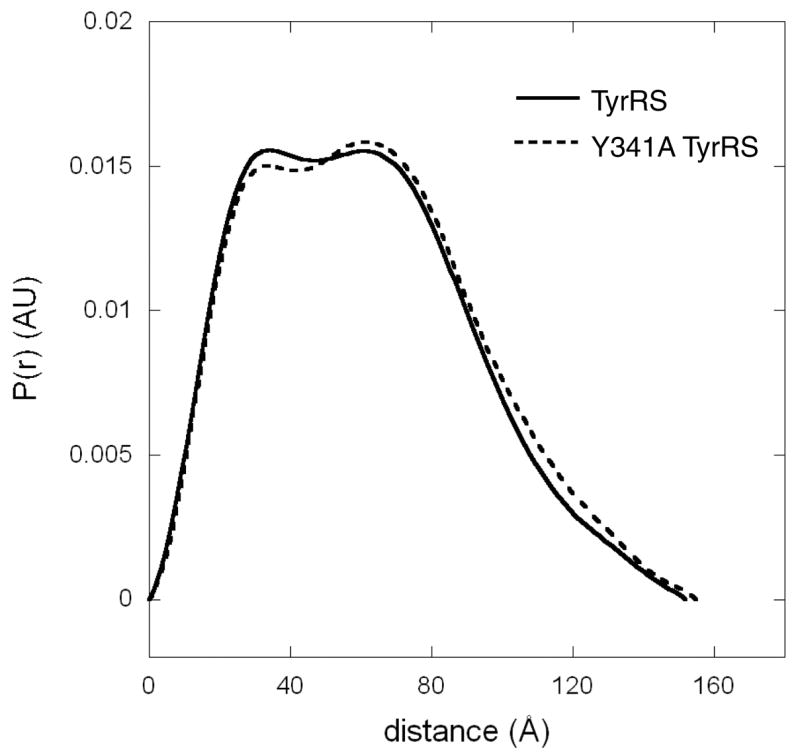
Electron pair distance distribution function generated by SAXS experiments for wild-type and Y341A TyrRS.
A more relaxed structure for the mutant protein was also confirmed from the Rg values obtained via P(r): Rg = 47.2 (±0.2) Å and Rg = 48.6 (±0.4) Å for the wild-type and Y341A TyrRS, respectively. Interestingly, the P(r) curves of the wild-type enzyme exhibited two peaks at around 30 and 60 Å electron-pair distances (Figure 3). The mutant protein had a lower 30 Å peak and a higher 60 Å peak, in addition, it had an increased-distance population between 90 and 140 Å, indicating that structural domains are likely to be more spread apart in the case of Y341A TyrRS. The positions and magnitudes of the peaks are independent of the Dmax value used in the analysis. Therefore, these observations further support the idea of a more open conformation for the mutant protein at the level of tertiary structure.
The Y341A mutation confers angiogenic activity to full-length TyrRS
The complex process of angiogenesis involves endothelial cell activation, proliferation, and migration. Previous work established that mini-TyrRS induced chemotaxis of endothelial cells in vitro, and also simulated angiogenesis in vivo [17]. To test our hypothesis that the Y341A substitution activates mini-TyrRS-like activity in full-length TyrRS, we first tested our mutant protein for stimulation of endothelial cell proliferation and migration in vitro, and then went on to test its activity in vivo in two animal models.
Mini-TyrRS-like activity for Y314A TyrRS in cell proliferation and migration assays
First, we investigated the stimulation of proliferation of bovine aortic endothelial cells (BAECs). For these studies, we used TyrRS, mini-TyrRS, and Y341A TyrRS. We also used Y341A mini-TyrRS as an additional control. As expected, mini-TyrRS stimulated the proliferation of BAECs, while full-length TyrRS did not. In addition, the Y341A mutation (to give Y341A mini-TyrRS) did not have much effect on the activity of mini-TyrRS. In contrast, the Y341A substitution activated full-length TyrRS for stimulation of cell proliferation (Figure 4A). The activity of Y341A TyrRS was comparable to that of mini-TyrRS.
Figure 4.
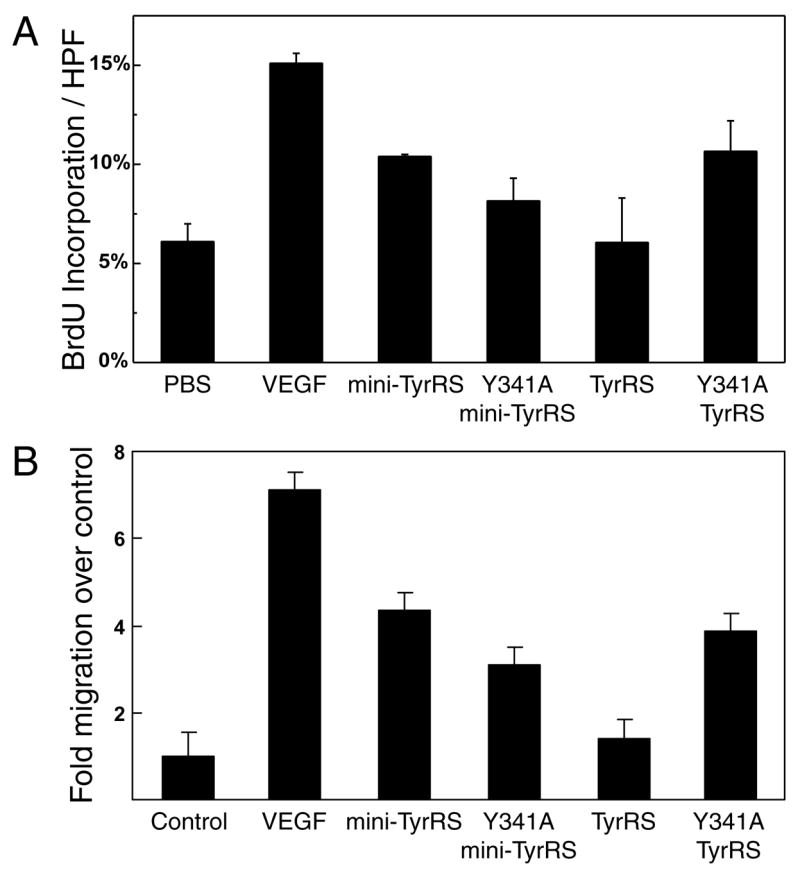
Endothelial cell proliferation and migration assays. (A) Y341A mutation activated full-length TyrRS for stimulating proliferation of bovine aortic endothelial cells. Cell proliferation was measured in five randomly chosen HPFs by the percentage of cells with positive bromodeoxyuridine (BrdU) incorporation. Samples were run in triplicate and mean ± s.d. is shown. (B) Y341A mutation activated full-length TyrRS for stimulating migration of human umbilical vein endothelial cells. Migration of untreated monolayers was normalized to 1. Data from eight experiments were collected for each sample and mean ± s.d. is shown.
Similar results were observed in an endothelial cell migration assay. In these experiments, wounds were created in monolayers of human umbilical vein endothelial cells (HUVECs). Our proteins were added in the media to test for their activities in wound closure by stimulating cell migration. Mini-TyrRS closed about 4-fold more area (Y341A mini-TyrRS closed about 3-fold more area) of the wound than did the control in 6 hours, whereas full-length TyrRS closed only slightly more relative to the control (Figure 4B). In contrast to TyrRS, Y341A TyrRS, like mini-TyrRS, closed 4-fold more area of the wound. Here again, the Y341A mutation activated TyrRS, in this case for endothelial cell migration.
Mini-TyrRS-like activity for Y314A TyrRS in animals
Next, we tested our proteins in vivo in the mouse matrigel (Figure 5A) and chick chorioallantoic membrane (CAM) (Figure 5B) assays for angiogenesis. In the mouse matrigel model, a collagen plug containing PBS or an added protein is injected subcutaneously. When t here is an added pro-angiogenic factor, blood vessels perfuse into the plug. Upon injection of a fluorescent endothelial-cell binding lectin, the blood vessel density in each plug can easily be quantified by spectrophotometric analysis.
Figure 5.
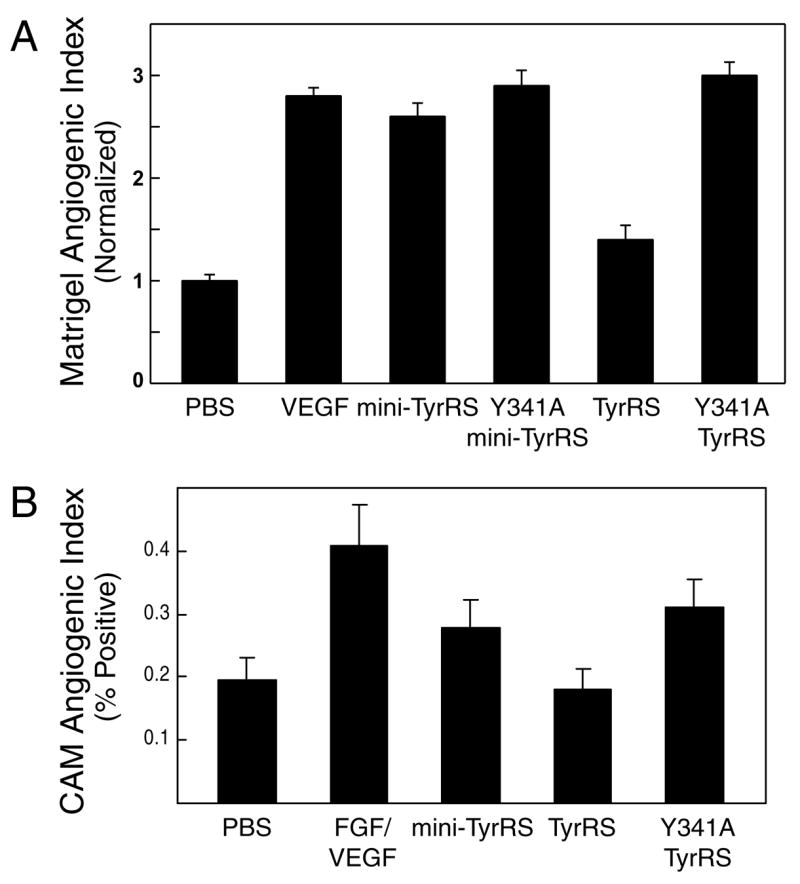
Mouse matrigel and chick chorioallantoic membrane angiogenesis assays. (A) Y341A mutation activated full-length TyrRS for angiogenesis in the mouse matrigel angiogenesis assay. The blood vessel content in the matrigel plug with PBS control was normalized to 1. Four mice were used for separate controls with PBS and VEGF and 6 mice were used for each of the TyrRS samples. Data show mean ± s.e.m. (B) Y341A mutation activated the full-length TyrRS for angiogenesis in chick chorioallantoic membrane angiogenesis assay. The plot shows mean of about 30 measurements for each sample ± s.e.m.
As previously reported [17] and confirmed here, in this model, VEGF and mini-TyrRS were both positive for pro-angiogenic activity. (The Y341A mutation in mini-TyrRS had no effect on its activity.) As expected and as also reported earlier, full-length TyrRS was inactive in this assay. However, the Y341A substitution activated TyrRS to give it an activity in the matrigel assay that was similar to that seen with mini-TyrRS (Figure 5A).
To confirm the results seen in the matrigel assay, we further tested the proteins in the CAM assay. Wild-type TyrRS was inactive, with levels of angiogenesis in the plug similar to that seen with PBS. However, Y341A TyrRS was pro-angiogenic, with an activity similar to mini-TyrRS (Figure 5B). Thus, these data independently support results from the mouse matrigel assay.
Angiogenic activity of Y341A TyrRS not from serendipitous proteolytic activation
If the Y341A mutation made TyrRS more susceptible to proteolysis, then the mini-TyrRS fragment of TyrRS might be serendipitously generated in the above assay systems we used to assess activity. In that scenario, the angiogenic activity we observed with Y341A TyrRS would come from proteolytically generated mini-TyrRS. To address this possibility, we looked more closely into our cell migration assay with HUVECs. We collected and analyzed media at the 6-hour time point of this assay, where we saw that only Y341A TyrRS, and not wild-type TyrRS, stimulated wound closure. Using western blot analysis with polyclonal anti-TyrRS antibodies, we found that the majority of the wild-type and Y341A TyrRS proteins were full-length. No more of the mini form was generated from Y341A than was produced from wild-type TyrRS (Figure 6A). These results provide no support for the idea that the activity of Y314A TyrRS is from serendipitous cleavage of the mutant versus the wild-type version of TyrRS.
Figure 6.

Angiogenic activity of Y341A TyrRS not from serendipitous proteolytic activation. (A) Less mini form of Y341A TyrRS than of TyrRS was generated in the media during the endothelial cell migration assay, as shown by western blot analysis with polyclonal antibody against TyrRS. This observation suggested that the cytokine activation of TyrRS by the Y341A mutation was not a result of proteolysis. (B) Time courses of plasmin and elastase digestions of both wild-type and the Y341A TyrRS. The Y341A TyrRS was as stable as the wild-type enzyme during the digestion. Arrows point to the mini form of TyrRS.
In addition, to see if Y341A TyrRS is inherently more fragile and susceptible to proteolysis, we tested whether plasmin or elastase could digest Y341A TyrRS more rapidly than it digested wild-type TyrRS. Each protein was incubated with a protease, and the time course of each digestion was monitored (Figure 6B). Plasmin digested away both full-length enzymes in 2 hours. For both wild-type and Y341A TyrRS, the mini-form was generated and cleared after 6 hours. (The C-domain was more resistant to plasmin digestion, so that even at 18 hours some C-domain remained.) An additional band, slightly smaller than mini-TyrRS, was observed in the digestion of Y341A TyrRS. This band, which was previously identified as comprised of residues 1-334 (Figure 2A), remained in the digestion mixture for more than 6 hours.
Consistently, for elastase digestions, Y341A TyrRS was cleared no faster than was wild-type TyrRS (Figure 6B). (As with the plasmin digestion, a fragment slightly smaller than mini-TyrRS remained for at least 6 hours. This fragment was identified as comprising residues 1-337 (Figure 2A).) Thus, with both plasmin and elastase digestions, no evidence for an intrinsically labile and protease-susceptible fragility of Y341A TyrRS could be obtained. While a shorter form of mini-TyrRS, which can potentially have mini-TyrRS-like cytokine activity, was generated by each protease acting on Y341A TyrRS and was more stable than mini-TyrRS in the in vitro digestion experiment (Figure 6B), our observation that the amounts of both mini-TyrRS and the smaller form of mini-TyrRS were minimal in the media of the HUVEC migration assay (Figure 6A) is consistent with the pro-angiogenic activity of Y341A TyrRS being intrinsic and arising from its having an accessible ELR motif.
EMAP II-like C-domain not activated by Y341A mutation
The C-domain of TyrRS is a homolog of EMAP II and has activities for stimulating production of specific cytokines, and for chemotaxis of leukocytes and monocytes, which are all masked in native, full-length TyrRS [16]. A heptapeptide unit presented on a β-barrel is critical for the chemotaxis activity [23] and this heptapeptide is buried in the operational model of dimeric human TyrRS, but exposed on the free monomer unit of the C-domain [26]. Our Y341A mutation was designed to produce a subtle change in conformation that specifically exposed the ELR motif of the mini-TyrRS portion of TyrRS. Because of the localized nature of the conformational change, as supported by the protease digestion study, and the subtlety of the overall conformational change suggested by small angle X-ray scattering analysis, we imagined that it was unlikely that the Y341A mutant would affect the region around the critical heptapeptide of the C-domain, and therefore the chemotaxis activity of the C-domain would not be activated by the Y341A mutation.
To further explore this question, we tested the C-domain activity in a cell migration assay, where only the C-domain, and not native TyrRS and mini-TyrRS, stimulate migration of monocytes. Mononuclear cells were purified from human blood over a histopaque gradient and allowed to migrate (in a Boyden chamber assay) under the influence of different concentrations of TyrRS, Y341ATyrRS, mini-TyrRS and mini-Y341ATyrRS. EMAP II and recombinant C-domain were used as positive controls, and both caused migration of monocytes in our assays (Figure 7). As expected, there was no migration under the influence of TyrRS and mini-TyrRS. Interestingly, Y341ATyrRS also did not stimulate migration of MPs. Thus, by this functional assay, the critical heptapeptide unit in the C-domain appears to still be masked in Y341A TyrRS. In addition, the ‘silence’ of the C-domain in Y341A TyrRS further supports the conclusions from Figure 6 that serendipitous cleavage of Y341A TyrRS is not responsible for its cytokine activation.
Figure 7.
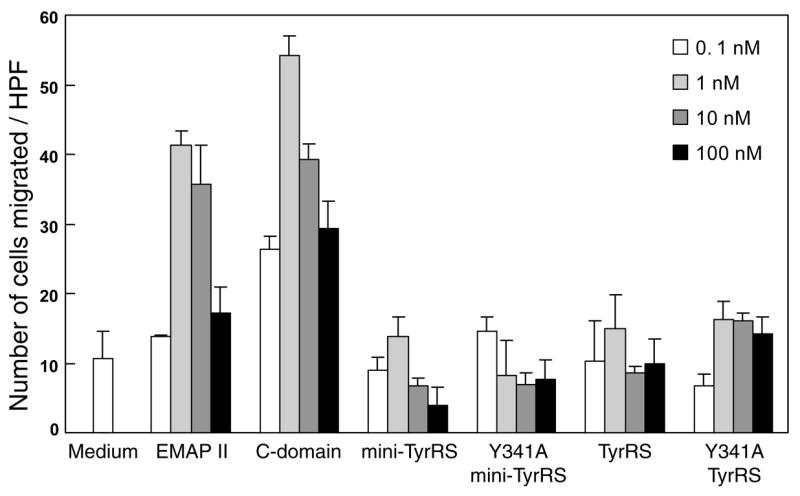
Monocyte chemotaxis assay. Each protein (0.1–100 nM) or medium alone was added to the lower compartment of chemotaxis chambers, and mononuclear cells (105 cells) were added to the upper compartment. Chambers was incubated for 3 hours and then migrating cells were counted in five HPFs in each case. Samples were run in triplicate and mean ± s.d. is shown.
Discussion
Given that dominant disease-causing mutations occur in genes for at least two human tRNA synthetases [3, 4], our work shows how in principle such mutations can occur. That is, for a tRNA synthetase that is also a procytokine, any mutation that serves to bypass the normal cytokine activation pathway (such as alternative splicing or proteolysis), like a mutation analogous to the Y341A allele of the gene encoding TyrRS, could in principle activate the imbedded cytokine activity and confer a constitutive gain-of-function.
Although dominant disease-causing mutations in genes for human tRNA synthetases are known, the alternative functions associated with disease are not well understood. For example, point mutations in human TyrRS and GlyRS are each causally associated with a distinct autosomal dominant form of Charcot-Marie-Tooth (CMT) disease [3, 4], the most common heritable peripheral neuropathy. Detailed studies of the aminoacylation activities of various CMT disease-causing mutant GlyRSs, and of a genetically constructed mouse model, have established that CMT disease can occur without defects in aminoacylation activity and, instead, associate the disease to changes in the dimer interface [10, 11, 30]. These changes in the dimer interface, in a way that is not known, may affect an alternative function of GlyRS that is associated with neurogenesis.
In the case of TyrRS, the disease-associated mutations are G41R and E196K, and deletion Δ153-156VKQV in the catalytic domain [3]. At least one of these mutant proteins (E196K) appears to be fully active in aminoacylation. Because the mutations are dominant, they are reminiscent of the work presented here, which demonstrates a mutational gain-of-cytokine function. However, in examining the locations of the CMT-causing mutations in the structure of TyrRS, no support could be found for the idea that the CMT-causing mutations would ‘open up’ the structure in the way that was seen with the Y341A substitution studied above. Thus, the dominant mutations connected to CMT disease may be unrelated to the cytokine activation and signaling pathways investigated here. Still, it remains of interest to test whether the CMT-causing mutations can be linked in any way to the cytokine activities known for human TyrRS and, conversely, whether the Y341A mutation would give rise to a CMT disease-like condition.
In addition to determining whether a gain-of-function mutation in a gene for a human tRNA synthetase can be created, another goal of our work was to understand the mechanism of activation of synthetase procytokines. Our data show how steric shielding-deshielding of a critical motif imbedded within TyrRS can regulate its cytokine activity. TrpRS, a close homolog of TyrRS, is activated by alternative splicing or natural proteolysis that removes a vertebrate-specific N-terminal extension of the polypeptide chain [20]. Thus, this extension is thought to regulate the cytokine activity. When the extension is removed, the resulting fragment, known as T2-TrpRS, acts through the VE-cadherin receptor on the surface of endothelial cells and triggers the Akt signaling pathway [31]. (The fragment has been used in animal models for macular degeneration and cancer, and in gene therapy applications [19, 32, 33].) Unlike TyrRS, where the ELR motif imbedded in the catalytic domain has been shown to be critical for the IL-8-like cytokine activity, the determinants in TrpRS needed for cytokine activity have not been identified. However, by analogy with the results presented here, we speculate that activation of the TrpRS procytokine may come from steric deshielding of a critical motif imbedded in the core protein. In this instance, the shield would be provided by the N-terminal extension.
Our work also showed that only subtle changes in structure are needed for cytokine activation. The protease probes (Figure 2) and small angle X-ray scattering (Figure 3) proved to be sufficiently sensitive to detect the alteration caused by the Y341A mutation. At the same time, the change in conformation was small enough that TyrRS was not destabilized and subjected to proteolysis (Figure 6). This conformational change has maintained most of the catalytic activity (see above). When the essential aminoacylation function and protein stability are both retained, mutations that activate a cytokine function can be vertically transmitted and accumulated in the population, where the mutant alleles may be associated with a non-lethal pathological condition. That the Y341A TyrRS example shows how cytokine activation can occur, without disruption of stability or the essential function in protein synthesis, makes plausible the idea that more examples of disease-associated mutations in genes for tRNA synthetases will be identified in the population.
Significance
This work addresses the question of the mechanism of how an expanded function of a tRNA synthetase is activated. A proposed model is supported by a series of biological assays and physical characterizations. The results also raise new possibilities for understanding why mutations in genes for tRNA synthetases are causally linked to human diseases. In this connection, the work is original in demonstrating the idea that gain-of-function can be achieved by mutation in these enzymes.
Experimental Procedures
Plasmid construction
Different mutation and deletion constructs of TyrRS were made using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Templates were pET20b(+) (Novagen, Madison, WI) vectors containing the wild-type TyrRS and mini-TyrRS, respectively. Synthetic oligonucleotides were purchased from Invitrogen Corporation (Carlsbad, CA). All proteins were expressed with a C-terminal His-tag to facilitate purification.
Protein production and endotoxin removal
Recombinant proteins were expressed in E.coli BL21-CodonPlus (DE3)-RIL cells (Stratagene). Cells were grown to an OD600 of 0.8, and induced for 3 hours with 1 mM isopropyl β-D-thiogalactopyranoside (Roche, Basel, Switzerland). Cells were pelleted and resuspended in Buffer A (20 mM Tris-HCl, pH 7.9, 30 mM imidizole, 500 mM NaCl). Following lysis by sonication, cell debris was separated by centrifugation at 74,000 x g for 30 min. The His-tagged proteins were purified by Ni-NTA affinity chromatography. Supernatant was loaded onto a Ni-NTA affinity column (Qiagen, Valencia, CA), washed with 100 mL buffer B (buffer A with 0.1% Triton-X114 (Sigma, St. Louis, MO)) and 150 mL buffer A. Proteins were eluted by a linear gradient of buffer A and buffer C (20 mM Tris-HCl, pH 7.9, 250 mM imidizole, 500 mM NaCl). Fractions containing >95% pure protein were pooled, concentrated and dialyzed into storage buffer (50% PBS pH 7.4, 50% glycerol, 2 mM DTT). Proteins used for small angle X-ray scattering analysis were further purified by mono Q and gel filtration chromatography. Protein concentration was determined by Bradford assay using the Bio-Rad Protein Assay reagent (Bio-Rad, Hercules, CA) with bovine serum albumin (Sigma, St. Louis, MO) as a standard. Endotoxin was removed using EndoTrap Red column (Lonza, Basel, Switzerland) from samples used for cell based assays and the angiogenesis assays, and endotoxin concentration was determined using Kinetic-QCL Limulus Amebocyte Lysate (LAL) assay (Lonza).
Protease digestion
Wild-type TyrRS and TyrRS-Y341A were mixed with plasmin or leukocyte elastase at a protein-to-protease ratio of about 32 μg per μg of plasmin, and 2500 μg per unit of the elastase. The mixtures were incubated at 20 °C for about 2 hours before the reactions were stopped by addition of formic acid to a final concentration of 0.1 %. The cleavage fragments in the mixtures were analyzed by MALDI TOF mass spectrometry and by N-terminal sequencing using Edman degradation.
Small angle X-ray scattering
Small angle X-ray scattering (SAXS) measurements were conducted for the wild-type TyrRS and Y341A TyrRS on beamline 4-2 at the Stanford Synchrotron Radiation Laboratory. X-ray scattering curves were measured for samples at various concentrations (2–20 mg/ml). The x-ray wavelength (λ) was 1.38 Å and the detector channel numbers were converted to the momentum transfer Q = 4π*sinθ/λ, where 2*θ is the scattering angle. The sample-to-distances of 2.5 m covered Q range 0.01–0.25 Å−1. The polycarbonate cell with mica windows was filled with a sample aliquot and held at 20 ºC throughout the measurement. A MarCCD165 detector was used throughout the data collection. A typical set of data collection consisted of 24 2-dimensional scattering images recorded in series for 10s each. A series of data were processed by MarParse [34] along with the matching buffer scattering data, typically recorded either immediately after or before the protein solution measurement, and scaled for the integrated beam intensity, azimuthally averaged, inspected for time-dependent changes which are usually caused by radiation damage, and subjected to statistical analysis and averaging. Higher statistical variations of the protein data (1.5 X) over the variation of the matching buffer data were allowed in the averaging. Any data frame that showed higher level of deviation with respect to the first protein scattering data frame beyond that level were rejected. The processed buffer scattering curves were subtracted from corresponding protein scattering curves after the above data processing.
The scaling of small- and high-angle data were performed by Primus [35], which was also used to compute Rg and I (Q=0) by Guinier plot in the Q*Rg range 0.45–1.3. The electron pair distance distribution function P(r) was obtained by the indirect Fourier transfer of the experimental data using GNOM [36].
BrdU proliferation assay
Bovine aortic endothelia cells (BAECs) (Lonza) were plated onto cover slips pre-coated with matrigel (BD Biosciences, Bedford, MA) in EGM-MV (Lonza) and grown until approximately 75% confluent in a humidified atmosphere with 5% CO2 at 37° C. Subsequently cells were washed and placed in DMEM (Invitrogen) low serum media supplemented with 0.2% FBS and 1% of a penicillin, streptomycin and glutamine solution (Invitrogen) for 24 hours before treatment with protein samples at a concentration of 1 μM. Cells were incubated with the proteins for 18 hours and then pulsed with BrdU (Amersham Pharmacia, Piscataway, NJ) for 30 minutes and fixed in a 4% paraformaldehyde solution. Proliferating cells were visualized using a primary rat anti-BrdU antibody (Accurate Chemicals & Scientific Corp., Westbury, NY) followed by an anti-rat Alexa-594 secondary antibody (Invitrogen). Nuclei were stained using DAPI (Invitrogen). Cover slips were then dried and mounted for analysis by fluorescent microscopy. Cell proliferation was assessed by counting the number of BrdU positive cells in 5 randomly chosen high-power fields (HPFs) and expressing this as a mean percentage of the total in these fields.
Endothelial cell migration assay
Human umbilical vein endothelial cells (HUVECs) (Lonza) were plated at a density of about 3 x 105 cells/well of a 6-well plate in EGM media (Lonza) containing 10% fetal bovine serum (FBS) and grown to confluent monolayers. Cells were starved in media without FBS for 16 hours and wounded across the well with a pipette tip. The wounded monolayers were washed twice with serum-free media to remove cell debris and cells allowed to migrate in the presence of media alone (negative control) and media containing TyrRS variants or VEGF (positive control). Images of the wound were taken at 0 and 6 hours post wounding and analyzed using ImageJ (National Institutes of Health, Bethesda, Maryland). The percentage of area closed at 6 hour with respect to the 0 hour area was calculated and used to calculate the fold migration for each sample over control.
Mononuclear cell migration assay
Human mononuclear cells were prepared from whole blood of normal healthy volunteers. Blood was centrifuged (700 × g) over Histopaque 1077 and 1119 (Sigma). The mononuclear cells so obtained were washed twice with PBS. The procedure was repeated twice and the cells resuspended in RPMI 1640 (ATCC, Manassas, VA) containing heat-inactivated fetal bovine serum (0.5%, Invitrogen) at a concentration of 2 × 106 cells/ml.
Cell migration was performed in a microchemotaxis chamber ChemoTX™ (Neuro Probe, Gaithersburg, MD) containing polycarbonate filters (5-μm pores) with polyvinylpyrrolidone (PVP) (Neuro Probe). A 50 μl suspension of cells in RPMI 1640 medium containing heat-inactivated fetal bovine serum (0.5%), was added/well to the upper chamber. Different concentrations (0.1–100 nM) of various TyrRS constructs and EMAPII (Peprotech, Rocky Hill, NJ) in RPMI 1640 (0.5% FBS) were added to the lower chamber, and cells allowed to migrate for 3 hours at 37 °C in a 5% CO2 incubator. After incubation, nonmigrating cells were removed by washing the membrane with PBS, and the membrane was fixed in methanol. Migrated cells were visualized by staining the membrane using Diff-Quik stain set (VWR International, West Chester, PA). Samples were run in triplicate and cells were counted in 5 high-power fields (HPFs) for each sample.
Murine matrigel angiogenesis assay
Athymic WEHI mice were subcutaneously implanted with 400 μL of growth-factor-depleted Matrigel (Becton Dickinson, Franklin Lakes, NJ) supplemented with PBS, 20 nM VEGF or 250 nM TyrRS, mini-TyrRS, Y341A TyrRS or Y341A mini-TyrRS. Five days later, the mice were injected intravenously with fluorescein-labeled endothelial binding lectin Griffonia (Bandeiraea) Simplicifolia I, isolectin B4 (GSL-B4) (Vector Laboratories, Burlingame, CA). Matrigel plugs were resected and homogenized in radioimmuno-precipitation (RIPA) buffer (10 mM sodium phosphate, pH 7.4, 150 mM sodium chloride, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate). Following homogenization, the fluorescein content of each plug was quantified by spectrophotometric analysis.
CAM angiogenesis assay
Chick chorioallantoic membrane (CAM) angiogenesis assays were performed at Innovascreen Inc. (New Glasgow, Canada) by the following procedure. Fertilized complement fixation for avian leukosis virus (COFAL)-negative eggs were incubated for 3.5 days at 38 °C / 60% humidity. Eggs were then opened and the embryos were transferred into sterile plastic weigh boats. The embryos were covered and incubated at 37.5 °C / 90% humidity. After five days, collagen/mesh implants containing 30 μl of PBS, VEGF / bFGF (0.15 / 0.50 μg) or TyrRS polypeptides (1 μM) were placed onto the CAM membrane of the embryos, and incubated for an additional 72 hours. The upper mesh layers of the implants were examined under a stereomicroscope and scored for the proportion of “boxes” (i.e., three dimensional regions defined by the mesh fibers), which contain a blood vessel relative to the total number of boxes.
Acknowledgments
This work was supported by grants GM 15539 and CA 92577 from the National Institutes of Health and by a grant from the National Foundation for Cancer Research. SAXS experiments were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 2.Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, Muravina TI, Serkov SV, Uziel G, Bugiani M, Schiffmann R, Krageloh-Mann I, Smeitink JA, Florentz C, Van Coster R, Pronk JC, van der Knaap MS. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 3.Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D'Hooghe M, Van Gerwen V, Callaerts P, Van Den Bosch L, Timmermans JP, Robberecht W, Gettemans J, Thevelein JM, De Jonghe P, Kremensky I, Timmerman V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 4.Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonellis A, Lee-Lin SQ, Wasterlain A, Leo P, Quezado M, Goldfarb LG, Myung K, Burgess S, Fischbeck KH, Green ED. Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. J Neurosci. 2006;26:10397–10406. doi: 10.1523/JNEUROSCI.1671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Bo R, Locatelli F, Corti S, Scarlato M, Ghezzi S, Prelle A, Fagiolari G, Moggio M, Carpo M, Bresolin N, Comi GP. Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology. 2006;66:752–754. doi: 10.1212/01.wnl.0000201275.18875.ac. [DOI] [PubMed] [Google Scholar]
- 7.Dubourg O, Azzedine H, Yaou RB, Pouget J, Barois A, Meininger V, Bouteiller D, Ruberg M, Brice A, LeGuern E. The G526R glycyl-tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology. 2006;66:1721–1726. doi: 10.1212/01.wnl.0000218304.02715.04. [DOI] [PubMed] [Google Scholar]
- 8.James PA, Cader MZ, Muntoni F, Childs AM, Crow YJ, Talbot K. Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology. 2006;67:1710–1712. doi: 10.1212/01.wnl.0000242619.52335.bc. [DOI] [PubMed] [Google Scholar]
- 9.Sivakumar K, Kyriakides T, Puls I, Nicholson GA, Funalot B, Antonellis A, Sambuughin N, Christodoulou K, Beggs JL, Zamba-Papanicolaou E, Ionasescu V, Dalakas MC, Green ED, Fischbeck KH, Goldfarb LG. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain. 2005;128:2304–2314. doi: 10.1093/brain/awh590. [DOI] [PubMed] [Google Scholar]
- 10.Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 11.Nangle LA, Zhang W, Xie W, Yang XL, Schimmel P. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc Natl Acad Sci USA. 2007;104:11239–11244. doi: 10.1073/pnas.0705055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinis SA, Plateau P, Cavarelli J, Florentz C. Aminoacyl-tRNA synthetases: a family of expanding functions. Mittelwihr, France, October 10–15, 1999 EMBO J. 1999;18:4591–4596. doi: 10.1093/emboj/18.17.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SG, Ewalt KL, Kim S. Functional expansion of aminoacyl-tRNA synthetases and their interacting factors: new perspectives on housekeepers. Trends Biochem Sci. 2005;30:569–574. doi: 10.1016/j.tibs.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Berger AC, Tang G, Alexander HR, Libutti SK. Endothelial monocyte-activating polypeptide II, a tumor-derived cytokine that plays an important role in inflammation, apoptosis, and angiogenesis. J Immunother (1997) 2000;23:519–527. doi: 10.1097/00002371-200009000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Lee YS, Han JM, Kang T, Park YI, Kim HM, Kim S. Antitumor activity of the novel human cytokine AIMP1 in an in vivo tumor model. Mol Cells. 2006;21:213–217. [PubMed] [Google Scholar]
- 16.Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science. 1999;284:147–151. doi: 10.1126/science.284.5411.147. [DOI] [PubMed] [Google Scholar]
- 17.Wakasugi K, Slike BM, Hood J, Ewalt KL, Cheresh DA, Schimmel P. Induction of angiogenesis by a fragment of human tyrosyl-tRNA synthetase. J Biol Chem. 2002;277:20124–20126. doi: 10.1074/jbc.C200126200. [DOI] [PubMed] [Google Scholar]
- 18.Yang XL, Schimmel P, Ewalt KL. Relationship of two human tRNA synthetases used in cell signaling. Trends Biochem Sci. 2004;29:250–256. doi: 10.1016/j.tibs.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Otani A, Slike BM, Dorrell MI, Hood J, Kinder K, Ewalt KL, Cheresh D, Schimmel P, Friedlander M. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc Natl Acad Sci USA. 2002;99:178–183. doi: 10.1073/pnas.012601899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wakasugi K, Slike BM, Hood J, Otani A, Ewalt KL, Friedlander M, Cheresh DA, Schimmel P. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc Natl Acad Sci USA. 2002;99:173–177. doi: 10.1073/pnas.012602099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webster T, Tsai H, Kula M, Mackie GA, Schimmel P. Specific sequence homology and three-dimensional structure of an aminoacyl transfer RNA synthetase. Science. 1984;226:1315–1317. doi: 10.1126/science.6390679. [DOI] [PubMed] [Google Scholar]
- 22.Kleeman TA, Wei D, Simpson KL, First EA. Human tyrosyl-tRNA synthetase shares amino acid sequence homology with a putative cytokine. J Biol Chem. 1997;272:14420–14425. doi: 10.1074/jbc.272.22.14420. [DOI] [PubMed] [Google Scholar]
- 23.Wakasugi K, Schimmel P. Highly differentiated motifs responsible for two cytokine activities of a split human tRNA synthetase. J Biol Chem. 1999;274:23155–23159. doi: 10.1074/jbc.274.33.23155. [DOI] [PubMed] [Google Scholar]
- 24.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 25.Yang XL, Skene RJ, McRee DE, Schimmel P. Crystal structure of a human aminoacyl-tRNA synthetase cytokine. Proc Natl Acad Sci USA. 2002;99:15369–15374. doi: 10.1073/pnas.242611799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang XL, Liu J, Skene RJ, McRee DE, Schimmel P. Crystal structure of an EMAP-II-like cytokine released from a human tRNA cytokine. Helvetica Chimica Acta. 2003;86:1246–1257. [Google Scholar]
- 27.Schimmel PR, Soll D. Aminoacyl-tRNA synthetases: general features and recognition of transfer RNAs. Annu Rev Biochem. 1979;48:601–648. doi: 10.1146/annurev.bi.48.070179.003125. [DOI] [PubMed] [Google Scholar]
- 28.Hu GF. Limited proteolysis of angiogenin by elastase is regulated by plasminogen. J Protein Chem. 1997;16:669–679. doi: 10.1023/a:1026302419881. [DOI] [PubMed] [Google Scholar]
- 29.Pepper MS. Extracellular proteolysis and angiogenesis. Thromb Haemost. 2001;86:346–355. [PubMed] [Google Scholar]
- 30.Xie W, Nangle LA, Zhang W, Schimmel P, Yang XL. Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase. Proc Natl Acad Sci USA. 2007;104:9976–9981. doi: 10.1073/pnas.0703908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tzima E, Reader JS, Irani-Tehrani M, Ewalt KL, Schwartz MA, Schimmel P. VE-cadherin links tRNA synthetase cytokine to anti-angiogenic function. J Biol Chem. 2005;280:2405–2408. doi: 10.1074/jbc.C400431200. [DOI] [PubMed] [Google Scholar]
- 32.Dorrell MI, Aguilar E, Scheppke L, Barnett FH, Friedlander M. Combination angiostatic therapy completely inhibits ocular and tumor angiogenesis. Proc Natl Acad Sci USA. 2007;104:967–972. doi: 10.1073/pnas.0607542104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otani A, Kinder K, Ewalt K, Otero FJ, Schimmel P, Friedlander M. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat Medicine. 2002;8:1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 34.Smolsky IL, Liu P, Niebuhr M, Ito K, Weiss TM, Tsuruta H. Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J Appl Cryst. 2007;40:s453–s458. [Google Scholar]
- 35.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 36.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]