

Abstract
Crohn’s disease and ulcerative colitis are chronic remitting and relapsing inflammatory bowel diseases. We present a typical case of Crohn’s disease in a young woman and discuss potential treatment options. Crohn’s disease and ulcerative colitis likely result from interaction of multiple genetic and environmental risk and protective factors. Both are diseases ultimately caused by immune dysregulation. Medical therapy is with mesalamine compounds, corticosteroids, immunomodulators and/or biologics that target TNFα signaling or α4-integrin-mediated trafficking. Investigational agents include those targeted against other cytokines and costimulatory molecules or designed to promote immune regulation such as exposure to helminths which is a focus of this review.
Introduction
Case Presentation
A 30-year-old woman with a history of Crohn’s disease (CD) was admitted to the hospital with fever, severe crampy abdominal pain, and diarrhea. The patient was first diagnosed with CD at the age of 26, when she developed intermittent bouts of sharp abdominal pain, accompanied by fevers and diarrhea. Initial colonoscopic examination revealed apthous ulceration at the terminal ileum and around the ileocecal junction. The patient was treated initially with oral prednisone (0.5 mg/kg/day) to good effect. She remained in remission on azathioprine (2.5 mg/kg/day) maintenance therapy for approximately four years, at which point she experienced a recurrence of symptoms, including frequent episodes of abdominal pain, severe diarrhea, dehydration and weight loss. Colonoscopy studies revealed discrete patchy segments of ulceration and inflamed mucosa throughout the colon (Figure 1). A biopsy revealed ulceration and mixed (lymphocytic and neutrophilic) infiltration, and deep inflammation of the intestinal mucosa. Her symptoms showed moderate improvement with administration of high dose corticosteroids (solumedrol 60mg/day IV), however she failed attempts at tapering the dose. The patient underwent an induction course of infliximab (5 mg/kg at 0, 2, and 6 weeks) without significant improvement. Azathioprine was discontinued and she is currently on a trial of weekly low-dose methotrexate. She may be a candidate for alternative biologics, surgery, or investigational therapies.
Figure 1. Colonoscopic view of the transverse colon in health (Normal) and disease (Crohn’s).
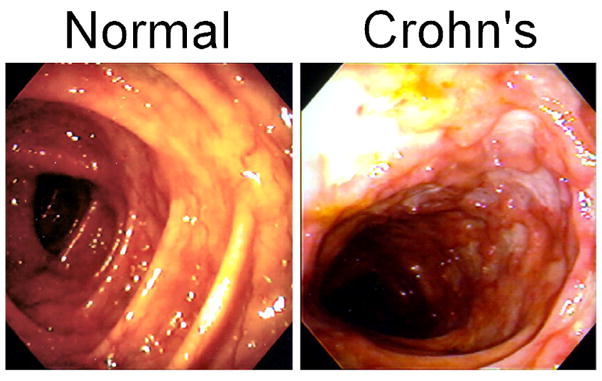
The normal colon shows regular haustra and a transparent intact mucosa. The colon from the patient with Crohn’s disease shows numerous deep ulcerations and areas of more normal appearing mucosa.
Discussion
Idiopathic Inflammatory Bowel Disease
Idiopathic Inflammatory Bowel Disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract that presents as either ulcerative colitis (UC) or Crohn’s disease (CD). UC and CD are both characterized by chronic remitting/ relapsing inflammation of the intestinal mucosa, often resulting in intermittent abdominal pain, fever, and diarrhea. Each disease also possesses distinguishing clinical, pathological, and endoscopic features [1]. Inflammation in UC typically involves the rectum, and extends continuously in a retrograde fashion. In severe cases, it can involve the entire colon. Endoscopic features include edema that obscures the normal vascular appearance, granular erythema and mucopurulent exudate, areas of extensive superficial ulceration, and the presence of inflammatory pseudopolyps. As illustrated in the case above, CD is characterized by sharply demarcated, non-contiguous inflammatory lesions that can become transmural (Figure 1). Non-caseating granulomas are occasionally present. While CD lesions may involve any segment of the gastrointestinal tract, they occur most commonly in the terminal ileum.
Genetics and Immunopathogenesis
Current evidence strongly suggests that IBD arises from a disruption of mucosal immune homeostasis in genetically susceptible individuals, resulting in altered processing of enteric antigens, pathogenic T cell activation, and chronic inflammation. The essential role of enteric microflora is supported by studies showing responsiveness of UC and CD to antibiotics and CD to fecal stream diversion, as well as experiments with induced mutant germ-free mice in which spontaneous colitis is dependent on reconstitution with normal luminal microflora [2;3]. Various innate, adaptive, and regulatory immune mechanisms have been implicated in IBD. These include dysregulated cellular stress responses, microbial recognition, autophagy, and processing of antigens by innate immune effector cells, pro-inflammatory CD4+ T cell polarization, and blunting of cytokine or T cell-driven tolerance [4]. Genetic factors contribute significantly to IBD pathogenesis. Approximately 5–10% of patients have at least one affected first degree relative and twin studies demonstrate a 50% concordance rate of CD among monozygotic twins, with lower rates (~18%) for UC [5–7]. In addition, specific genetic correlates have recently been identified in IBD, shedding new light on the underlying mechanisms involved and providing a useful framework for future research into the pathogenesis of these diseases. Nevertheless, like many complex-trait diseases, no single genetic factor determines development of IBD. Rather, a collection of environmental and genetic factors must coincide to produce the disease.
The first specific gene unequivocally associated with IBD was the NOD2/ CARD15 gene for CD [8;9]. Multiple genome-wide analyses have confirmed that specific NOD2 polymorphisms increase the risk for developing ileocolonic CD. NOD2 codes for a cytosolic microbial molecular pattern-recognition protein, which is part of a larger class of broadly expressed innate immune receptors (toll- and nod-like receptors). In Caucasian populations, compound heterozygotes and homozygotes for NOD2 mutations carry an odds ratio of 17.1 for CD, while simple heterozygosity carries an odds ratio of 2.4 [10]. The NOD2 gene product recognizes the bacterial protein muramyl dipeptide (MDP), regulates NFκB and MAP kinase signaling pathways [11]. The exact mechanism by which NOD2 variants contribute to disruption of intestinal immune homeostasis and precipitation of CD is not entirely clear. NOD2 variants may increase the risk of CD by causing hyporeactivity of certain innate responses, thereby forcing excess responses in other pathways disrupting homeostatic mechanisms. Alternatively, CD-related NOD2 mutations may impair negative regulation of TLR-mediated responses to enteric microflora, promoting excessive inflammation. Recently, antigen-presenting cells from mice carrying CD-associated NOD2 polymorphisms were found to exhibit resistance to the normal IRF4-mediated tolerizing effects of MDP-stimulation [12].
Genome wide association searches have found multiple other loci contributing to the risk of developing CD (Table 1). Currently, at least 30 loci show unequivocal association with CD risk, 2 show highly probable association, and another 8 show likely association [13]. Other searches have found additional loci [14]. Some CD loci also influence the risk for ulcerative colitis [15]. Identified gene products include NOD2, XBP1, ATG16L1, IRGM, and IL23R.
Table 1.
Genetic loci with identified or candidate genes associated with the risk of developing Crohn’s Disease
| Locus | Gene/Candidate | Function |
|---|---|---|
| Loci with confirmed or with highly probable contribution to CD risk | ||
| 16q12 | NOD2/CARD15 | MDP sensor (recognition of bacteria) |
| 2q37 | ATG16L1 | Autophagy |
| 5q33 | IRGM | Initiates autophagy of intracellular bacteria |
| 22q12 | XBP1 | ER stress response |
| 18p11 | PTPN2 | Cell signaling (tyrosine kinase), growth factor stimulation |
| 12q12 | MUC19, LRRK2 | Mucus protein, Unknown (Parkinson’s disease, autophagy) |
| 5p13 | PTGER4 (EP4) | Prostaglandin receptor |
| 9q32 | TNFSF15 | Induces endothelial cell apoptosis |
| 10q21 | ZNF365 | Unknown (zinc-finger protein) |
| 1p13 | PTPN22 | Cell signaling (tyrosine kinase) associates with CBL |
| 1q23 | ITLN1 | Galactose-binding lectin (recognition of bacteria) |
| 6p22 | CDKAL1 | Unknown (regulation of a cyclin-dependent kinase) |
| 6q27 | CCR6 | Chemokine receptor |
| 9p24 | JAK2 | Cell signaling (tyrosine kinase), cytokine stimulation |
| 11q13 | C11orf30 | Unknown (oncogene) |
| 17q21 | ORMDL3 | Unknown (also associated with asthma risk) |
| 17q21 | STAT3 | Cell signaling, cytokine stimulation |
| 21q22 | ICOSLG | ICOS ligand (costimulation) |
| Loci with confirmed or with highly probable contribution to CD and UC risk | ||
| 1p31 | IL23R | IL23 receptor |
| 5q33 | IL12B (IL12p40) | Component of IL12 and IL23 |
| 6p21 | MHC genes | Epitope selection |
| 10q24 | Nkx2-3 | Regulates epithelial cell and lymphocyte development |
| 3p21 | MST1 | Cytokine, macrophage stimulatory protein |
| Loci with likely contribution to CD risk | ||
| 2p23 | GCKR | Cell signaling (e.g. Wnt signaling) |
| 2p16 | PUS10 | Unknown (tRNA pseudouridine synthesis) |
| 17q12 | CCL2, CCL7 | C-C chemokines, macrophage recruitment |
| 6p25 | LYRM4 | Unknown (protein folding) |
| 6p25 | SLC22A23 | Organic ion transporter |
| 2q11 | IL18RAP | IL18 receptor component |
Crohn’s disease and UC are organ-specific inflammatory diseases. NOD2 is expressed by intestinal Paneth cells which produce bacteriocidal α-defensins. NOD2 variants confer risk only for the subtypes of Crohn’s disease that involve the ileum where Paneth cells are numerous. In addition to the NOD2 effects discussed above, impairment of Paneth cell function may explain the organ-specific distribution of inflammation associated with NOD2 variants. Intestinal epithelial cells are components of the innate immune system. Recently, variations in the gene for XBP1, a transcription factor that directs endoplasmic-reticulum (ER) stress responses, were found to probably contribute to the risk of CD [16]. Intestinal epithelial cells are highly secretory and are susceptible to ER stress. Mice that lack XBP1 specifically in intestinal epithelial cells develop enteritis, lack Paneth cells, zand have fewer goblet cells [16]. This suggests that disturbed epithelial cell function can contribute to CD risk by impairing innate defenses.
Further evidence implicating innate immune mechanisms in CD is the identification of autophagy-related gene ATG16L1 as a susceptibility variant for CD [17]. ATG16L1 codes for part of a multimeric protein complex that is involved in degradation of cytoplasmic material. Autophagy is both an important cellular homeostatic function and an evolutionarily conserved innate defense mechanism against invading viruses and intracellular bacteria or parasites [18]. The exact role of autophagosomal mutations in CD pathogenesis is not yet clear, but initial evidence from experiments using S. typhimurium and intestinal epithelial cells have shown that altered gene products from ATG16L1 impair pathogen clearance and elimination of intracellular bacteria [19]. IRGM is another autophagy gene product involved in the isolation and degradation of intracellular bacteria [20] that is associated with CD risk. These findings highlight the role of innate mechanisms in mucosal immune homeostasis. Interestingly, recent studies using murine macrophages have revealed considerable overlap between TLR-signaling pathways in phagocytosis and autophagy, and that autophagy is itself specifically inducible with LPS via a TLR4-mediated pathway [21;22]. In addition to it’s innate immune functions, autophagic processing helps direct cytosolic antigens toward MHC class II presentation [23] and T helper cell responses.
Classically, the inflammatory profiles of CD and UC differ in that CD is characterized by Th1-mediated inflammation while UC displays Th2-like pathology. Early evidence from mouse models of CD revealed IL-12 to be key a mediator of Th1-directed mucosal inflammation [24]. Ablation of IL-12 signaling with gene deletion or targeted antibodies improved intestinal inflammation in mice [25]. However, the functional role of IL-12 in IBD pathogenesis has been reconsidered in light of recent findings from functional and genome-wide analyses that implicate IL23 and its receptor gene (IL23R) in CD [26]. IL-23 is a pro-inflammatory heterodimeric cytokine with a unique p19 subunit and a p40 subunit shared in common with IL-12. IL-23 participates in multiple inflammatory pathways [27]. In particular, IL-23 has been demonstrated to upregulate and maintain (but not induce) expression of IL-17 by a distinct subset of pro-inflammatory CD4+ Th17 cells [28]. Evidence from functional studies in vitro and in vivo have shown that IL-17 induces expression of multiple pro-inflammatory chemokines and matrix metalloproteins and is negatively regulated by Th1 and Th2 cytokines [29]. Elevated levels of IL-17 expression have been demonstrated in serum and intestinal mucosa of patients with active CD and UC [30].
Many of the proinflammatory processes previously ascribed to IL-12 may in fact be attributable to IL-23. This suspicion has been born out in a number of studies demonstrating an essential and independent role for the IL-23/IL-17 cytokine axis in models of colitis, encephalitis, and arthritis [31–33]. Experiments using murine models of colitis show that selective targeting of the IL-23p19 subunit with monoclonal antibodies attenuates intestinal inflammation and inhibits spontaneous colitis in the context of IL-10 deficiency. In contrast, targeted depletion of IL-12p35 has little effect on development of innate or T cell-mediated intestinal inflammation [31;34] . Mouse models suggested that antibodies specific to IL-23 and IL-12p40 could be effective in the treatment of intestinal inflammation [35]. This finding has been translated into clinical trials of monoclonal anti-IL-12p40 antibody in mild to moderate CD [36;37].
Epidemiology
Despite the promising advances in our understanding of the genetic basis of IBD, the polymorphisms identified thus far only account for 10–20% of the overall disease risk in CD [13]. The genetic contribution is less for UC. This suggests that multiple genetic influences and non-genetic contributions have yet to be identified. The epidemiology of IBD strongly suggests an environmental contribution to disease expression. Approximately 1–2 million people have CD or UC in the US and Canada [38]. Multiple studies have demonstrated lower rates of UC after appendectomy or in people that smoke, and a positive correlation between smoking and CD [39–41]. Thus, life events alter disease risk.
The highest historical rates of IBD are observed in industrialized countries of North America, the UK, and Europe. Although the disease is considered rare in tropical regions of the world, rising rates of IBD have been documented in India and East Asia in recent decades [42;43]. Moreover, first and second generation immigrants coming from areas of low incidence to areas of high incidence acquire levels of risk similar or higher than that of their adopted country [44;45]. These findings suggest that the varying rates of IBD observed among racial and ethnic groups reflect shared environmental influences [46]. Indeed, it appears that as countries develop economically, the risk for IBD increases [38;47].
Economic development brings many changes, but one of the most dramatic is change in hygienic practice. In addition to basic plumbing and sewage treatment, development results in improved food processing, cement side walks, and access to a daunting array of cleaning products. Increasing hygiene limits exposure to previously ubiquitous infectious agents such as helminths. Helminths are parasitic worms that infect humans throughout the world, although exposure is concentrated in tropical areas and places with poor sanitation and high levels of poverty [48]. Helminths are the class of organisms most dramatically restricted by economic development. For example, prevalence of Trichuris trichiura infections in South Korean schoolchildren fell from 74.2% in 1969 to 0.02% in 2004 [49]. Concurrently, the prevalence of IBD increased substantially in Seoul, South Korea from 1986 through 2000 [43]. Epidemiological data suggest that the prevalence of helminth colonization is inversely related to economic and human development indices as well as rates of IBD, multiple sclerosis, asthma and allergic disorders [50–53]. These observations provide circumstantial evidence that helminthic infection could be protective against autoimmune inflammatory diseases, including IBD [54].
Helminths are complex organisms with long lifespans. They have developed strategies for evading attack by the immune system of their host, such as molecular mimicry, degradation of host immunoglobulins, blocking of pro-inflammatory cytokines, and induction of regulatory T- and B-cell responses [55]. A recent study of Cameroon school age children evaluating peripheral blood lymphocyte cytokine production found that levels of regulatory cytokines IL-10 and TGF-β correlated directly to intensity of infection with Ascaris lumbricoides and Trichuris trichiura and inversely with total immune reactivity [56]. It is likely that induction of immune hyporesponsiveness helps protect the parasite from eradication by the host. However, it is also probable that the host benefits from the helminth-induced immune regulation as it could prevent excessive and potentially pathogenic inflammation [53;54;57].
Specific immunomodulatory effects of helminthic infection have been characterized in mouse models of inflammation. Mice exposed to a range of helminths (e.g. Schistosoma. mansoni, Trichinella spiralis, Hymenolepis diminuta or Heligmosomoides polygyrus) are protected from colitis induced by rectal challenge with di- or trinitrobenzene sulfonic acid [58–61]. Colonization with these helminths inhibits the expression of the pro-inflammatory cytokines IFNγ, IL-12p40, and IL-17A, while augmenting expression of regulatory factors such as IL-10, TGFβ, and FoxP3+ T cells [58;60–63]. The induction of multiple regulatory circuits permits suppression of colitis even in the congenital absence of IL-10 [64]. However, congenital loss of STAT6 [58] or T cell TGFβ signaling (Ince, unpublished observation) or acute loss of IL-10 signaling [59] circumvents helminth-associated protection from colitis. Thus, at least some regulatory circuits are non-redundant. Specific critical regulatory pathways induced by helminth exposure may differ by helminth species, mouse strain, and inflammatory model. Helminths also protect from inflammation in murine models of type 1 diabetes, reactive airway disease, and multiple sclerosis [65–71]. Immunomodulatory effects similar to those found in colonized mice have been observed in people with helminth infections possibly suppressing ulcerative colitis [72], allergen reactivity [73], or multiple sclerosis [74].
If helminth exposure offsets a genetic predisposition toward IBD, then eradication of helminths would result increased IBD prevalence. Genomic surveys suggest that variation in IL12p40, IL23R, and CCR6 (expressed on Th17 cells [75]) contribute to the risk of CD (Table 1). In mice, helminth colonization suppresses mucosal IL12p40, IL23 and IL17 expression [63]. If this effect occurs in people, then helminth exposure could obscure any genetic tendency toward CD conferred by variation in these genes. A proper ER stress response, regulated in part by XBP1, is critical for epithelial cell innate immune function. Helminths promote IL-10 production and IL-10 modulates intestinal epithelial cell ER stress responses [76]. Elevated IL-10 could obscure a XBP1-mediated genetic tendency toward dysregulated ER stress response in a subset of patients with CD. Helminths induce alternatively activated macrophages [77] as does MST1 [78]. Helminthic induction of alternatively-activated macrophages could prevent effects due to variation in MST1. Further speculation about the effects of helminths on genetic predisposition toward IBD is possible but obviously all of this awaits verification.
Treatment
The patient in the case presented above received an initial course of oral prednisone, followed by long-term remission on azathoprine maintenance therapy. Corticosteroids and mesalamine are standard first line therapies for induction of remission in mild to moderate IBD. In one recent analysis 44% of patients with CD required corticosteroids for induction of remission, with 58% achieving complete remission at 4 weeks and 32% showing prolonged response at 1 year; 28% of cases became steroid-dependent and 40% required surgery [79]. Mesalamine medications differ with respect to mechanism and timing of 5-aminosalicylic acid release [80], and are more efficacious in UC than CD [81]. In addition to inhibiting cyclooxygenase and lipoxygenase, mesalamine likely functions as an agonist of peroxisome proliferator-activated receptor γ (PPAR-γ). PPAR-γ inhibits NFκB-dependent inflammatory pathways [82].
The effectiveness of cytotoxic immunosuppressant therapies with azathioprine, 6-mercaptopurine, or methotrexate for induction and maintenance of remission in CD is well established [83]. However, while they have been shown to reduce steroid-dependence and maintain remission it is not clear that they reduce the need for surgery in CD [84]. The thiopurines also are effective for UC. Thiopurines have a narrow therapeutic window and must be dosed properly to be effective. Azathioprine is metabolized to 6-mercaptopurine and then to 6-thioguanine nucleotides (6-TG), which interfere with proliferation and promote apoptosis of activated lymphocytes. Rare patients with thiopurine methyltransferase (TMPT) deficiency are at high risk for myelosuppression. While this risk has prompted recommendations to test for TMPT expression prior to starting treatment with a thiopurine, most patients that develop significant myelosuppression have normal TMPT genotypes. Thus, close monitoring is required regardless of TMPT test result [85]. Mesalamine can inhibit TMPT promoting myelotoxicity. Methotrexate is an immunomodulator that is efficacious in CD, but less so for UC [86;87]. It inhibits dihydrofolate reductase and thymidylate synthase to suppress lymphocyte proliferation. Since this patient has not yet been treated with methotrexate, this drug remains an option.
After four years of remission, the patient experienced an exacerbation of her CD, which became corticosteroid dependent and significantly more severe than her initial presentation. She subsequently underwent a course of infliximab therapy, with unsatisfactory results. Infliximab is a chimeric monoclonal antibody (mAb) to TNFa. Multiple randomized clinical trials have shown that infliximab or adalimumab, another anti-TNFα mAb, are effective therapeutic options for many patients whose symptoms no longer respond to maintenance therapy or become steroid-dependent despite use of immunomodulators [88]. However, use of this class of drugs is complicated by development of specific anti-immunoglobulin antibodies as well as increased susceptibility to opportunistic infection [89]. While anti-TNFα mAb show improved effectiveness relative to previous therapies, long-term response rates are limited to about 50% of those who initially respond to anti-TNFa agents [88]. Certolizumab pegol is a humanized Fab fragment of an anti-TNFa mAb, conjugated with polyethylene glycol. Early studies reported a 64% response rate to initial induction at 6 weeks, with 40% remaining in remission at 26 weeks [90].
Patients with CD that becomes refractory to multiple interventions may have “fixed” disease due to extensive scaring, strictures and/or fistulae. These anatomic changes do not reverse with medications and often require surgery. Historically, about 80% of patients with CD require surgery within 10 years of diagnosis. This patient’s response to high dose corticosteroids suggests that she has an ongoing inflammatory component to her disease. However, she should be evaluated for fixed anatomic change. She may become responsive to medications once a dominant stricture or fistula is resected. Patients with IBD can also become truly refractory to medications. In such cases, a treatment option for UC is total colectomy, which is “curative” for ongoing inflammation. CD can affect any part of the GI tract, so cannot be permanently cured by surgery.
Another therapy for refractory CD is natalizumab, a humanized antibody that blocks α4 integrin-mediated adhesion [91]. Sustained remission rates with natalizumab are significantly better than placebo (26% vs. 16%). Natalizumab may increase the risk for progressive multifocal leukoencephalopathy. To address this concern, patients taking natalizumab must be enrolled in the TOUCH prescribing program and not be given additional immunomodulators. Recently two patients on natalizumab monotherapy for multiple sclerosis were reported to have developed PML. With over 12,000 patients having been treated with natalizumab for at least one year, development of PML is a rare complication. However, the risk/benefit profile of natalizumab must be addressed for each patient.
Other potential treatment options under active investigation, and recently reviewed in [92], include monoclonal antibodies to cytokines IL-12/23p40 (ustekinumab [37] and ABT-874) and IL-6 (tocilizumab); antibodies to T cell surface receptors such as CD25 (basiliximab); fusion proteins that block costimulatory receptor function such as CD80/86 (abatacept); small molecules or antibodies to block chemokines or adhesion molecules; and mucosal delivery of recombinant human regulatory cytokines like IL-10 [92]. Although patients need referrals to specialized care centers to access these investigational therapies, it is exciting to have a number of treatments “in the pipeline”.
Helminths and helminth-associated molecules are under active investigation as potential therapy for CD and UC. Two clinical trials have suggested that colonization with Trichuris suis is effective for reducing disease activity in both CD and UC [93;94]. T. suis is a porcine whipworm closely related to the human whipworm T. trichuria. It is an attractive candidate as a therapeutic organism since it only briefly colonizes human hosts, does not multiply or migrate out of the intestine, and it is not known to cause disease. The results of the initial studies were promising. Of the 29 patients with CD who received T. suis ova (2500 ova, every 3 weeks for 24 weeks), 79% responded with significant reduction in symptoms [94]. In a double-blind placebo-controlled trial of 54 patients with active UC, 43.3% of the patients given T. suis ova (2500 ova every 2 weeks for 12 weeks) showed improvement, compared to 16.7% of those given placebo (p < 0.04) [93]. Additional studies in UC, CD, multiple sclerosis, and allergic rhinitis are underway or planned for the near future.
Conclusions
Novel genetic analyses and insights into the molecular mechanisms of intestinal inflammation have shed considerable light recently on the underlying processes that give rise to IBD. It is clear that multiple stimulatory and regulatory circuits are involved in maintaining the balance between host defense and pathogenic inflammation in the gut. Patients with IBD require medications that broadly suppress immune and inflammatory pathways. Currently, these medications have significant adverse effects. Patients, like the one in the vignette can loose responsiveness to a previously effective regimen. Novel medications targeted to specific pathways are being developed to provide alternatives and hopefully reduce complications. In addition, an intervention based on the epidemiology of IBD (helminthic therapy) is being clinically evaluated.
Acknowledgments
Supported by grants from the Department of Veterans Affairs Office of Research and Development, the Crohn’s and Colitis Foundation of America, and the Doris Duke Clinical Research Fellowship Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Podolsky DK. Inflammatory bowel disease. NEnglJ Med. 2002;347:417–29. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 2.Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol Rev. 2005;206:260–76. doi: 10.1111/j.0105-2896.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 3.Winslet MC, Allan A, Poxon V, Youngs D, Keighley MR. Faecal diversion for Crohn's colitis: a model to study the role of the faecal stream in the inflammatory process. Gut. 1994;35:236–42. doi: 10.1136/gut.35.2.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 5.Halfvarson J, Bodin L, Tysk C, Lindberg E, Jarnerot G. Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology. 2003;124:1767–73. doi: 10.1016/s0016-5085(03)00385-8. [DOI] [PubMed] [Google Scholar]
- 6.Jess T, Riis L, Jespersgaard C, et al. Disease concordance, zygosity, and NOD2/CARD15 status: follow-up of a population-based cohort of Danish twins with inflammatory bowel disease. AmJ Gastroenterol. 2005;100:2486–92. doi: 10.1111/j.1572-0241.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 7.Orholm M, Munkholm P, Langholz E, Nielsen OH, Sorensen IA, Binder V. Familial occurrence of inflammatory bowel disease. New England Journal of Medicine. 1991;324:84–8. doi: 10.1056/NEJM199101103240203. [DOI] [PubMed] [Google Scholar]
- 8.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 9.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 10.Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn's disease risk and phenotype in diverse populations: a metaanalysis. AmJ Gastroenterol. 2004;99:2393–404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 11.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. NatRev Immunol. 2008;8:458–66. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe T, Asano N, Murray PJ, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–59. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barmada MM, Brant SR, Nicolae DL, et al. A genome scan in 260 inflammatory bowel disease-affected relative pairs. InflammBowel Dis. 2004;10:513–20. doi: 10.1097/00054725-200409000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Fisher SA, Tremelling M, Anderson CA, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn's disease. Nat Genet. 2008;40:710–2. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–56. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 18.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–77. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–41. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 21.Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menendez-Benito V, Neefjes J. Autophagy in MHC class II presentation: sampling from within. Immunity. 2007;26:1–3. doi: 10.1016/j.immuni.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Becker C, Wirtz S, Neurath MF. Stepwise regulation of TH1 responses in autoimmunity: IL-12-related cytokines and their receptors. Inflamm Bowel Dis. 2005;11:755–64. doi: 10.1097/01.mib.0000172808.03877.4d. [DOI] [PubMed] [Google Scholar]
- 25.Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. Journal of Experimental Medicine. 1995;182:1281–90. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Langrish CL, McKenzie B, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. JClin Invest. 2006;116:1317–26. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. AnnuRevImmunol. 2007;25:821–52. 821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 29.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. AnnuRevImmunol. 2007;25:821–52. 821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 30.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. Journal of Clinical Investigation. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 33.Murphy CA, Langrish CL, Chen Y, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. JExp Med. 2003;198:1951–7. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uhlig HH, McKenzie BS, Hue S, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–18. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 35.Elson CO, Cong Y, Weaver CT, et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–70. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 36.Mannon PJ, Fuss IJ, Mayer L, et al. Anti-interleukin-12 antibody for active Crohn's disease. NEnglJ Med. 2004;351:2069–79. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 37.Sandborn WJ, Feagan BG, Fedorak RN, et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 2008;135:1130–41. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 38.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–17. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 39.Koutroubakis IE, Vlachonikolis IG, Kouroumalis EA. Role of appendicitis and appendectomy in the pathogenesis of ulcerative colitis: a critical review. InflammBowel Dis. 2002;8:277–86. doi: 10.1097/00054725-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 40.Calkins BM. A meta-analysis of the role of smoking in inflammatory bowel disease. Digestive Diseases & Sciences. 1989;34:1841–54. doi: 10.1007/BF01536701. [DOI] [PubMed] [Google Scholar]
- 41.Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clin Proc. 2006;81:1462–71. doi: 10.4065/81.11.1462. [DOI] [PubMed] [Google Scholar]
- 42.Sood A, Midha V. Epidemiology of inflammatory bowel disease in Asia. Indian J Gastroenterol. 2007;26:285–9. [PubMed] [Google Scholar]
- 43.Ouyang Q, Tandon R, Goh KL, Ooi CJ, Ogata H, Fiocchi C. The emergence of inflammatory bowel disease in the Asian Pacific region. CurrOpin Gastroenterol. 2005;21:408–13. [PubMed] [Google Scholar]
- 44.Carr I, Mayberry JF. The effects of migration on ulcerative colitis: a three-year prospective study among Europeans and first- and second- generation South Asians in Leicester (1991–1994) American Journal of Gastroenterology. 1999;94:2918–22. doi: 10.1111/j.1572-0241.1999.01438.x. [DOI] [PubMed] [Google Scholar]
- 45.Odes HS, Locker C, Neumann L, et al. Epidemiology of Crohn's disease in southern Israel. American Journal of Gastroenterology. 1994;89:1859–62. [PubMed] [Google Scholar]
- 46.Green C, Elliott L, Beaudoin C, Bernstein CN. A population-based ecologic study of inflammatory bowel disease: searching for etiologic clues. Am J Epidemiol. 2006;164:615–23. doi: 10.1093/aje/kwj260. [DOI] [PubMed] [Google Scholar]
- 47.Lakatos L, Mester G, Erdelyi Z, et al. Striking elevation in incidence and prevalence of inflammatory bowel disease in a province of western Hungary between 1977–2001. World Journal of Gastroenterology. 2004;10:404–9. doi: 10.3748/wjg.v10.i3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bethony J, Brooker S, Albonico M, et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet. 2006;367:1521–32. doi: 10.1016/S0140-6736(06)68653-4. [DOI] [PubMed] [Google Scholar]
- 49.Hong ST, Chai JY, Choi MH, Huh S, Rim HJ, Lee SH. A successful experience of soil-transmitted helminth control in the Republic of Korea. Korean J Parasitol. 2006;44:177–85. doi: 10.3347/kjp.2006.44.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Silva NR, Brooker S, Hotez PJ, Montresor A, Engels D, Savioli L. Soil-transmitted helminth infections: updating the global picture. Trends Parasitol. 2003;19:547–51. doi: 10.1016/j.pt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 51.de Silva HJ, de Silva NR, de Silva AP, Jewell DP. Emergence of inflammatory bowel disease 'beyond the West': do prosperity and improved hygiene have a role? TransRSocTropMed Hyg. 2008;102:857–60. doi: 10.1016/j.trstmh.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 52.Maizels RM. Infections and allergy - helminths, hygiene and host immune regulation. CurrOpin Immunol. 2005;17:656–61. doi: 10.1016/j.coi.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 53.Elliott DE, Summers RW, Weinstock JV. Helminths as governors of immune-mediated inflammation. International Journal for Parasitology. 2007;37:457–64. doi: 10.1016/j.ijpara.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 54.Elliott DE, Urban JFJ, Argo CK, Weinstock JV. Does the failure to acquire helminthic parasites predispose to Crohn's disease? FASEB Journal. 2000;14:1848–55. doi: 10.1096/fj.99-0885hyp. [DOI] [PubMed] [Google Scholar]
- 55.Maizels RM, Yazdanbakhsh M. Immune regulation by helminth parasites: cellular and molecular mechanisms. Nature Reviews. Immunology. 2003;3:733–44. doi: 10.1038/nri1183. [DOI] [PubMed] [Google Scholar]
- 56.Turner JD, Jackson JA, Faulkner H, et al. Intensity of intestinal infection with multiple worm species is related to regulatory cytokine output and immune hyporesponsiveness. JInfect Dis. 2008;197:1204–12. doi: 10.1086/586717. [DOI] [PubMed] [Google Scholar]
- 57.van Riet E, Hartgers FC, Yazdanbakhsh M. Chronic helminth infections induce immunomodulation: consequences and mechanisms. Immunobiology. 2007;212:475–90. doi: 10.1016/j.imbio.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 58.Elliott D, Li J, Blum A, et al. Exposure to Schistosome Eggs protects mice from TNBS-induced colitis. American Journal of Physiology. 2003;284:G385–G391. doi: 10.1152/ajpgi.00049.2002. [DOI] [PubMed] [Google Scholar]
- 59.Hunter MM, Wang A, Hirota CL, McKay DM. Neutralizing anti-IL-10 antibody blocks the protective effect of tapeworm infection in a murine model of chemically induced colitis. Journal of Immunology. 2005;174(11):7368–75. doi: 10.4049/jimmunol.174.11.7368. [DOI] [PubMed] [Google Scholar]
- 60.Khan WI, Blennerhasset PA, Varghese AK, et al. Intestinal nematode infection ameliorates experimental colitis in mice. Infection & Immunity. 2002;70:5931–7. doi: 10.1128/IAI.70.11.5931-5937.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Setiawan T, Metwali A, Blum AM, et al. Heligmosomoides polygyrus promotes regulatory T cell cytokine production in normal distal murine intestine. Infect. Immun. 2007;75:4655–63. doi: 10.1128/IAI.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Metwali A, Setiawan T, Blum AM, et al. Induction of CD8+ regulatory T cells in the intestine by Heligmosomoides polygyrus infection. AmJ Physiol Gastrointest Liver Physiol. 2006;291:G253–G259. doi: 10.1152/ajpgi.00409.2005. [DOI] [PubMed] [Google Scholar]
- 63.Elliott DE, Metwali A, Leung J, et al. Colonization with Heligmosomoides polygyrus suppresses mucosal IL-17 production. J Immunol. 2008;181:2414–9. doi: 10.4049/jimmunol.181.4.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elliott DE, Setiawan T, Metwali A, Blum A, Urban JF, Jr, Weinstock JV. Heligmosomoides polygyrus inhibits established colitis in IL-10-deficient mice. European Journal of Immunology. 2004;34:2690–8. doi: 10.1002/eji.200324833. [DOI] [PubMed] [Google Scholar]
- 65.Zaccone P, Fehervari Z, Jones FM, et al. Schistosoma mansoni antigens modulate the activity of the innate immune response and prevent onset of type 1 diabetes. EurJ Immunol. 2003;33:1439–49. doi: 10.1002/eji.200323910. [DOI] [PubMed] [Google Scholar]
- 66.Saunders KA, Raine T, Cooke A, Lawrence CE. Inhibition of autoimmune type 1 diabetes by gastrointestinal helminth infection. Infect Immun. 2007;75:397–407. doi: 10.1128/IAI.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kitagaki K, Businga TR, Racila D, Elliott DE, Weinstock JV, Kline JN. Intestinal helminths protect in a murine model of asthma. J Immunol. 2006;177:1628–35. doi: 10.4049/jimmunol.177.3.1628. [DOI] [PubMed] [Google Scholar]
- 68.Wilson MS, Taylor MD, Balic A, Finney CA, Lamb JR, Maizels RM. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. Journal of Experimental Medicine. 2005;202:1199–212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mangan NE, van RN, McKenzie AN, Fallon PG. Helminth-modified pulmonary immune response protects mice from allergen-induced airway hyperresponsiveness. J Immunol. 2006;176:138–47. doi: 10.4049/jimmunol.176.1.138. [DOI] [PubMed] [Google Scholar]
- 70.La Flamme AC, Ruddenklau K, Backstrom BT. Schistosomiasis decreases central nervous system inflammation and alters the progression of experimental autoimmune encephalomyelitis. Infection & Immunity. 2003;71:4996–5004. doi: 10.1128/IAI.71.9.4996-5004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sewell D, Qing Z, Reinke E, et al. Immunomodulation of experimental autoimmune encephalomyelitis by helminth ova immunization. International Immunology. 2003;15:59–69. doi: 10.1093/intimm/dxg012. [DOI] [PubMed] [Google Scholar]
- 72.Büning J, Homann N, von Smolinski D, et al. Helminths as Governors of Inflammatory Bowel Disease. Gut. 2008;57:1182–3. doi: 10.1136/gut.2008.152355. [DOI] [PubMed] [Google Scholar]
- 73.Rodrigues LC, Newcombe PJ, Cunha SS, et al. Early infection with Trichuris trichiura and allergen skin test reactivity in later childhood. ClinExpAllergy. 2008 doi: 10.1111/j.1365-2222.2008.03027.x. [DOI] [PubMed] [Google Scholar]
- 74.Correale J, Farez M. Association between parasite infection and immune responses in multiple sclerosis. Ann Neurol. 2007;61:97–108. doi: 10.1002/ana.21067. [DOI] [PubMed] [Google Scholar]
- 75.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 76.Shkoda A, Ruiz PA, Daniel H, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 77.Reyes JL, Terrazas LI. The divergent roles of alternatively activated macrophages in helminthic infections. Parasite Immunol. 2007;29:609–19. doi: 10.1111/j.1365-3024.2007.00973.x. [DOI] [PubMed] [Google Scholar]
- 78.Morrison AC, Correll PH. Activation of the stem cell-derived tyrosine kinase/RON receptor tyrosine kinase by macrophage-stimulating protein results in the induction of arginase activity in murine peritoneal macrophages. J Immunol. 2002;168:853–60. doi: 10.4049/jimmunol.168.2.853. [DOI] [PubMed] [Google Scholar]
- 79.Faubion WA, Jr, Loftus EV, Jr, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology. 2001;121:255–60. doi: 10.1053/gast.2001.26279. [DOI] [PubMed] [Google Scholar]
- 80.Sutherland L, Macdonald JK. Oral 5-aminosalicylic acid for maintenance of remission in ulcerative colitis. CochraneDatabaseSystRev. 2006;%19:CD000544. doi: 10.1002/14651858.CD000544.pub2. [DOI] [PubMed] [Google Scholar]
- 81.van Bodegraven AA, Mulder CJ. Indications for 5-aminosalicylate in inflammatory bowel disease: is the body of evidence complete? World J. Gastroenterol. 2006;12:6115–23. doi: 10.3748/wjg.v12.i38.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rousseaux C, Lefebvre B, Dubuquoy L, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. JExp Med. 2005;201:1205–15. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–57. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 84.Cosnes J, Nion-Larmurier I, Beaugerie L, Afchain P, Tiret E, Gendre JP. Impact of the increasing use of immunosuppressants in Crohn's disease on the need for intestinal surgery. Gut. 2005;54:237–41. doi: 10.1136/gut.2004.045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Colombel JF, Ferrari N, Debuysere H, et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn's disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025–30. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]
- 86.Feagan BG, Rochon J, Fedorak RN, et al. Methotrexate for the treatment of Crohn's disease. The North American Crohn's Study Group Investigators. NEnglJ Med. 1995;332:292–7. doi: 10.1056/NEJM199502023320503. [DOI] [PubMed] [Google Scholar]
- 87.Chande N, Macdonald JK, McDonald JW. Methotrexate for induction of remission in ulcerative colitis. CochraneDatabaseSystRev. 2007:CD006618. doi: 10.1002/14651858.CD006618.pub2. [DOI] [PubMed] [Google Scholar]
- 88.Behm BW, Bickston SJ. Tumor necrosis factor-alpha antibody for maintenance of remission in Crohn's disease. CochraneDatabaseSystRev. 2008:CD006893. doi: 10.1002/14651858.CD006893. [DOI] [PubMed] [Google Scholar]
- 89.Rutgeerts P, Van AG, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–610. doi: 10.1053/j.gastro.2004.02.070. [DOI] [PubMed] [Google Scholar]
- 90.Schreiber S, Khaliq-Kareemi M, Lawrance IC, et al. Maintenance therapy with certolizumab pegol for Crohn's disease. NEnglJMed. 2007;%19;357:239–50. doi: 10.1056/NEJMoa062897. [DOI] [PubMed] [Google Scholar]
- 91.Targan SR, Feagan BG, Fedorak RN, et al. Natalizumab for the treatment of active Crohn's disease: results of the ENCORE Trial. Gastroenterology. 2007;132:1672–83. doi: 10.1053/j.gastro.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 92.Peyrin-Biroulet L, Desreumaux P, Sandborn WJ, Colombel JF. Crohn's disease: beyond antagonists of tumour necrosis factor. Lancet. 2008;372:67–81. doi: 10.1016/S0140-6736(08)60995-2. [DOI] [PubMed] [Google Scholar]
- 93.Summers RW, Elliott DE, Urban JF, Jr, Thompson RA, Weinstock JV. Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology. 2005;128:825–32. doi: 10.1053/j.gastro.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 94.Summers RW, Elliott DE, Urban JF, Jr, Thompson R, Weinstock JV. Trichuris suis therapy in Crohn's disease. Gut. 2005;54:87–90. doi: 10.1136/gut.2004.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]