

Abstract
We report the computer-aided design, chemical synthesis, and biological evaluation of a novel family of δ opioid receptor (DOR) antagonists containing a 1,2,4-triazole core structure that are structurally distinct from other known opioid receptor active ligands. Among those δ antagonists sharing this core structure, 8 exhibited strong binding affinity (Ki = 50 nM) for the DOR and appreciable selectivity for δ over μ and opioid receptors (δ/μ = 80; δ/κ > 200).
Opioid analgesics are the mainstay for treatment of moderate to severe pain. Research on opioids and their receptors has remained active over the past decade.1 Three opioid receptor subtypes, designated as δ, κ, and μ, have been identified in the central nervous system (CNS) and periphery2,3 and are products of three distinct and extensively studied genes. Recent evidence suggests that subtype-selective opioid receptor agonists and antagonists offer great potential as therapeutic agents devoid of the numerous adverse side effects (e.g., respiratory depression, physical dependence, and gastrointestinal effects) associated with morphine.4 In particular, δ-selective antagonists have been shown to modulate the development of tolerance5,6 and dependence on μ agonists such as morphine,7 to offset the behavioral effects of drugs of abuse such as cocaine,8 and to elicit favorable immunomodulatory9 and emotional effects.10 On the other hand, δ-selective agonists have been shown to elicit the prototypical analgesic effects of clinically available opioids.4 They may also provide unique benefits as cardioprotective and neuroprotective agents11 and as treatments for depression and anxiety.12,13
In view of their broad range of pharmacological applications, the δ-selective opioids have attracted interest in our laboratory and elsewhere. Given the paucity of high-quality X-ray crystal structure data for GPCRs such as the opioid receptor, our drug design strategy has relied on ligand-based molecular modeling approaches. An additional component of our drug discovery paradigm is the proprietary Shape Signatures computational tool that provides unique capabilities for scaffold hopping in the search for new lead compounds.14,15
A three-point pharmacophore was extracted by overlaying a series of high-affinity opioid receptor ligands including the δ-antagonist naltrindole.16 This pharmacophore model (Figure 1, gray) comprised the basic nitrogen atom, the centroid of the phenol ring (A), and the centroid of the hydrophobic ring (B). Virtual screening of an in-house database of ∼1.2 million commercially available small-molecule chemicals was conducted to identify structures matching this three-point pharmacophore. Additional molecular models were developed for a distinct series of DOR-selective agonists17 and antagonists18 to demonstrate the structural requirement for δ selectivity. Promising chemical entities were then subjected to filters using an expanded Lipinski rule of five19 hierarchical scheme. The substituted 1,2,4-triazoles (Figure 1) emerged from this scheme as an interesting core structural framework for our DOR active agents. In selecting appropriate substitution patterns for the 1,2,4-triazole ring to confer δ binding affinity and selectivity, we exploited the “message–address” concept20,21 associated with classical morphine-like opioids. For instance, a sterically bulky group (e.g., tert-butyl) was attached to the B aryl group to mimic the δ “address” in our 1,2,4-triazoles. Several di- and trisubstituted 1,2,4-triazoles (Table 1) were selected for chemical synthesis and biological evaluation. Structural alignment of naltrindole and 8 in the conformation adopted in its X-ray crystal structure reveals good overlap between the tert-butyl group of 8 and the δ “address” of naltrindole (Figure 2, Supporting Information).
Figure 1.
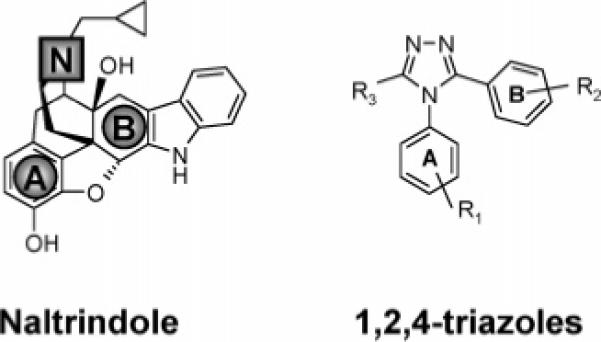
Comparison of the structures of naltrindole and the present 1,2,4-triazoles.
Table 1.
Structures and Opioid Receptor Binding Affinities for Substituted 1,2,4-Triazoles
| % inhibitiona |
Ki (nM)b |
selectivity ratio |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | δ | μ | κ | δ | μ | κ | δ/μ | δ/κ |
| 1 | 3-OCH3 | 3-tert-butyl | H | 31 | 11 | 5 | >10000 | >10000 | >10000 | na | |
| 2 | 3-OH | 3-tert-butyl | H | 91 | 78 | 66 | 230 | 850 | 1500 | 3.70 | 6.52 |
| 3 | 3-OCH3 | 4-tert-butyl | H | 8 | 9 | 8 | >10000 | >10000 | >10000 | na | |
| 4 | 3-OH | 4-tert-butyl | H | 54 | 16 | 16 | ∼10000 | >10000 | >10000 | na | |
| 5 | 3-OH | 3-phenyl | H | 84 | 84 | 42 | 140 | 1000 | >10000 | 7.14 | >71.4 |
| 6 | 3-OH | 4-phenyl | H | 75 | 60 | 46 | 1500 | >10000 | >10000 | >6.66 | >6.66 |
| 7 | 3-OH | 3,4-(CH=CH)2 | H | 68 | 48 | 27 | 2100 | >10000 | >10000 | >4.76 | >4.76 |
| 8 | 3-OH | 4-tert-butyl | N(CH3)2 | 94 | 32 | 6 | 50 | 4000 | >10000 | 80 | >200 |
| 9 | 3-OCH3 | 4-tert-butyl | N(CH3)2 | 28 | 50 | 3 | >10000 | >10000 | >10000 | na | |
| 10 | 3-OH | 3-tert-butyl | N(CH3)2 | 76 | 19 | 6 | 1050 | >10000 | >10000 | >9.5 | >9.5 |
| 11 | 3-OH | 3-phenyl | N(CH3)2 | 86 | 23 | 8 | 150 | >10000 | >10000 | >66.6 | >66.6 |
| 12 | 3-OH | 4-phenyl | N(CH3)2 | 82 | 15 | 11 | 130 | >10000 | >10000 | >76.9 | >76.9 |
| 13 | 3-OH | 3,4-(CH=CH)2 | N(CH3)2 | 68 | 48 | 27 | 480 | >10000 | >10000 | >20.8 | >20.8 |
| 14 | 4-OH | 4-tert-butyl | N(CH3)2 | 27 | 8 | 21 | >10000 | >10000 | >10000 | na | |
| 15 | 3-OH | 4-tert-butyl | N(CH2CH2)2NCH3 | 18 | 5 | 12 | >10000 | >10000 | >10000 | na | |
| 16 | 3-OH | 3-tert-butyl | CH2N(CH3)2 | 65 | 27 | 10 | 2600 | >10000 | >10000 | >3.86 | >3.86 |
| 17 | 3-OH | 4-tert-butyl | CH2N(CH3)2 | 88 | 20 | 9 | 460 | >10000 | >10000 | >21.6 | >21.6 |
| 18 | 3-OH | 4-tert-butyl | (CH2)2N(CH3)2 | 90 | 23 | 19 | 900 | >10000 | >10000 | >11.1 | >11.1 |
Compounds, initially screened at 10 μM, are expressed as percentage inhibition of the reference compound which is normalized to 100%. (−)-[9−3H]bremazocine was used as the radiolabeled ligand.
Inhibitory effect to (−)-[9−3H]bremazocine on membranes isolated from HEK 293 cells stably transfected with mouse (δ and μ) or rat (κ) opioid receptors. Ki values are the average of two to three independent experiments.
Figure 2.

Up-regulation results of compounds 2 and 8.
Three separate reaction schemes were developed for the synthesis of the 1,2,4-triazoles (Scheme 1) with thioamides as key intermediates. In most cases, thioamides were synthesized from the corresponding amides.22,23 For 1, 2, 10, and 16, a coupling reaction of arylmagnesium reagents with isothiocyanates was conducted to synthesize the thioamides.24,25 Amidrazones could be efficiently prepared by reaction of thioamides with excess hydrazine at room temperature. Cyclization of amidrazones with different reagents led to products with methoxyl groups at the R2 positions. 1−7 were obtained using trimethyl orthoformate as the cyclization reagent,26 while 8−15 involved cyclization of amidrazones with phosgeninium salts (Viehe's salts), which were easily synthesized from the corresponding amines.27 Compounds 16 and 17 were synthesized from 1 and 3, respectively, through reaction with Eschenmoser's salt.28,29 Compound 18 was prepared directly by condensation of amidrazone with 3-N,N-dimethylaminopropionic acid hydrochloride in the presence of dicyclohexyldiimide (DCC) (Scheme 1). For most of the products, the final step of cleaving methoxyl groups at R2 was completed easily by reaction with BBr3 in dichloromethane. Where hydrolysis of the methoxyl group was incomplete using the above BBr3 procedure, excess NaSH was added to achieve ether cleavage in acceptable yields.
Scheme 1.

General Synthesis of Substituted 1,2,4-Triazolesa
Initially, 1−4 were synthesized to evaluate the feasibility of our approach (Table 1). Radioligand binding assays revealed that 2 binds to all three opioid receptors with Ki values of 230 (δ), 850 (μ), and 1500 (κ) nM, respectively. As anticipated, it exhibited some subtype selectivity for the δ over μ and κ opioid receptors. Structural analogues (5−18) were synthesized in order to increase the δ binding affinity and selectivity (Table 1). Several of the subject compounds (e.g., 5, 8, 11, 12) exhibited selectivity for the δ over μ and κ opioid receptors, which concurs with our initial design strategy to confer δ selectivity. The inhibitory activity was much greater at all three opioid receptors for compounds with R1 = OH (2 and 4) compared with R1 = OCH3 (1 and 3). In fact, the latter compounds showed very limited inhibitory activity for any of the opioid receptors even at 10 μM. Comparison of 8 and 14 indicates that the binding affinity for all three opioid receptor subtypes was virtually abolished when the hydroxyl substituent at R1 is moved from the meta to para position on the aromatic ring. Although this single example precludes making generalizations, the strong preference for the meta over para phenolic moiety is consistent with the familiar SAR of morphine-like opioids.30–33
For 1−7, R2 substitutions were preferred at the meta position over the para position (e.g., Ki(δ) = 230 nM for 2 vs ∼10 000 nM for 4). For compounds with R3 substitutions, namely 8−17, the opposite trend was observed in cases exhibiting an appreciable affinity difference (see 8 vs 10). Compound 8 (Ki(δ) = 50 nM), with R2 = p-tert-butyl and R3 = N(CH3)2, yielded the best results overall among this first generation of triazole-based opioid receptor active agents in terms of δ binding affinity and subtype selectivity. It is worth noting that introduction of groups more highly constrained than tert-butyl at R2 failed to increase binding affinity for the δ receptor. For example, the δ binding affinity was poorer for 11, 12 and 13 (Ki = 150, 130, and 480 nM, respectively) than for 8 (Ki = 50 nM).
The functional activity of our substituted 1,2,4-triazoles on the opioid receptors was determined by receptor up-regulation assays. Incubation of the δ opioid receptor with 2 and 8 produced a sharp increase in receptor expression, suggesting that the subject compounds are δ opioid antagonists (Figure 2). Interestingly, 8 exhibited >3-fold up-regulation of the δ opioid receptor in this assay. The pharmacological significance of this observation is currently under investigation in our laboratory.
In fact, a N,N-dimethylamino group at R3 did produce a sharp increase in binding affinity to the δ receptor. Compare, for example, the Ki(δ) of 8 (50 nM, R3 = N(CH3)2) with its simple homologue 4 (∼10 000 nM, R3 = H). One might reasonably attribute the greater activity of 8 over 4 to the strong basicity of the N atom at R3. Nevertheless, 8 is only slightly more basic than 4 (pKa(pred) = 3.36 vs 2.18).17 Ab initio quantum mechanical calculations on 8 at the HF/6−31G** level of theory, in vacuum and aqueous (implicit solvation) conditions, indicated that the most basic atom is not the N in R3 = N(CH3)2 (i.e., Nsub) but rather N1 or N2 in the triazole ring (Table 2). Among the four N atoms in 8, the rank of basicity is N1 ∼ N2 > Nsub > N4. These results suggest that Nsub is less basic than the triazole-ring atoms N1 and N2, although it should be restated that all of the N atoms in 8 are weakly basic. It is evident that the basicity of the N(CH3)2 group is mitigated by its strong conjugation with the triazole ring. One might suspect that disrupting this conjugation by extension of the substituent group would afford a basic N atom and thereby enhance binding affinity. However, 17 (R3 = CH2N(CH3)2) and 18 (R3 = (CH2)2N(CH3)2) showed >6-fold decrease in binding affinity to the DOR compared with 8.
Table 2.
Relative Energies of Protonationa Obtained from HF/6−31G** ab Initio Calculations on Compound 8 Assuming Vacuum and Aqueous Conditions
In units of kcal/mol.
In conclusion, we report here a novel family of δ-selective opioid receptor antagonists containing the 1,2,4-triazole core structure. The subject compounds are chemically and structurally distinct from the classical opioids such as morphine and other known small-molecule opioids (e.g., (+)-4-[(α)R)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC80)). Moreover, these compounds are synthetically accessible as pure compounds in high yield and, uncommon among opioids, lack chiral centers. Compound 8, the most active among this first generation of substituted 1,2,4-triazoles, exhibited strong binding affinity (Ki = 50 nM) and appreciable selectivity (selectivity ratio: δ/μ = 80; δ/κ > 200) for the δ opioid receptor. The weak basicity of 8 (pKa(pred) = 3.36) favors the neutral (unprotonated) form under physiological conditions (pH 7.4). Virtually all known opioids, whether agonists or antagonists, contain at least one basic N atom. The only exception to our knowledge is the agonist salvinorin A, a natural compound extracted from S. divinorum,34 and a series of cyclic peptides reported by Schiller et al.35 that act as δ and μ receptor antagonists. The present compounds thus represent the first nonpeptidic δ-selective opioid antagonists lacking a basic N atom.
Acknowledgment
Funding for this research was provided to W.J.W. by the Biotechnology Research & Development Corporation (Peoria, IL) and to R.D.H. by the National Institute on Drug Abuse (Grant DA09113). The authors also thank Dr. John Duchek of Tyco-Mallinckrodt, Inc. (St. Louis, MO) for fruitful discussions. Access to the computational facilities at the UMDNJ Informatics Institute, supported in part by the National Library of Medicine (Grant G08 LM6230−07), is also gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures for the synthesis of all new compounds, details on the molecular modeling and in vitro assays, and the X-ray crystal structure of 8 together with the crystallographic structural data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ananthan S, Khare NK, Saini SK, Seitz LE, Bartlett JL, Davis P, Dersch CM, Porreca F, Rothman RB, Bilsky EJ. Identification of opioid ligands possessing mixed micro agonist/δ antagonist activity among pyridomorphinans derived from naloxone, oxymorphone, and hydromorphone [correction of hydropmorphone]. J. Med. Chem. 2004;47:1400–1412. doi: 10.1021/jm030311v. [DOI] [PubMed] [Google Scholar]
- 2.Pert CB, Snyder SH. Opiate receptor: demonstration in nervous tissue. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- 3.Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- 4.Bilsky EJ, Calderon SN, Wang T, Bernstein RN, Davis P, Hruby VJ, McNutt RW, Rothman RB, Rice KC, Porreca F. SNC 80, a selective, nonpeptidic and systemically active opioid delta agonist. J. Pharmacol. Exp. Ther. 1995;273:359–366. [PubMed] [Google Scholar]
- 5.Kest B, Lee CE, McLemore GL, Inturrisi CE. An antisense oligodeoxynucleotide to the delta opioid receptor (DOR-1) inhibits morphine tolerance and acute dependence in mice. Brain Res. Bull. 1996;39:185–188. doi: 10.1016/0361-9230(95)02092-6. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, Pintar JE. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24:243–252. doi: 10.1016/s0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 7.Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991;258:299–303. [PubMed] [Google Scholar]
- 8.Reid LD, Hubbell CL, Glaccum MB, Bilsky EJ, Portoghese PS, Porreca F. Naltrindole, an opioid delta receptor antagonist, blocks cocaine-induced facilitation of responding for rewarding brain stimulation. Life Sci. 1993;52:PL67–PL71. doi: 10.1016/0024-3205(93)90084-g. [DOI] [PubMed] [Google Scholar]
- 9.House RV, Thomas PT, Kozak JT, Bhargava HN. Suppression of immune function by non-peptidic delta opioid receptor antagonists. Neurosci. Lett. 1995;198:119–122. doi: 10.1016/0304-3940(95)11983-4. [DOI] [PubMed] [Google Scholar]
- 10.Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gaveriaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat. Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- 11.Barry U, Zuo Z. Opioids: old drugs for potential new applications. Curr. Pharm. Des. 2005;11:1343–1350. doi: 10.2174/1381612053507459. [DOI] [PubMed] [Google Scholar]
- 12.Torregrossa MM, Isgor C, Folk JE, Rice KC, Watson SJ, Woods JH. The delta-opioid receptor agonist (+)BW373U86 regulates BDNF mRNA expression in rats. Neuropsychopharmacology. 2004;29:649–659. doi: 10.1038/sj.npp.1300345. [DOI] [PubMed] [Google Scholar]
- 13.Nieto MM, Guen SL, Kieffer BL, Roques BP, Noble F. Physiological control of emotion-related behaviors by endogenous enkephalins involves essentially the delta opioid receptors. Neuro-science. 2005;135:305–313. doi: 10.1016/j.neuroscience.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 14.Nagarajan K, Zauhar R, Welsh WJ. Enrichment of ligands for the serotonin receptor using the Shape Signatures approach. J. Chem. Inf. Model. 2005;45:49–57. doi: 10.1021/ci049746x. [DOI] [PubMed] [Google Scholar]
- 15.Zauhar RJ, Moyna G, Tian L, Li Z, Welsh WJ. Shape signatures: a new approach to computer-aided ligand- and receptor-based drug design. J. Med. Chem. 2003;46:5674–5690. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 16.Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- 17.Peng Y, Keenan SM, Zhang Q, Welsh WJ. 3D-QSAR comparative molecular field analysis on opioid receptor agonists SNC80 and its analogs. J. Mol. Graphics Modell. 2005;24:72–80. doi: 10.1016/j.jmgm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Peng Y, Keenan SM, Zhang Q, Kholodovych V, Welsh WJ. 3D-QSAR comparative molecular field analysis on opioid receptor antagonists: pooling data from different studies. J. Med. Chem. 2005;48:1620–1629. doi: 10.1021/jm049117e. [DOI] [PubMed] [Google Scholar]
- 19.Lipinski C. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 20.Portoghese PS. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci. 1989;10:230–235. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
- 21.Schwyzer R. ACTH: a short introductory review. Ann. N. Y. Acad. Sci. 1977;297:3–26. doi: 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- 22.Curphey TJ. Thionation with the reagent combination of phosphorus pentasulfide and hexamethyldisiloxane. J. Org. Chem. 2002;67:6461–6473. doi: 10.1021/jo0256742. [DOI] [PubMed] [Google Scholar]
- 23.Metzner P, Thuillier A. Sulfur Reagents in Organic Synthesis. Academic Press; New York: 1994. pp. 44–46. [Google Scholar]
- 24.Dohle W, Lindsay DM, Knochel P. Copper-mediated cross-coupling of functionalized arylmagnesium reagents with functionalized alkyl and benzylic halides. Org. Lett. 2001;3:2871–2873. doi: 10.1021/ol0163272. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Q, Peng Y, Welsh WJ. Efficient synthesis of N,N-dialkylamino-1,2,4-triazoles. Manuscript in preparation.
- 26.Larsen SD, DiPaolo BA. Traceless solid-phase synthesis of 1,2,4-triazoles using a novel amine resin. Org. Lett. 2001;3:3341–3344. doi: 10.1021/ol016578a. [DOI] [PubMed] [Google Scholar]
- 27.Morris J, Wishika DG, Fang YA. Novel synthesis of 2-aminochromones via phosgeniminium salts. J. Org. Chem. 1992;57:6502–6508. [Google Scholar]
- 28.Gall M, Kamdar BV, Lipton MF, Chidester CG, DuChamp DJ. Mannich reaction of heterocycles with dimethyl(methylen)-ammonium chloride: a high yield, one-step conversion of estazolam to adinazolam. J. Heterocycl. Chem. 1988;25:1649–1661. [Google Scholar]
- 29.Arend M, Westermann B, Risch N. Modern variants of the mannich reaction. Angew. Chem., Int. Ed. 1998;37:1044–1070. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 30.Greiner E, Spetea M, Krassnig R, Schullner F, Aceto M, Harris LS, Traynor JR, Woods JH, Coop A, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 18. N-Substituted 14-phenylpropyloxymorphinan-6-ones with unanticipated agonist properties: extending the scope of common structure–activity relationships. J. Med. Chem. 2003;46:1758–1763. doi: 10.1021/jm021118o. [DOI] [PubMed] [Google Scholar]
- 31.Coop A, Rothman RB, Dersch C, Partilla J, Porreca F, Davis P, Jacobson AE, Rice KC. δ Opioid affinity and selectivity of 4-hydroxy-3-methoxyindolomorphinan analogues related to naltrindole. J. Med. Chem. 1999;42:1673–1679. doi: 10.1021/jm9807003. [DOI] [PubMed] [Google Scholar]
- 32.Portoghese PS, Sultana M, Takemori AE. Design of peptidomimetic delta opioid receptor antagonists using the message–address concept. J. Med. Chem. 1990;33:1714–1720. doi: 10.1021/jm00168a028. [DOI] [PubMed] [Google Scholar]
- 33.Ullrich T, Dersch CM, Rothman RB, Jacobson AE, Rice KC. Derivatives of 17-(2-methylallyl)-substituted noroxymorphone: variation of the delta address and its effects on affinity and selectivity for the delta opioid receptor. Bioorg. Med. Chem. Lett. 2001;11:2883–2885. doi: 10.1016/s0960-894x(01)00580-7. [DOI] [PubMed] [Google Scholar]
- 34.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc. Natl. Acad. Sci. U S A. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiller PW, Berezowska I, Nguyen TM, Schmidt R, Lemieux C, Chung NN, Falcone-Hindley ML, Yao W, Liu J, Iwama S, Smith AB, 3rd, Hirschmann R. Novel ligands lacking a positive charge for the δ- and μ-opioid receptors. J. Med. Chem. 2000;43:551–559. doi: 10.1021/jm990461z. [DOI] [PubMed] [Google Scholar]