

Abstract
The adipocytes synthesize and store triglycerides as lipid droplets surrounded by various proteins and phospholipids at its surface. Recently, the molecular basis of some of the genetic syndromes of lipodystrophies has been elucidated and some of these genetic loci have been found to contribute to lipid droplet formation in adipocytes. The two main types of genetic lipodystrophies are congenital generalized lipodystrophy (CGL) and familial partial lipodystrophy (FPL). So far, three CGL loci: 1-acylglycerol-3-phosphate-O-acyltransferase 2 (AGPAT2), Berardinelli-Seip Congenital Lipodystrophy 2 (BSCL2) and caveolin 1 (CAV1) and four FPL loci: lamin A/C (LMNA), peroxisome proliferator-activated receptor γ (PPARG), v-AKT murine thymoma oncogene homolog 2 (AKT2) and zinc metalloprotease (ZMPSTE24), have been identified. AGPAT2 plays a critical role in the synthesis of glycerophospholipids and triglycerides required for lipid droplet formation. Another protein, seipin (encoded by BSCL2 gene), has been found to induce lipid droplet fusion. CAV1 is an integral component of caveolae and might contribute towards lipid droplet formation. PPARγ and AKT2 play important role in adipogenesis and lipid synthesis. In this review, we discuss and speculate about the contribution of various lipodystrophy genes and their products in the lipid droplet formation.
Keywords: Lipodystrophy, AGPAT2, BSCL2, CAV1, LMNA, PPARG, AKT2, ZMPSTE24, Lipid droplet, Acyltransferases
The disorders of lipodystrophies have been known for more than a century. The first one was initially known as lipodystrophia progressiva or Barraquer-Simons syndrome (now called acquired partial lipodystrophy)[1, 2]. Since then many other acquired and genetic syndromes of lipodystrophy have been reported, the most recent being the one induced by protease-inhibitors based highly active antiretroviral therapy in patients infected with human immunodeficiency virus [3]. All the disorders are characterized by selective loss of body fat although the extent of fat loss varies. If the fat loss is significant, patients develop insulin resistance and its complications such as, diabetes, dyslipidemia, hepatic steatosis, acanthosis nigricans, polycystic ovarian disease and hypertension [1, 4]. A substantial progress has been made recently in understanding the molecular defects in patients with genetic forms of lipodystrophies, which will be reviewed in brief here. Readers are referred to more detailed recent reviews on the subject [5, 6]. Acquired lipodystrophies have been reviewed recently in several other publications [2, 4, 7-9]. In this review, we will speculate about the role of some of the lipodystrophy loci in the formation of lipid droplets (also called lipid bodies) in the cells.
The two most common phenotypes observed among patients with genetic lipodystrophies are: a. generalized loss of body fat occurring at birth which is called congenital generalized lipodystrophy (CGL, Berardinelli-Seip syndrome) or partial loss of body fat generally occurring later in life either during childhood or puberty called familial partial lipodystrophy (FPL) (Fig. 1). So far, three genetic loci have been reported for CGL, whereas for FPL, four loci have been discovered. Besides these, there are some other uncommon phenotypes for which the genetic basis remains to be elucidated
Fig. 1. Phenotypes of Congenital generalized lipodystrophy and familial partial lipodystrophy of the Dunnigan variety.
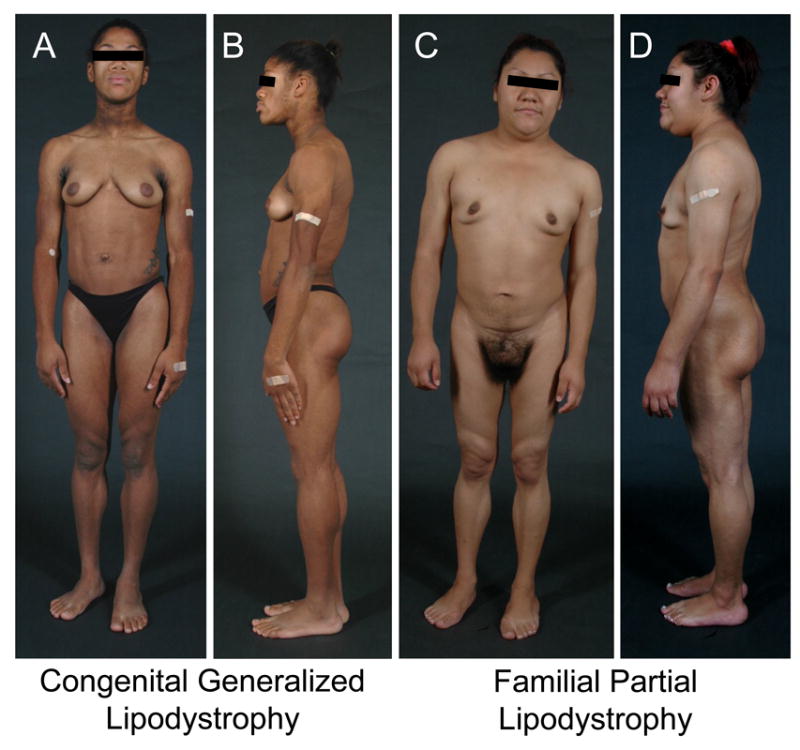
A. and B. Front and lateral views of a 19-year-old female of African-American origin with congenital generalized lipodystrophy, type 1 due to 1-acylglycerol-3-phosphate acyltransferase 2 (AGPAT2) homozygous mutation. She has generalized lack of body fat, marked muscularity, acanthosis nigricans in the neck and axillae and acromegaloid features and umbilical prominence. She developed diabetes at the age of 14 years and severe hypertriglyceridemia was noted 15 years of age.
C and D. Front and lateral views of a 24-year-old Hispanic woman with familial partial lipodystrophy of the Dunnigan variety due to heterozygous missense mutation in the Lamin A/C (LMNA) gene. She had fat loss the upper and lower extremities and trunk at puberty and also accumulated excess fat in the face, submental, supraclavicular and vulvar regions. She had mild acanthosis nigricans in the neck and axillae.
Congenital Generalized Lipodystrophy (CGL)
This rare autosomal recessive disorder is usually recognized at birth or shortly thereafter because of near total lack of body fat and increased muscular appearance of neonates. The children with this disorder undergo rapid growth and have markedly increased appetite. Acanthosis nigricans manifests later. Liver enlargement due to fatty deposition can be seen early in life and can lead to cirrhosis later. Women with CGL may have hirsutism, clitoromegaly, oligoamenorrhea and polycystic ovaries. After pubertal development, some patients develop focal lytic lesions in the long bones. Hypertrophic cardiomyopathy and mild mental retardation are seen in some patients [10-12]. Metabolic complications can be seen early and hypertriglyceridemia and diabetes are difficult to manage. Patients typically have markedly low serum levels of leptin and adiponectin [13].
To understand the molecular basis of this disorder, two groups independently pursued positional cloning approach that led to identification of two loci: 1-acylglycerol-3-phosphate-O-acyltransferase 2 (AGPAT2) gene on chromosome 9q34 for CGL, type 1 [14, 15] and Berardinelli-Seip Congenital Lipodystrophy 2 (BSCL2) gene on chromosome 11q13 for CGL, type 2. [16] Cardiomyopathy and mild mental retardation is more prevalent in CGL, type 2 patients,[11, 12, 15] whereas focal lytic lesions in long bones are more prevalent in those with CGL, type 1 [17]. Patients with CGL type 1 lose all metabolically active adipose tissue present in most subcutaneous areas, intraabdominal and intrathoracic regions, and bone marrow but have well-preserved mechanical adipose tissue depots located in the palms, soles, under the scalp, retro-orbital and peri-articular regions. On the other hand, patients with CGL type 2 lose both types of adipose tissue [17-19].
Only recently, a single patient from Brazil with a complex phenotype was reported to harbor homozygous null mutation in caveolin 1 (CAV1) gene. This patient had some distinct clinical features such as well-preserved bone marrow fat, and lack of lytic lesions in the long bones [20]. She had preservation of “mechanical” adipose tissue in the retro-orbital region, peri-articular region and in the palms and soles; but the scalp fat was decreased. She had short stature, primary amenorrhea, hypocalcemia and hypomagnesemia, which were attributed to vitamin D resistance [20].
Still there is a possibility of cloning additional loci for CGL as some affected patients do not reveal mutations in any of these genes and their pedigrees do not show linkage to these loci [11, 21]. In one of our pedigrees, two siblings with CGL also have congenital muscular dystrophy, not reported previously [22].
CGL1 locus: AGPAT2
The AGPATs are acyltransferases which catalyze esterification of a fatty acid to lysophosphatidic acid (LPA or 1-acylglycerol-3-phosphate) in order to covert it to phosphatidic acid (PA or 1,2 diacylglycerol-3-phosphate). This is a key intermediate step during biosynthesis of glycerophospholipids and triglycerides.[23] (Figure 2). Based on structural homology to the major isoforms, AGPAT1 and AGPAT2, at least seven other proteins have been designated as AGPATs. However, documentation of AGPAT activity, i.e., conversion of LPA to PA, has not been performed for many of these isoforms. Furthermore, some AGPAT isoforms have been found to have other enzymatic activities and have been reannotated as glycerol phosphate acyltransferase 4 (GPAT4 instead of AGPAT6) or acyl-CoA:lysophosphatidylethanolamine acyltransferase 2 (LPEAT2 instead of AGPAT7) [24-26]. All the isoforms studied until now localize to the ER, where the formation of lipid droplet is initiated. It remains unclear if all of these or only a few are involved in the formation of lipid droplet. AGPAT2 mRNA has been shown to be highly expressed in the mouse fibroblast, 3T3-L1 cells and the human omental adipose tissue. AGPAT2 protein has 278 amino acids and has two highly conserved domains, NHXXXXD and EGTR, required for the enzymatic activity.[27, 28] However, study of a few naturally occurring AGPAT2 mutants reveals important role of the carboxy-terminus for enzymatic activity as well [29]. Reduced AGPAT2 activity in adipose tissue, thus may result in lipodystrophy either due to lack of triglyceride synthesis or due to abnormal adipocyte function from lack of phospholipids [23].
Fig. 2. The triglyceride and glycerophospholipid biosynthetic pathway in the adipose tissue.

Adipose tissue requires glycerol-3-phosphate as the initial substrate for triglyceride and glycerophospholipid biosynthesis. Initially, glycerol-3-phosphate is acylated using fatty acyl coenzyme A (FA-CoA) at the sn-1 position by the class of enzymes called glycerol-3-phosphate acyltransferases (GPATs), and forms 1-acylglycerol-3-phosphate or lysophosphatidic acid (LPA). Further acylation of LPA at the sn-2 position by the enzymes called 1-acylglycerol-3-phosphate acyltransferases (AGPATs or LPAATs) results in formation of phosphatidic acid (PA). Phosphatidic acid phosphatases then remove the phosphate group from PA to produce diacylglycerol (DAG). Further acylation of DAG at the sn-3 position by the enzymes called diacylglycerol acyltransferases (DGATs) finally produces triacylglycerol (TG). The synthesis of glycerophospholipids uses the intermediates, PA and DAG. Phosphatidylinositol and cardiolipin can be formed from PA, whereas, phosphatidylcholine, phosphatidylethanolamine and phosphatidylserine can be synthesized from DAG.
CGL2 locus: BSCL2
The BSCL2 encodes the protein seipin, which was initially proposed to be a 398 amino acid protein [16]. However, homology search predicts a protein of 462 amino acids [30, 31],. Seipin localizes to endoplasmic membrane [30-33] whose physiological function is just beginning to be unraveled. Seipin has a CAAX motif at the C-terminus which could undergo posttranslational processing including prenylation. It also has a canonical N-glycosylation site N-X-S/T (N-V-S at position 88-90, numbering is based on initial amino acid sequence). Interestingly, heterozygous mutations in this glycosylation site have recently been associated with autosomal dominant motor neuron diseases called Silver syndrome, spastic paraplegia 17, and distal hereditary motor neuropathy type V [32]. BSCL2 mRNA is highly expressed in the brain and testis as well as in the adipose tissue [16, 30, 34].
CGL3 locus: CAV1
Caveolins are integral components of caveolae, which are specialized plasma membrane microdomains seen on electron microscopy as 50-100 nm vesicular invaginations. Adipocyte membranes have abundance of caveolae which increase by 10-fold during adipocyte differentiation [35]. CAV1 is the major fatty acid-binding protein which resides on the adipocyte membranes and translocates to lipid droplets in response to increased levels of free fatty acids [36-38]. Thus, lack of CAV1 function may result in lipodystrophy by affecting adipocyte differentiation, lipid transport through caveolae and disruption of lipid droplet formation.
Biology of lipid droplets and the role of CGL loci
In mammals, neutral lipids are synthesized and stored in specialized cells, called adipocytes (Figure 3). The synthesis of TG begins in the lumen of ER such that as the lipid droplets increase in size, the ER leaflet facing the cytoplasm begins to bulge, surrounded by the ER membrane. The neutral lipids are hydrophobic in nature and are thus coated with various molecules which have hydrophobic and hydrophilic ends. Proteins of PAT class, (named after perilipin, adipocyte differentiation related protein and tail interacting protein 47) fit these criteria and thus surround the lipid droplets [39]. In addition, glycerophospholipids, such as phosphatidylcholine (PC), phosphatidylethanolamine (PE) and phosphatidylserine (PS), also form the outer core of the lipid droplets [39].
Fig. 3. Schematics of lipid droplet formation in adipocyte.

Panel A shows the progressive formation of lipid droplet (LD) at the endoplasmic reticulum in normal cells. Shown also are the enzymes involved in the synthesis of triglycerides, although it is unclear if all the enzymes of the pathway are present at the LD. The small LDs in adipocytes fuse to form one or more large LDs. Shown also are proteins of PAT class which decorate the LD surface. Panel B shows the reduced triglyceride synthesis due to deficiency of one of the key enzymes, AGPAT2 in patients with congenital generalized lipodystrophy, type 1. Fusion of LDs may still occur, but at considerably reduced rate. It is likely that the LDs may be totally devoid of TG (shown in white) or minimal TG synthesis may occur utilizing other AGPAT isoforms. Other possibilities (not shown) are that LDs may not form due to lack of synthesis of LD surface glycerophospholipids or there may be total lack of adipocyte development due to lack of phospholipid synthesis required for formation of cell membrane and other organelles. In panel C, where the cells lack expression of seipin as happens in patients with congenital lipodystrophy, type 2, fusion of LDs may not occur, however, synthesis of triglyceride may still continue resulting in several small LDs instead of one or more large LDs.
As shown schematically (TG synthesis pathway, Figure 2), the enzyme AGPAT2 is at the critical junction of glycerophospholipid and TG synthesis, thus loss of AGPAT2 may initially restrict the lipid droplet formation due to decrease phospholipid synthesis (partially active AGPAT2 mutants) or completely, if deleted.
The role of another protein, seipin, is still not fully appreciated. It has no known functional domain to indicate its function. Its only predicted partner is a midasin – a AAA ATPase protein of variety of cellular function [30]. However, recently, two groups have reported that seipin homologue in the yeast is required for lipid droplet assembly or maintenance [40, 41]. Loss of yeast homolog for seipin leads to decreased fusions of lipid bodies and thus loss of formation of one enlarged lipid body seen in adipocytes. Thus one function of the seipin appears to be controlling the dynamics of lipid body size. Another series of experiments in yeast showed that the seipin is required for the lipid bodies formation and that mutant yeast leads to aberrant lipid bodies. This study also showed that the BSCL2 null mutant fibroblast showed small lipid droplet compared to wild type fibroblast. Although in yeast, loss of seipin homologue leads to decreased “fused lipid bodies”, in human fibroblasts, opposite effect was observed [41]. Although, these studies now reveal its function in lipid droplets in yeast, its precise role in human adipose tissue is still far from clear. Does seipin have a role in cellular differentiation? Answer to this question awaits further experiments. Recently, Payne et al. [34] have shown that BSCL2 expression is strongly induced during adipocyte differentiation and is essential for adipogenesis. Since nearly all BSCL2 mutations causing CGL have been null mutations, one can propose that BSCL2 mutations cause lipodystrophy either by affecting adipocyte differentiation or by affecting lipid droplet formation in adipocytes.
Caveolin-1, one of the major membrane protein of caveolae, has also been linked to lipid droplet formation. CAV1 is a major fatty acid-binding protein on the plasma membranes of the adipocytes and translocates to lipid droplets in response to excess free fatty acids [36], suggesting its role in the transport or storage of free fatty acids and triglycerides in lipid droplets [37, 38]. Caveolin deficiency results in fewer lipid droplets in mouse embryonic fibroblasts [36] and hepatocytes [42] than those seen in the wild type cells. Thus, lipodystrophy in the patient with CAV1 homozygous mutation [43] may be due to lack or disruption of lipid droplet formation.
Familial Partial Lipodystrophy (FPL)
These rare varieties are characterized by variable loss of body fat from the extremities as well as from the truncal region which usually occurs during childhood or puberty as in the Dunnigan variety, which is due to missense lamin A/C (LMNA) mutations and reportedly at variable time in patients with peroxisome proliferator-activated receptor gamma (PPARG) mutations. During childhood these patients do not show a lipodystrophy phenotype. The associated metabolic complications also develop later in life. Affected females can be easily recognized but it is difficult to diagnose men affected with FPL. Three loci, LMNA, PPARG and v-AKT murine thymoma oncogene homolog 2 (AKT2) have been identified for autosomal dominant types of FPL [44-46] and for the autosomal recessive variety associated with mandibuloacral dysplasia, two loci, LMNA and zinc metalloprotease (ZMPSTE24) have been identified [47, 48]. While LMNA was identified using the positional cloning approach [44, 49] other loci were identified mainly using candidate gene approach.
The fat loss from the extremities occurs gradually at the time of puberty in patients with FPL, Dunnigan variety and some patients at the same time, gain excess fat at the face, chin (“double chin’), neck (‘Cushingoid appearance with buffalo hump’) and in females in the vulvar region [50]. Women are more severely affected with metabolic complications such as diabetes, dyslipidemia and coronary heart disease than men [51-53]. Some women develop acanthosis nigricans, hirsutism, menstrual abnormalities, and polycystic ovaries. Occasional patients also develop multisystem dystrophy including cardiomyopathy which manifests with conduction system disturbances and congestive heart failure [54].
Only about twenty patients with FPL due to heterozygous mutations in PPARG have been reported so far and thus it is either much less common than FPL, Dunnigan variety [55] or is less recognized likely due to milder phenotype. The onset of lipodystrophy has been reported to be from 2nd decade to later in life. Fat loss affects the distal extremities more than the proximal extremities. While slight reduction in facial fat has been reported, some patients have excess fat in the face. Only one pedigree has been reported with a missense mutation in (AKT2) gene [46]. Detailed phenotyping was not conducted in the affected subjects thus how the phenotype differs from that observed in FPL due to LMNA and PPARG mutations is not clear. Besides LMNA, PPARG and AKT2, additional loci are likely as many FPL patients do not reveal any mutations in these genes [45, 56].
FPL has also been reported in patients with a complex phenotype of mandibuloacral dysplasia, a rare autosomal recessive disorder. Patients are normal at birth but soon develop hypoplasia of the mandible and clavicles, and acro-osteolysis of the terminal phalanges [57]. They may also have delayed closure of cranial sutures, joint contractures, mottled cutaneous pigmentation and short stature. Features of accelerated aging such bird like facies, high-pitched voice and alopecia can also be evident. Patients either develop partial loss of subcutaneous fat from the extremities or a more generalized fat loss involving the face, trunk and extremities [57]. While more than 30 patients have been reported to have homozygous or compound heterozygous mutations in LMNA, only 5 patients have been known to have ZMPSTE24 mutations [58]. Additional loci for FPL associated with mandibuloacral dysplasia are likely as some patients do not have any variants in either LMNA or ZMPSTE24 [59].
FPL1 locus: LMNA
LMNA encodes two major proteins, prelamin A, and lamin C, and two minor proteins, lamin AΔ10 and C2, by alternative splicing. The mature lamin A is formed after successive post-translational modification of its precursor, prelamin A, a CAAX motif protein whereas the truncated short form, lamin C, does not undergo post-translational modification. This process involves farnesylation, O-methylation and proteolysis. Zinc metalloproteinase (ZMPSTE24) is essential for proteolytic processing of prelamin A to mature lamin A. The lamins belong to the intermediate filament family of structural proteins and form hetero- or homo-dimeric coiled-coil structures in the nuclear lamina which is located inside the inner membrane of the nuclear envelope [60, 61]. Lamins interact with chromatin and other nuclear lamina proteins such as emerin, several forms of lamin associated proteins, nesprin and other nucleoplasm proteins. Thus, missense mutations may affect nuclear function and may resulting in apoptosis and premature cell death of adipocytes, thus causing lipodystrophy. There may also be cellular toxicity and premature cell aging related to accumulation of prelamin A [62] Most of the mutations, which are associated with FPL, are clustered in the immunoglobulin G fold (IgG) region. IgG domains are known to be associated with variety of proteins and thus, lack of such interactions might lead to the defects in either adipogenesis or neutral lipid synthesis. Lamins A and C are ubiquitously expressed proteins and therefore why specific mutations affect predominantly adipocytes only and not other cells, remains unclear. Since all cells do not express all the proteins, we can only speculate that these mutations lead to the loss of protein-protein interaction essential for the function and synthesis of adipocytes and lipids. Furthermore, why only some adipocytes from certain areas of the body are lost, and not others, remains unknown.
FPL2 locus: PPARG
Given the critical role of PPARγ in adipogenesis and its high expression in the adipose tissue, dominant negative missense mutations may cause lipodystrophy by affecting adipogenesis [63]. However, why loss of fat is restricted to some areas of the body and not others remains unclear.
FPL3 locus: AKT2
AKT2, also known as protein kinase B (PKB), is a phosphoinositide-dependent serine/threonine kinase and is involved in post-receptor insulin signaling. Loss of adipose tissue in patients with AKT2 mutations may either be due to reduced adipocyte differentiation or dysfunctional post-receptor insulin signaling [46].
FPL4 locus: ZMPSTE24
In patients with ZMPSTE24 deficiency cellular accumulation of prelamin A and/or lack of mature lamin A may be responsible for phenotypic features including lipodystrophy [64].
Other Types
Besides the predominant subtypes, CGL and FPL, there are other relatively uncommon varieties, such as lipodystrophy associated with SHORT Syndrome, and neonatal progeroid syndrome (also called Wiedemann-Rautenstrauch syndrome [65-69]. The genetic bases of these varieties remain to be elucidated.
Patients with Hutchinson-Gilford progeria syndrome and atypical progeroid syndrome, due to heterozygous missense mutations in LMNA gene also have been reported to have a progressive and generalized loss of body fat during childhood [70-73].
Hegele et al. [74] reported variants in lamin B2 (LMNB2) gene in 4 patients with “acquired partial lipodystrophy” (Barraquer-Simons syndrome) but the pattern of fat loss affecting the knees, thighs and gluteal region was very atypical for acquired partial lipodystrophy which mainly affects the head, neck, trunk and upper extremities and spares the lower extremities [2]. None of them had complement 3 deficiency or complement 3 nephritic factor, three had DM, and all four had hypertriglyceridemia, which are not characteristic features of acquired partial lipodystrophy. Finally, no segregation of these variants in family members was reported. No pictures of these patients were published to determine what exact lipodystrophy pattern was associated with the LMNB2 variants. Thus, without confirmation, this association of LMNB2 variants with acquired partial lipodystrophy is highly unlikely.
Recently, Cao et al [75] reported heterozygous CAV1 mutations, I134fsdelA-X137 and -88delC, in patients with partial lipodystrophy and hypertriglyceridemia. The I134fsdelA-X137 mutation was present in a 28-year-old female and her 55-year-old father with lipodystrophy affecting the face and arms, neurodegeneration and congenital cataracts [76]. On the other hand, the -88delC mutation was present in a 35-year-old male with lipodystrophy affecting the upper and lower extremities. However, whether the -88delC mutation in the 5′ untranslated region affected the transcription of CAV1 gene was not demonstrated. Furthermore, Ae Kim et al. [43] reported that none of the three confirmed subjects harboring the heterozygous null mutation, G28X, in CAV1, had lipodystrophy, hyperinsulinemia or hypertriglyceridemia. Furthermore, the Cav1+/- mice do not show any phenotype [77]. Thus, whether heterozygous mutations in CAV1 in humans have any functional consequences remains unclear.
Perspective
The synthesis of lipid droplets has been intensively studied in the adipocytes. As we indicated earlier, CGL loci, AGPAT2, BSCL2 and CAV1, play an important role in lipid droplet formation in the adipocytes. On the other hand, of the known FPL loci, only PPARG and AKT2 have well-documented role in adipogenesis, and perhaps in lipogenesis (lipid synthesis) as well. There is no experimental evidence to suggest the role of lamin A/C and ZMPSTE24 in lipid droplet formation but instead may be important for adipocyte survival.
The Oil-red-O staining of the mouse and human fatty liver shows intense uniformly distributed staining and lipid droplets which tend to be smaller than those in adipocytes [78]. How do the liver cells (hepatocytes) synthesize and store excess triglycerides remains an enigma. Do hepatocytes synthesize several small lipid droplets which do not fuse to form one giant lipid droplet as observed in adipocytes? If so, is this due to lack of expression of certain proteins which help protect the highly hydrophobic surface from the hydrophilic cytoplasmic environment or lack of proteins which help in fusion of small lipid droplets? In fact, seipin may be one of those protein involved in lipid droplet fusion. Accumulation of TG in muscles is the least understood in terms of its mechanism. The formation of the intramyocellular lipid droplet, its surface proteins and phospholipids is still a mystery, yet is an important mechanism involved in peripheral insulin resistance.
Another area of future investigation is related to the identification of the role of all the enzymes including acyltransferase(s) involved in the actual synthesis of TG which forms the core of the lipid droplets. Further information about subcellular localization of various enzymes involved in TG biosynthesis, i.e., GPAT, AGPAT, PAP and DGAT and whether they co-localize and whether they are juxtaposed to one another at various organelles will allow us to understand which isoforms are involved in the synthesis of TG and which are involved in the synthesis of glycerophospholipids. Recent immunofluorescence imaging of lipid droplets have revealed the heterogeneity of the lipid droplets in the cellular pool within the cell, informing us that study of lipid droplet may be cell specific and should be studied tissue wise [79]. This might help understanding the most important medical challenge of our times, i.e., insulin resistance in the liver, muscle and adipose tissue.
Acknowledgments
The authors were partly supported by the National Institutes of Health grants, R01-DK54387, UL1-RR024982, and by the Southwestern Medical Foundation. The authors thank Crystal Nielsen and Sarah Gilmore for graphics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Garg A. Lipodystrophies. Am J Med. 2000;108:143–152. doi: 10.1016/s0002-9343(99)00414-3. [DOI] [PubMed] [Google Scholar]
- 2.Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine. 2004;83:18–34. doi: 10.1097/01.md.0000111061.69212.59. [DOI] [PubMed] [Google Scholar]
- 3.Carr A. HIV protease inhibitor-related lipodystrophy syndrome. Clinical Infectious Diseases. 2000;30 2:S135–142. doi: 10.1086/313854. [DOI] [PubMed] [Google Scholar]
- 4.Garg A. Acquired and genetic lipodystrophies. N Engl J Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal AK, Garg A. Genetic basis of lipodystrophies and management of metabolic complications. Annu Rev Med. 2006;57:297–311. doi: 10.1146/annurev.med.57.022605.114424. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal AK, Garg A. Genetic disorders of adipose tissue development, differentiation, and death. Annu Rev Genomics Hum Genet. 2006;7:175–199. doi: 10.1146/annurev.genom.7.080505.115715. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Misra A, Garg A. Lipodystrophy in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab. 2002;87:4845–4856. doi: 10.1210/jc.2002-020794. [DOI] [PubMed] [Google Scholar]
- 8.Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine. 2003;82:129–146. doi: 10.1097/00005792-200303000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Grinspoon S, Carr A. Cardiovascular risk and body-fat abnormalities in HIV-infected adults. N Engl J Med. 2005;352:48–62. doi: 10.1056/NEJMra041811. [DOI] [PubMed] [Google Scholar]
- 10.Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy) Acta Paediatrica Supplement. 1996;413:2–28. doi: 10.1111/j.1651-2227.1996.tb14262.x. [DOI] [PubMed] [Google Scholar]
- 11.Agarwal AK, Simha V, Oral EA, Moran SA, Gorden P, O'Rahilly S, Zaidi Z, Gurakan F, Arslanian SA, Klar A, Ricker A, White NH, Bindl L, Herbst K, Kennel K, Patel SB, Al-Gazali L, Garg A. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88:4840–4847. doi: 10.1210/jc.2003-030855. [DOI] [PubMed] [Google Scholar]
- 12.Van Maldergem L, Magre J, Khallouf TE, Gedde-Dahl T, Jr, Delepine M, Trygstad O, Seemanova E, Stephenson T, Albott CS, Bonnici F, Panz VR, Medina JL, Bogalho P, Huet F, Savasta S, Verloes A, Robert JJ, Loret H, De Kerdanet M, Tubiana-Rufi N, Megarbane A, Maassen J, Polak M, Lacombe D, Kahn CR, Silveira EL, D'Abronzo FH, Grigorescu F, Lathrop M, Capeau J, O'Rahilly S. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39:722–733. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87:2395–2398. doi: 10.1210/jcem.87.5.8624. [DOI] [PubMed] [Google Scholar]
- 14.Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, Kocak N, O'Rahilly S, Taylor SI, Patel SB, Bowcock AM. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84:3390–3394. doi: 10.1210/jcem.84.9.6103. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal AK, Arioglu E, de Almeida S, Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31:21–23. doi: 10.1038/ng880. [DOI] [PubMed] [Google Scholar]
- 16.Magre J, Delepine M, Khallouf E, Gedde-Dahl T, Jr, Van Maldergem L, Sobel E, Papp J, Meier M, Megarbane A, Bachy A, Verloes A, d'Abronzo FH, Seemanova E, Assan R, Baudic N, Bourut C, Czernichow P, Huet F, Grigorescu F, de Kerdanet M, Lacombe D, Labrune P, Lanza M, Loret H, Matsuda F, Navarro J, Nivelon-Chevalier A, Polak M, Robert JJ, Tric P, Tubiana-Rufi N, Vigouroux C, Weissenbach J, Savasta S, Maassen JA, Trygstad O, Bogalho P, Freitas P, Medina JL, Bonnicci F, Joffe BI, Loyson G, Panz VR, Raal FJ, O'Rahilly S, Stephenson T, Kahn CR, Lathrop M, Capeau J. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28:365–370. doi: 10.1038/ng585. [DOI] [PubMed] [Google Scholar]
- 17.Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy due to mutations in the AGPAT2 or Seipin genes. J Clin Endocrinol Metab. 2003;88:5433–5437. doi: 10.1210/jc.2003-030835. [DOI] [PubMed] [Google Scholar]
- 18.Garg A, Fleckenstein JL, Peshock RM, Grundy SM. Peculiar distribution of adipose tissue in patients with congenital generalized lipodystrophy. J Clin Endocrinol Metab. 1992;75:358–361. doi: 10.1210/jcem.75.2.1639935. [DOI] [PubMed] [Google Scholar]
- 19.Ebihara K, Kusakabe T, Masuzaki H, Kobayashi N, Tanaka T, Chusho H, Miyanaga F, Miyazawa T, Hayashi T, Hosoda K, Ogawa Y, Nakao K. Gene and phenotype analysis of congenital generalized lipodystrophy in Japanese: a novel homozygous nonsense mutation in seipin gene. J Clin Endocrinol Metab. 2004;89:2360–2364. doi: 10.1210/jc.2003-031211. [DOI] [PubMed] [Google Scholar]
- 20.Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O'Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93:1129–1134. doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- 21.Magre J, Delepine M, Van Maldergem L, Robert JJ, Maassen JA, Meier M, Panz VR, Kim CA, Tubiana-Rufi N, Czernichow P, Seemanova E, Buchanan CR, Lacombe D, Vigouroux C, Lascols O, Kahn CR, Capeau J, Lathrop M. Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes. 2003;52:1573–1578. doi: 10.2337/diabetes.52.6.1573. [DOI] [PubMed] [Google Scholar]
- 22.Simha V, Agarwal AK, Aronin PA, Iannaccone ST, Garg A. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet A. 2008;146A:2318–2326. doi: 10.1002/ajmg.a.32457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agarwal AK, Barnes RI, Garg A. Genetic basis of congenital generalized lipodystrophy. Int J Obes Relat Metab Disord. 2004;28:336–339. doi: 10.1038/sj.ijo.0802487. [DOI] [PubMed] [Google Scholar]
- 24.Chen YQ, Kuo MS, Li S, Bui HH, Peake DA, Sanders PE, Thibodeaux SJ, Chu S, Qian YW, Zhao Y, Bredt DS, Moller DE, Konrad RJ, Beigneux AP, Young SG, Cao G. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. J Biol Chem. 2008;283:10048–10057. doi: 10.1074/jbc.M708151200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagle CA, Vergnes L, Dejong H, Wang S, Lewin TM, Reue K, Coleman RA. Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4, as the enzyme deficient in Agpat6-/- mice. J Lipid Res. 2008;49:823–831. doi: 10.1194/jlr.M700592-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao J, Shan D, Revett T, Li D, Wu L, Liu W, Tobin JF, Gimeno RE. Molecular identification of a novel mammalian brain isoform of acyl-CoA:lysophospholipid acyltransferase with prominent ethanolamine lysophospholipid acylating activity, LPEAT2. J Biol Chem. 2008;283:19049–19057. doi: 10.1074/jbc.M800364200. [DOI] [PubMed] [Google Scholar]
- 27.Leung DW. The structure and functions of human lysophosphatidic acid acyltransferases. Front Biosci. 2001;6:d944–953. doi: 10.2741/leung. [DOI] [PubMed] [Google Scholar]
- 28.Lewin TM, Wang P, Coleman RA. Analysis of amino acid motifs diagnostic for the sn-glycerol-3-phosphate acyltransferase reaction. Biochemistry. 1999;38:5764–5771. doi: 10.1021/bi982805d. [DOI] [PubMed] [Google Scholar]
- 29.Haque W, Garg A, Agarwal AK. Enzymatic activity of naturally occurring 1-acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital generalized lipodystrophy. Biochem Biophys Res Commun. 2005;327:446–453. doi: 10.1016/j.bbrc.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 30.Agarwal AK, Garg A. Seipin: a mysterious protein. Trends Mol Med. 2004;10:440–444. doi: 10.1016/j.molmed.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Lundin C, Nordstrom R, Wagner K, Windpassinger C, Andersson H, von Heijne G, Nilsson I. Membrane topology of the human seipin protein. FEBS Lett. 2006;580:2281–2284. doi: 10.1016/j.febslet.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 32.Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, Horl G, Malli R, Reed JA, Dierick I, Verpoorten N, Warner TT, Proukakis C, Van den Bergh P, Verellen C, Van Maldergem L, Merlini L, De Jonghe P, Timmerman V, Crosby AH, Wagner K. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004;36:271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 33.Ito D, Fujisawa T, Iida H, Suzuki N. Characterization of seipin/BSCL2, a protein associated with spastic paraplegia 17. Neurobiol Dis. 2008;31:266–277. doi: 10.1016/j.nbd.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Payne VA, Grimsey N, Tuthill A, Virtue S, Gray SL, Dalla Nora E, Semple RK, O'Rahilly S, Rochford JJ. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes. 2008;57:2055–2060. doi: 10.2337/db08-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan JY, Carpentier JL, van Obberghen E, Grunfeld C, Gorden P, Orci L. Morphological changes of the 3T3-L1 fibroblast plasma membrane upon differentiation to the adipocyte form. J Cell Sci. 1983;61:219–230. doi: 10.1242/jcs.61.1.219. [DOI] [PubMed] [Google Scholar]
- 36.Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes. 2004;53:1261–1270. doi: 10.2337/diabetes.53.5.1261. [DOI] [PubMed] [Google Scholar]
- 37.Le Lay S, Hajduch E, Lindsay MR, Le Liepvre X, Thiele C, Ferre P, Parton RG, Kurzchalia T, Simons K, Dugail I. Cholesterol-induced caveolin targeting to lipid droplets in adipocytes: a role for caveolar endocytosis. Traffic (Copenhagen, Denmark) 2006;7:549–561. doi: 10.1111/j.1600-0854.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- 38.Ostermeyer AG, Paci JM, Zeng Y, Lublin DM, Munro S, Brown DA. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J Cell Biol. 2001;152:1071–1078. doi: 10.1083/jcb.152.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olofsson SO, Bostrom P, Andersson L, Rutberg M, Levin M, Perman J, Boren J. Triglyceride containing lipid droplets and lipid droplet-associated proteins. Curr Opin Lipidol. 2008;19:441–447. doi: 10.1097/MOL.0b013e32830dd09b. [DOI] [PubMed] [Google Scholar]
- 40.Fei W, Shui G, Gaeta B, Du X, Kuerschner L, Li P, Brown AJ, Wenk MR, Parton RG, Yang H. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J Cell Biol. 2008;180:473–482. doi: 10.1083/jcb.200711136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RG, Goodman JM. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A. 2007;104:20890–20895. doi: 10.1073/pnas.0704154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandez MA, Albor C, Ingelmo-Torres M, Nixon SJ, Ferguson C, Kurzchalia T, Tebar F, Enrich C, Parton RG, Pol A. Caveolin-1 is essential for liver regeneration. Science. 2006;313:1628–1632. doi: 10.1126/science.1130773. [DOI] [PubMed] [Google Scholar]
- 43.Ae Kim C, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O'Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J. Association of a homozygous nonsense Caveolin-1 mutation with Berardinelli-Seip Congenital Lipodystrophy. J Clin Endocrinol Metab. 2008 doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- 44.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109–112. doi: 10.1093/hmg/9.1.109. [DOI] [PubMed] [Google Scholar]
- 45.Agarwal AK, Garg A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-γ gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab. 2002;87:408–411. doi: 10.1210/jcem.87.1.8290. [DOI] [PubMed] [Google Scholar]
- 46.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, Dunger DB, Barford D, Umpleby AM, Wareham NJ, Davies HA, Schafer AJ, Stoffel M, O'Rahilly S, Barroso I. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71:426–431. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 49.Peters JM, Barnes R, Bennett L, Gitomer WM, Bowcock AM, Garg A. Localization of the gene for familial partial lipodystrophy (Dunnigan variety) to chromosome 1q21-22. Nat Genet. 1998;18:292–295. doi: 10.1038/ng0398-292. [DOI] [PubMed] [Google Scholar]
- 50.Garg A, Peshock RM, Fleckenstein JL. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety) J Clin Endocrinol Metab. 1999;84:170–174. doi: 10.1210/jcem.84.1.5383. [DOI] [PubMed] [Google Scholar]
- 51.Garg A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety) J Clin Endocrinol Metab. 2000;85:1776–1782. doi: 10.1210/jcem.85.5.6605. [DOI] [PubMed] [Google Scholar]
- 52.Haque WA, Oral EA, Dietz K, Bowcock AM, Agarwal AK, Garg A. Risk factors for diabetes mellitus in familial partial lipodystrophy, Dunnigan variety. Diabetes Care. 2003;26:1350–1355. doi: 10.2337/diacare.26.5.1350. [DOI] [PubMed] [Google Scholar]
- 53.Hegele RA. Premature atherosclerosis associated with monogenic insulin resistance. Circulation. 2001;103:2225–2229. doi: 10.1161/01.cir.103.18.2225. [DOI] [PubMed] [Google Scholar]
- 54.Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino terminal head and alpha-helical rod domain of the lamin A/C (LMNA) gene. Am J Med. 2002;112:549–555. doi: 10.1016/s0002-9343(02)01070-7. [DOI] [PubMed] [Google Scholar]
- 55.Semple RK, Chatterjee VK, O'Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116:581–589. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herbst KL, Tannock LR, Deeb SS, Purnell JQ, Brunzell JD, Chait A. Kobberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care. 2003;26:1819–1824. doi: 10.2337/diacare.26.6.1819. [DOI] [PubMed] [Google Scholar]
- 57.Simha V, Garg A. Body fat distribution and metabolic derangements in patients with familial partial lipodystrophy associated with mandibuloacral dysplasia. J Clin Endocrinol Metab. 2002;87:776–785. doi: 10.1210/jcem.87.2.8258. [DOI] [PubMed] [Google Scholar]
- 58.Miyoshi Y, Akagi M, Agarwal AK, Namba N, Kato-Nishimura K, Mohri I, Yamagata M, Nakajima S, Mushiake S, Shima M, Auchus RJ, Taniike M, Garg A, Ozono K. Severe mandibuloacral dysplasia caused by novel compound heterozygous ZMPSTE24 mutations in two Japanese siblings. Clin Genet. 2008;73:535–544. doi: 10.1111/j.1399-0004.2008.00992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simha V, Agarwal AK, Oral EA, Fryns JP, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab. 2003;88:2821–2824. doi: 10.1210/jc.2002-021575. [DOI] [PubMed] [Google Scholar]
- 60.Worman HJ. Nuclear envelope proteins and human disease. Symp Soc Exp Biol. 2004:41–55. [PubMed] [Google Scholar]
- 61.Muchir A, Worman HJ. The nuclear envelope and human disease. Physiology (Bethesda) 2004;19:309–314. doi: 10.1152/physiol.00022.2004. [DOI] [PubMed] [Google Scholar]
- 62.Caron M, Auclair M, Donadille B, Bereziat V, Guerci B, Laville M, Narbonne H, Bodemer C, Lascols O, Capeau J, Vigouroux C. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14:1759–1767. doi: 10.1038/sj.cdd.4402197. [DOI] [PubMed] [Google Scholar]
- 63.Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 64.Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin A. J Cell Sci. 1994;107(Pt 1):61–67. doi: 10.1242/jcs.107.1.61. [DOI] [PubMed] [Google Scholar]
- 65.Koenig R, Brendel L, Fuchs S. SHORT syndrome. Clin Dysmorphol. 2003;12:45–49. doi: 10.1097/00019605-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 66.Lipson AH, Cowell C, Gorlin RJ. The SHORT syndrome: further delineation and natural history. J Med Genet. 1989;26:473–475. doi: 10.1136/jmg.26.7.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rautenstrauch T, Snigula F, Krieg T, Gay S, Muller PK. Progeria: a cell culture study and clinical report of familial incidence. Eur J Pediatr. 1971;124:101–111. doi: 10.1007/BF00477545. [DOI] [PubMed] [Google Scholar]
- 68.Wiedemann HR. An unidentified neonatal progeroid syndrome: follow-up report. Eur J Pediatr. 1979;130:65–70. doi: 10.1007/BF00441901. [DOI] [PubMed] [Google Scholar]
- 69.Pivnick EK, Angle B, Kaufman RA, Hall BD, Pitukcheewanont P, Hersh JH, Fowlkes JL, Sanders LP, O'Brien JM, Carroll GS, Gunther WM, Morrow HG, Burghen GA, Ward JC. Neonatal progeroid (Wiedemann-Rautenstrauch) syndrome: report of five new cases and review. Am J Med Genet. 2000;90:131–140. [PubMed] [Google Scholar]
- 70.Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–2624. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 71.Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO, 3rd, Gahl WA, Introne WJ. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J. LMNA mutations in atypical Werner's syndrome. Lancet. 2003;362:440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- 73.Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, Christin-Maitre S. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocrinol Metab. 2003;88:1006–1013. doi: 10.1210/jc.2002-021506. [DOI] [PubMed] [Google Scholar]
- 74.Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79:383–389. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cao H, Alston L, Ruschman J, Hegele RA. Heterozygous CAV1 frameshift mutations (OMIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids in health and disease. 2008;7:3. doi: 10.1186/1476-511X-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berger JR, Oral EA, Taylor SI. Familial lipodystrophy associated with neurodegeneration and congenital cataracts. Neurology. 2002;58:43–47. doi: 10.1212/wnl.58.1.43. [DOI] [PubMed] [Google Scholar]
- 77.Razani B, Lisanti MP. Caveolin-deficient mice: insights into caveolar function human disease. J Clin Invest. 2001;108:1553–1561. doi: 10.1172/JCI14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murphy DJ, Vance J. Mechanisms of lipid-body formation. Trends Biochem Sci. 1999;24:109–115. doi: 10.1016/s0968-0004(98)01349-8. [DOI] [PubMed] [Google Scholar]
- 79.Thiele C, Spandl J. Cell biology of lipid droplets. Curr Opin Cell Biol. 2008;20:378–385. doi: 10.1016/j.ceb.2008.05.009. [DOI] [PubMed] [Google Scholar]