

Abstract
The 2.4 Å crystal structure of the β2-adrenergic receptor (β2AR) in complex with the high-affinity inverse agonist (-)-carazolol provides a detailed structural framework for the analysis of ligand recognition by adrenergic receptors. Insights into agonist binding and the corresponding conformational changes triggering GPCR activation mechanism are of special interest. Here we show that while the carazolol pocket captured in the β2AR crystal structure accommodates (-)-isoproterenol and other agonists without steric clashes, a finite movement of the flexible extracellular part of TM-V helix (TM-Ve) obtained by receptor optimization in the presence of docked ligand can further improve the calculated binding affinities for agonist compounds. Tilting of TM-Ve towards the receptor axis provides a more complete description of polar receptor/ligand interactions for full and partial agonists, by enabling optimal engagement of agonists with two experimentally identified anchor sites, formed by Asp113/Asn312 and Ser203/Ser204/Ser207 side chains. Further, receptor models incorporating a flexible TM-V backbone allow reliable prediction of binding affinities for a set of diverse ligands, suggesting potential utility of this approach to design of effective and subtype-specific agonists for adrenergic receptors. Systematic differences in capacity of partial, full and inverse agonists to induce TM-V helix tilt in the β2AR model suggest potential role of TM-V as a conformational “rheostat” involved in the whole spectrum of β2AR responses to small molecule signals.
Keywords: Adrenergic, GPCR, G-protein, agonist, antagonist, activation, flexible docking, binding energy, conformational change
Introduction
A diverse family of more than 800 heptahelical G-protein coupled receptors (GPCRs) plays a critical role in recognition of neurotransmitters, cytokines, hormones, light and other extracellular signals and comprises targets for about a half of existing drugs (Lagerstrom and Schioth 2008; Tyndall and Sandilya 2005). While detailed knowledge of ligand-receptor interactions would be instrumental in design of new and improved clinical candidates, the insight into spatial structure of GPCR has been limited to ab initio models (Goddard and Abrol 2007) or models based on rhodopsin crystal structure (Palczewski and others 2000). The first high resolution crystal structure of a GPCR with diffusible ligand has been determined recently for β2-adrenergic receptor (β2AR) (Cherezov and others 2007; Rosenbaum and others 2007) revealing in atomic detail the binding interactions for an inverse agonist (-)-carazolol (PDB code 2RH1). This structure, followed by other co-crystals of β2AR (Hanson and others 2008b) and β1AR (Warne and others 2008) with the antagonists (-)-timolol (PDB code 3D4S) and (-)-cyanopindolol (PDB code 2VT4) respectively, represents a leap forward in the understanding of the inactive state of adrenergic GPCRs. At the same time, its potential utility as a structural template for the prediction of agonist binding conformations and relative affinities requires careful assessment (Kobilka and Schertler 2008). Indeed, the binding of agonists, especially full agonists, is expected to be accompanied by some conformational changes in the β2AR, which are reflected in highly synergistic contributions of the agonist functional groups in the receptor binding and activation (Del Carmine and others 2004; Liapakis and others 2000; Liapakis and others 2004). Kinetics studies (Kobilka 2007; Swaminath and others 2004) also support existence of early intermediate states in the β2AR induced by agonist recognition and binding. Fast initial adjustments of the receptor to an agonists binding may be followed by slower downstream changes potentially involving a rotamer “toggle switch” (Shi and others 2002) and movements of TM domains (Schwartz and others 2006)), leading to GPCR activation.
Extensive mutation analysis has established that a full β2AR agonist isoproterenol and related catecholamine compounds engage specific amino acid side chains of transmembrane (TM) helical domains III, V, VI and VII (Hannawacker and others 2002; Kobilka 2007; Liapakis and others 2000; Sato and others 1999; Strader and others 1989; Xhaard and others 2006) in a pocket that largely overlaps with the binding pocket of carazolol in the β2AR co-crystal (Cherezov and others 2007). Some of these receptor interactions are common for agonists and antagonists alike, while the others are specific to agonists only. Thus, most β-adrenergic agonists and antagonists have a positively charged amine or ethanolamine groups (“tails”) which has been shown to interact with the receptor anchor site formed by Asp1133.32 and Asn3127.39 side chains (Strader and others 1988; Suryanarayana and Kobilka 1993) (superscript numbering according to ref.(Ballesteros and Weinstein 1995)). However, a strong polar interaction network between the catechol functional group (“head”) and another anchor site formed by serines Ser2035.42, Ser2045.43, Ser2075.46 on TM-V is specific for agonists only; this network is most extensive for “full” agonists, conferring maximal activity of the β2AR. Both “tail” and “head” anchor interactions are critical for agonist specific β2AR activation, as removal of any of the corresponding polar moieties in the agonists or in the receptor has been shown to dramatically reduce agonist activity (Ambrosio and others 2000; Liapakis and others 2004; Strader and others 1989). Initial analysis of agonist binding geometry in ref. (Rosenbaum and others 2007) however, shows that when ethanolamine tail of isoproterenol is superimposed onto the corresponding atoms of carazolol in the β2AR crystal structure, the cathechol hydroxyls of the agonist are too distant from the TM-V serines to form the anchor hydrogen bonds. These simple geometry considerations suggest that some adjustments in the binding pocket, and specifically in the TM5 domain, may be required to achieve optimal agonist binding (V.K., V.C, M.A.H., C.B.R and R.A. unpublished)(Warne and others 2008).
Herein we report the results of more rigorous, energy-based conformational modeling of representative antagonists and agonists (Figure 1), which supports agonist-specific changes in the β2AR binding pocket. The fully flexible ligands were docked into the pocket using three distinct approaches, in which β2AR was represented as 1) a rigid receptor derived from the pdb-deposited coordinates of the heavy atoms (2RH1), 2) a receptor with flexible side chains in the binding pocket, and 3) a receptor with flexibility in side chain conformation and limited flexibility in the protein backbone of the TM-V helix. Using the first and the second approaches we demonstrate that the binding pocket conformation of the carazolol-β2AR complex is sterically compatible with isoptorerenol and other full agonists, and that binding can be somewhat improved through rotamer changes in Ser2035.42, Ser2045.43 and Ser2075.46 side chains. However, optimal engagement of both experimentally determined ligand/receptor anchor interactions cannot be achieved without additional flexibility in the β2AR backbone, as in the third approach. The predicted backbone movements comprise a finite (∼2Å) “inward” tilt of the mobile extracellular segment of TM-V α-helix (TM-Ve), which improves the calculated binding affinity for full agonists as much as ∼1000 fold. Further, incorporation of a flexible TM-V backbone allows consistent prediction of experimental binding affinities for a diverse set of β2AR ligands from full agonists to inverse agonists. Interestingly, the optimal TM-Ve tilt and the accompanying improvement in calculated binding affinity are less pronounced for partial agonists; the movement is completely abolished for the antagonists and inverse agonists studied here. This strongly differentiating response to agonist and antagonist binding suggests a potential role of TM-Ve tilt as a regulator in the initial stages of the signal transduction mechanism in β2AR and closely related aminergic receptors.
Fig 1.

Chemical structures of β2AR ligands used in this study. Note that (S)-β-OH chiral center of (-)-carazolol is sterically equivalent to (R)-β-OH of (-)-isoproterenol and other chiral agonists shown. To avoid confusion we will use only (-) or (+) notion to indicate chirality of ligands in this paper.
Results
Energy based refinement of the β2AR model for antagonists/inverse agonist binding
To establish the suitability of the β2AR crystal structure for the prediction of ligand conformations, we started with docking of (-)-carazolol and several other antagonists/inverse agonists into “rigid” and “flexible side chain” models of the receptor. As shown in Figure 2A, redocking of (-)-carazolol into the rigid model of β2AR (PDB code 2rh1) consistently reproduced the X-ray coordinates of the ligand heavy atoms with RMSD ∼ 0.3 Å. Similarly, docking into the β2AR model with flexible side chains resulted in (-)-carazolol geometry nearly identical to the crystal structure (RMSD ∼ 0.25 Å). The β2AR binding pocket conformation was also preserved, with the exception of two serine side chains in the TM-V domain (Figure 2B). Most notably, the energy-optimized rotamer of Ser2035.42 side chain significantly improved hydrogen bonding to carbazole with N-O distance reduced from 3.3 Å to 2.7 Å, and also allowed formation of a hydrogen bond to Tyr1995.38 backbone carbonyl. The latter type of Ser/Thr side chain to main chain H-bonds (Oγ(i)--O(i-4)), frequently found in the middle of α-helices is considered as an important protein stabilizing interaction (Ballesteros and others 2000; Eswar and Ramakrishnan 2000). The predicted rotamer of another, Ser2045.43 side chain supported an improved geometry of hydrogen bonding with Ala2005.39 backbone. Thus, this model suggests a possibility of hydrogen bond pairing between Ser2045.43 hydroxyl and Asn2936.55 side chain nitrogen, albeit with a marginal O-N distance (3.3.Å). The optimized rotamers for both Ser2035.42 and Ser2045.43 side chains are consistent with the electron densities observed for the β2AR-carazolo crystal structure (Cherezov and others 2007). Moreover, the predicted configuration of Ser2045.43 side chain and its interaction network can be found in the β2AR crystal structure with timolol (PDB code 3D4S)..
Fig 2.
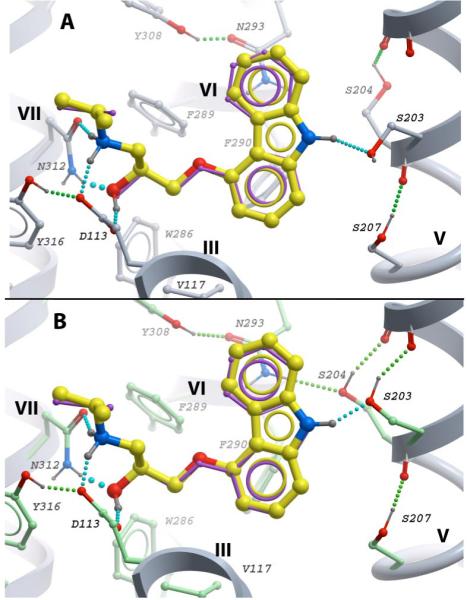
Predicted conformations of (-)-carazolol in (A) rigid and (B) flexible side chain β2AR models. The protein models are shown as grey backbone ribbon; β2AR side chains in direct contact with carazolol are shown as sticks, except for Tyr1995.38, Val1143.33 and Phe1935.32 in front of the ligand, which are omitted for clarity. Rigid side chains are shown as sticks with grey carbon atoms, flexible side chains with green carbon atoms. The exact position of (-)-carazolol in the crystal structure is shown by thin purple sticks, the predicted ligand poses are shown by thicker sticks with yellow carbon atoms. Ligand-receptor hydrogen bonds are shown by chains of cyan balls; intracellular hydrogen bonds for the binding pocket side chains are shown as chains of green balls. The models and graphics in Fig 2 and other figures in this study are generated with ICM-Pro software (Molsoft LLC).
Docking of other antagonists/inverse agonists in the β2AR flexible side chain model also resulted in ligand conformations similar to (-)-carazolol, with some notable variations. Thus, for the (+)-carazolol stereoisomer model, the switch from (-) to (+) stereoisomer of the ethanolamine “tail” resulted in partial loss of hydrogen bonding network with Asp1133.32/Asn3127.39 anchor site, which is in line with a substantial (∼20 fold) stereo-selectivity towards (-)-carazolol. (All-atom models for these and other ligands can be found in Supplementary Materials in pdb format)
Full agonist (-)-isoproterenol binding to the β2AR receptor
β2AR agonists are expected to bind to different conformational states of receptor than antagonists or inverse agonists. Therefore, one of the questions we need to answer is whether binding of catecholamine agonists such as (-)-isoproterenol is sterically compatible with the carazolol-bound conformational state of the β2AR captured in the crystal structure (Cherezov and others 2007). The second question is whether specific changes in β2AR model side chain or backbone conformation can be identified that substantially improve agonist binding affinity and its interactions with known anchor sites.
(-)-Isoproterenol in the rigid and flexible side chain β2AR models
Docking into the rigid model of the β2AR consistently yielded a single low energy conformation of (-)-isoproterenol (Figure 3A), where the ligand fitted into the carazolol-binding pocket without any apparent steric clashes. Moreover, a detailed analysis of (-)-isoproterenol interactions shows energetically favorable contribution to the ligand binding energy for all 14 contact residues in the “rigid” β2AR pocket. At the same time the model in Figure 3A shows that when the ethanolamine “tail” of (-)-isoproterenol is anchored at the Asp1133.32/Asn3127.39 anchor site, its catechol “head” is unable to reach hydroxyls of Ser2035.42, Ser2045.43, and Ser2075.46 in TM-V. This is also consistent with the observation by Rosenbaum et al based on a simple superposition of ethanolamine “tails” of carazolol and isoproterenol in the β2AR binding pocket (Rosenbaum and others 2007).
Fig 3.

Structural modeling of agonist (-)-isoproterenol in the β2AR binding site. View point and color scheme as in Figure 2. A) The best energy conformation of (-)-isoproterenol, optimized in the rigid β2AR model. Distances from catechol hydroxyls to TM-V serines are shown as red dashed lines. B) One of the two local minima conformations for (-)-isoproterenol in β2AR model with flexible side chains; optimal hydrogen bond network with TM-V serines shown C) An alternative local minimum conformation of (-)-isoproterenol with optimal interaction network between ethanolamine tail and Asp1133.32/ Asn3127.39 anchor site. Distances to TM-V serines are shown as red dashed lines.
Docking of (-)-isoproterenol in the flexible side chain model of β2AR provides more rigorous evaluation of agonist interactions with the receptor anchor sites (Figures 3B and 3C). The results show that although rotameric adjustments in the TM-V serines and other residues in the vicinity of the binding site substantially improve binding of (-)-isoproterenol, the formation of optimal hydrogen bonding interactions between the ligand and both interaction sites remains physically impossible. Interestingly, instead of one global energy minimum, the docking into the flexible side chain model of the β2AR resulted in two clusters of (-)-isoproterenol conformations. The majority of docking runs (15 out of 20) yielded a low energy pose shown in Figure 3B, where (-)-isoproterenol is shifted towards TM-V, allowing the catechol “head” to form a strong hydrogen bond network with the TM-V serines. However, this position of the ligand forced a rearrangement and partial loss of the ethanolamine group interactions with the Asp1133.32-Asn3127.39 site. In contrast, in the remaining five runs of the docking procedure the complex fully retained the interaction network for the isoproterenol ethanolamine group with the Asp1133.32/Asn3127.39 anchor, while stretching the ligand catechol group toward the TM-V serines (Figure 3C). Despite the agonist being almost perfectly aligned between the anchor sites, the distances between the catechol and the hydroxyls of TM-V serines in this conformation were still too great to allow formation of a hydrogen bond network. This analysis demonstrates that side chain flexibility alone does not provide enough adjustment in the β2AR model to satisfy the experimental constraints associated with the binding of full agonists to the two anchor sites simultaneously. Therefore, some changes in the protein backbone conformation, as compared to β2AR-carazolol complex, are required to engage both functional groups of the full agonist.
TM-V helix tilt upon (-)-isoproterenol binding
Because one of the two critical anchor sites for agonist binding is located on the extracellular part of TM-V, the optimal binding arrangement may be achieved by movement of the TM-V domain towards Asp1133.32/Asn3127.39 on TM-III/TM-VII. The extracellular α-helical segment of TM-V (residues 197-204, further referred to as “TM-Ve”) is flanked by a relatively flexible loop EL2 and a helical “bulge” above the highly conserved Pro2115.50 residue, which suggests an enhanced mobility of TM-Ve. The possibility of an inward movement of TM-Ve is further supported by previous studies reporting conformational flexibility in this region (Javitch and others 2002; Javitch and others 1995) (Chelikani and others 2007). In contrast, a significant movement of helices TM-III/TM-VII towards TM-V appears to be less likely, as TM-III is known to form a tight folding core of β2AR together with helices TM-I/TM-II/TM-IV (Chelikani and others 2007).
To analyze a possibility of TM-Ve movement, we developed a conformational model of the β2AR with backbone flexibility introduced in a portion of the extracellular loop EL2 (residues 191-196) and in the proline-induced bulge of TM-V (residues 205-210), as described in Methods. The backbone flexibility, combined with flexibility of side chains in the proximity of the binding site, allow for substantial motility of the TM-Ve segment. Docking of (-)-isoproterenol into this β2AR model consistently resulted in a receptor conformation with an inward tilt of the TM-Ve helix, as shown in Figure 4. The TM-Ve tilt corresponds to an approximately 2 Å shift of the TM-V anchor site toward helices TM-III/TM-VII, as measured for Ser2035.42 Cα atom. Analysis of H-bonding distances in this model shows that such TM-Ve tilt is sufficient to bring the two anchor sites of the binding pocket within optimal distance for interactions with both the catechol and ethanolamine groups of isoproterenol simultaneously (see Table 1). The move of the TM-V extracellular domain toward the ligand binding pocket does not result in any serious steric overlaps with other helices of the protein, and is accompanied by only minor adjustment of the flexible side chains on the helix-helix interfaces. The resulting improvement in (-)-isoproterenol binding affinity upon TM-Ve tilt was predicted to be as high as 3 pKd, i.e. about 1000-fold.
Fig 4.
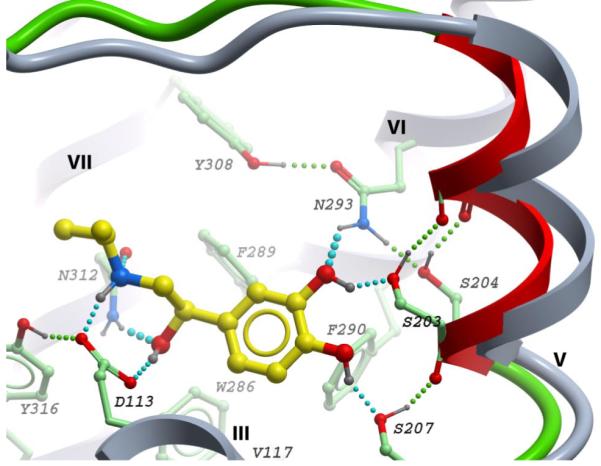
The best energy conformation of (-)-isoproterenol in β2AR model with backbone flexibility (colored green) in the TM-V proline-induced kink (residues 205-210) and a portion of EL2 loop (191-196). The extracellular portion of TM-V (TM-Ve) is shown as red ribbon. The original position of TM-Ve and other static helices are shown as grey ribbon, flexible side chains in proximity of the binding site have carbon atoms colored green. Supplementary materials contain 3D atomic coordinates for this model, as well as for β2AR models with other ligands studied in this work.
Table 1.
Receptor-ligand hydrogen bond distances for different conformations of the β2AR- (-)-isoproterenol complex. The columns correspond to models in Figure 3B, Figure 3C and Figure 4 respectively. Distances, not compatible with hydrogen bonding are highlighted by bold type
| Hydrogen bonds (β2AR : Isoproterenol) | Donor-Acceptor Distance, Å | ||
|---|---|---|---|
| Catechol-anchored | Amino-anchored | Flexible TM-V model | |
| OD1 (Asp113) - N+ (amino) | 2.7 | 2.7 | 2.9 |
| OD2(Asp113) - O (β-OH) | 2.5 | 2.7 | 2.7 |
| OD1 (Asn312) - N+ (amino) | 4.4 | 2.9 | 3.1 |
| ND2 (Asn312) - O (β-OH) | 6.0 | 3.2 | 3.2 |
| OG(Ser207) - O (para) | 2.7 | 3.3 | 2.6 |
| OG(Ser203) - O (para) | 3.0 | 4.5 | 3.0 |
| OG(Ser203) - O (meta) | 2.7 | 5.1 | 2.6 |
| OG(Ser204) - O (meta) | 3.5 | 4.1 | 3.9 |
The conformational model of the β2AR-isoproterenol complex consistently reproduced H-bonding of catechol meta-OH with Ser2035.42 and para-OH with Ser2075.46 side chains, as previously derived from mutation studies (Liapakis and others 2000; Strader and others 1989). At the same time, the model predicts an alternative interaction network for Ser2045.43 side chain, where instead of directly engaging (-)-isoproterenol meta-OH (Strader and others 1989), the Ser2045.43 hydroxyl is involved in two intramolecular hydrogen bonds. This intramolecular network, also involving the Tyr3087.35 and Asn2936.55 side chains and Ala2005.39 main chain (e.g. Figure 2C) can be found in the crystal structure of the β2AR-timolol complex (Hanson and others 2008a). The modeling results also suggested that (-)-isoproterenol binding and the corresponding TM-Ve tilt can significantly improve this interaction network by reducing the donor-acceptor distance in Asn2936.55-Ser2045.43 hydrogen bond from 3.3 to 2.6 Å (compare Figures 2C and 4).
TM-V movement and binding affinity predictions for a diverse set of β2AR agonists
The β2AR conformational models, both with rigid and with flexible TM-V backbone, were applied to predict binding poses and affinities for a set of diverse β2AR agonists. Interestingly, TM-Ve flexibility produced a marked improvement in the calculated binding affinities for agonist compounds while having little effect on antagonist binding affinities (results summarized in Table 2). Full agonists were all predicted to have 2 - 3 pKd (100-1000 fold) improved binding affinities upon a significant (1.6 - 2.2 Å) inward shift of the TM-Ve anchor site. In contrast, for the antagonist and the inverse agonist complexes the predicted position of TM-Ve remains close to the β2AR-carazolol crystal structure with little change in the calculated binding affinity. For partial agonists the amount of TM-Ve tilt and its effect on binding affinities are intermediate, with the smallest affinity difference predicted for MAPE at 0.5 pKd.
Table 2.
Predicted ligand binding affinities for β2AR conformational models with/without TM-V backbone flexibility and corresponding shift of TM-V anchor site. Calculations for TA-2005 performed with the wild-type β2AR and two Tyr3087.35 mutants
| Ligand | Predicted affinitya pKdPred | Predicted TM-V anchor shift, Å | Predicted pKd gain due to TM-V shift | Measured Affinity, pKd | |
|---|---|---|---|---|---|
| Rigid TM-V | Flexible TM-V | ||||
| Inverse agonists and antagonists | |||||
| (-)-Carazolol | 11.0 | 11.0 | 0.0 | 0.0 | 10.7b |
| (+)-Carazolol | 9.2 | 9.2 | 0.0 | 0.0 | 9.3b |
| (-)-Propranolol | 8.2 | 8.2 | 0.0 | 0.0 | 9.3c |
| (-)-Pindolol | 8.3 | 8.7 | 0.2 | 0.3 | 9.3c |
| Full agonists | |||||
| (-)-Isoproterenol | 4.8 | 7.8 | 2.0 | 3.0 | 7.5c |
| (+)-Isoproterenol | 3.9 | 6.5 | 1.6 | 2.5 | 5.7c |
| (-)-Epinephrine | 2.6 | 5.7 | 2.0 | 3.1 | 6.5c |
| (-)-Norepinephrine | 1.7 | 4.0 | 1.6 | 2.3 | 5.4c |
| (R,R)-TA-2005 | 5.1 | 8.3 | 2.2 | 3.2 | 8.3d |
| (R,R)-TA-2005 (Y308F) | 4.1 | 7.6 | 1.9 | 3.5 | 7.2d |
| (R,R)-TA-2005 (Y308A) | 3.8 | 7.1 | 2.1 | 3.3 | 7.1d |
| Partial agonists | |||||
| (-)-Salbutamol | 5.8 | 7.1 | 1.1 | 1.3 | 6.4c |
| Dopamine | 2.7 | 4.1 | 1.4 | 1.4 | 4.1c |
| MAPE | 3.2 | 3.7 | 1.8 | 0.5 | 4.6c |
| (-)-Clenbuterol | 6.2 | 7.0 | 0.9 | 0.9 | 7.5c |
| (-)-Isopropyl-norsynephrine | 4.6 | 6.2 | 1.7 | 1.6 | 5.4c |
| Adrenalone | 4.5 | 4.7 | 1.6 | 0.1 | 5.1c |
Calculated as predicted binding energy normalized to the measured affinity scale: pKdpred = 0.313*ΔGpred - 5.8
data in ref. (Manalan and others 1981)
data in ref. (Del Carmine and others)
data in ref. (Kikkawa and others 1998)
The correlation of predicted and experimental binding affinities for the diverse set of full, partial and inverse agonists/antagonists is illustrated in Figure 5A. Note, that while the model with the rigid TM-V backbone has rather poor accuracy of the binding affinity predictions (R2∼0.75 and RMSD∼ 1.3 pKd), the flexible TM-V model improves accuracy to R2=0.89 and RMSD ∼ 0.7 pKd.
Fig 5.

A) Comparison of the predicted and measured ligand binding affinities (pKd), as listed in Table 3 of the paper. Affinities are shown for β2AR conformational models with rigid (brown crosses) and flexible (blue diamonds) TM-V backbone. The arrows illustrate improvements in the ligand binding affinity from the rigid TM-V model to the flexible TM-V model for agonists isoproterenol (dark blue), epinephrine (light blue), dopamine (brown) and MAPE (plum). Accuracy of affinity predictions estimated as R2=0.75, RMSE=1.3 pKd for the rigid backbone model, and R2=0.89, RMSE=0.7 pKd for the flexible TM-V model.
B) Binding affinities for inverse agonist carazolol, full agonists (-)-isoproterenol and (-)-epinephrine, as well as partial agonists dopamine and MAPE, calculated as a function of TM-V anchor site shift. Results of single-point analysis for agonists from Table 3 are shown by bigger shapes.
For carazolol and a few representative agonists we also performed a more detailed conformational analysis, by evaluating ligand binding in β2AR models with a range of fixed positions of the TM-Ve helix. The resulting dependencies of the binding affinity as a function of the TM-Ve shift in Figure 5B are in good agreement with the “single point” data n Figure 5A. While calculated affinity of (-)-carazolol steadily decreases with the TM-V anchor shifting inward, the affinity curves for agonists show a range of improved affinity values. Note, that full agonists (-)-isoproterenol and (-)-epinephrine have similar shapes of the affinity curve with maxima at about 1.8-2.2 Å shift of the TM-Ve anchor, this distinct shape may reflect engagement of individual anchor residues in the binding site. Partial agonist dopamine has a relatively flat affinity profile between 0.9 and 1.4 Å shifts of the TM-Ve, while MAPE affinity is almost independent from the TM-Ve movements in the whole range studied. Further details of the 3D conformations and affinities for several representative full and partial agonists are presented in the following sections (the 3D models of these ligand-β2AR complexes can also be found in Supplementary Materials).
Full agonist binding
The endogenous adrenergic agonists (-)-epinephrine and (-)-norepinephrine are close chemical analogues of isoproterenol, with either a methyl group or a proton, respectively, at the tip of the ethanolamine “tail”. Both compounds promote full activation of the receptor, although with higher EC50 values than isoproterenol. Based on biochemical data, both ligands are expected to interact with the same anchor sites of β2AR as isoproterenol (Liapakis and others 2004). Indeed, docking of (-)-epinephrine and (-)-norepinephrine in the β2AR model with flexible TM-V backbone consistently predicts binding poses and interaction patterns very similar to those of (-)-isoproterenol, with the corresponding heavy atom RMSDs of 0.2 Å and 0.5 Å respectively. A slightly smaller shift of the TM-V anchor site found for (-)-norepinephrine complex, ∼1.6 Å, can be attributed to its smaller “tail” which allows more adjustment in the position of the anchor side chains Asp1133.32 and Asn3127.39. Also, lack of hydrophobic interaction with the aromatic system of Trp1093.28 is apparently responsible for reduced affinity of these ligands as compared to isoproterenol; this effect was quantitatively predicted by the corresponding models, as shown in Table 2.
Partial agonist binding
Dopamine, salbutamol and MAPE (halostachine) are partial agonists of the β2AR, and may interact differently with the receptor, perhaps stabilizing different conformational states as compared to the full agonists (Baker 2005; Kikkawa and others 1997; Seifert and others 2001; Swaminath and others 2005). As illustrated in Figure 6A, dopamine is predicted to bind to the same binding pocket as isoproterenol, forming hydrogen bond networks with TM-V serines through its catechol head. In the absence of the alkyl-hydroxyl moiety, the ligand’s tail binds only through the amino group interactions with the Asp1133.32/Asn3127.39 anchor. The spatial position of dopamine amino tail in the anchor site is not as well defined as for ethanolamine tail common for other ligands. This difference is reflected in a wider peak of the affinity profile in Figure 5B, smaller TM-Ve movement and smaller energy gain, as compared to (-)-isoproterenol (see Table 2).
Fig 6.

Predicted dopamine (A), salbutamol (B) and MAPE (C) binding into β2AR model with flexible side chains. Color scheme as in Fig 4.
Salbutamol docking to the flexible TM-V model of the β2AR (Figure 6B) indicates that the replacement of the ligand’s meta-OH with hydroxymethyl group can dramatically modify its interaction pattern with TM-V serines. Thus, the model suggests that in contrast to catechol meta-OH, the meta-hydroxymethyl group of salbutamol may serve as a hydrogen bond acceptor for both Ser2035.42 and Ser2045.43. The formation of these two H-bonds, though, is offset by the loss of two intramolecular side-chain to main-chain H-bonds, and therefore it is unlikely to contribute significantly to the overall ligand binding affinity. The modified aromatic head of salbutamol also allows an adjustment of the molecule and its ethanolamine tail position toward the TM-III/TM-VII anchor. This, in turn results in a smaller optimal shift of TM-Ve anchor site (0.7 Å) as compared to the value predicted for isoproterenol-bound β2AR. A close similarity of salbutamol to isoproterenol and the observed consistency of the docking poses suggest that salbutamol can occupy the same pocket as the other agonists in our study. The previously observed non-competitive binding of catechol molecules to salbutamol-β2AR complex (Kobilka 2007) can possibly be explained by an alternative binding pose of the catechol itself, which is a small and non-specific compound.
MAPE is a partial β2AR agonist lacking both hydroxyls in the aromatic “head” of the molecule, and thus unable to form any hydrogen bonds with TM-V serines. Though the MAPE binding pose in the model in Figure 6C leaves a space for TM-V inward tilt, the corresponding gain in ligand binding affinity was predicted to be small (>0.5 pKd), as compared to ∼2.7 pKd for (-)-isoproterenol (see Figure 5B and Table 2).
Binding and selectivity of a non-catechol full agonist TA-2005
The above analysis was performed for a representative set of full and partial agonists of the β2AR with relatively broad specificity across the β-adrenergic family. However, the molecular basis of agonist selectivity for the β-adrenergic receptors is of special interest for clinical applications, where β1AR antagonists (“β-blockers”) are widely used for treatment of heart disease and β2AR agonists are indicated for asthma (Baker 2005). A comparison of residues in the β2AR binding pocket with the equivalent positions in the other members of the β-adrenergic family reveals that the residues predicted to interact with the endogenous agonists are highly conserved. The one non-conserved residue, Tyr3087.35 (Phe359 in β1AR), has been previously implicated in the high binding selectivity of the non-catechol full agonist TA-2005 for the β2AR (Kikkawa and others 1997; Kikkawa and others 1998).
To analyze the basis for TA-2005 specificity at the structural level, we performed docking of the ligand to β2AR in the framework of the flexible TM-V backbone model described above. The lowest energy conformation of the complex (Figure 7) has a hydrogen bond between the oxygen of the p-methoxyphenyl group of TA-2005 and Tyr3087.35, in addition to the anchor interactions with Asp1133.32/Asn3127.39 and Ser2035.42/Ser2075.46/Ser2045.43 sites similar to those predicted for isoproterenol. The model suggests that 8-hydroxy-carbostyril functional “head” can be as effective as the catechol moiety in its interaction with TM-V serines, and that optimal binding of TA-2005 requires a similar shift of the TM-V anchor site (2.2 Å) as for (-)-isoproterenol in the Figure 4 model.
Fig 7.
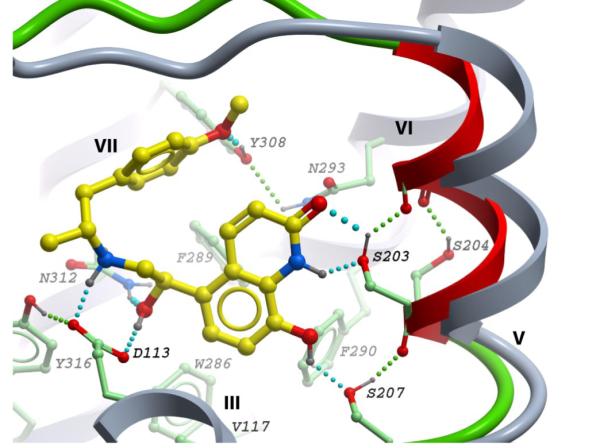
Predicted binding of TA-2005 into β2AR model with flexible side chains and TM-V backbone shift. Color scheme as in Fig 4.
The described model also provides a reasonable explanation of TA-2005 selectivity to β2AR, predicting approximately 1 pKd affinity drop for Y308F and Y308A mutations, as shown in Table 2 (Kikkawa and others 1998). Although other mechanisms can be also involved in selectivity for this and other β-adrenergic agonists, this example suggests a potential utility of the model in structure-based discovery of new subtype specific β-adrenergic ligands.
Discussion
The high resolution crystal structure of the β2AR complex with inverse agonist (-)-carazolol (Cherezov and others 2007) provides a solid template for analysis of the β2AR interactions with different classes of ligands. Although largely consistent with previous knowledge, the results of structure-based modeling of (-)-isoproterenol and other agonists suggests some new details of binding to the β2AR and associated conformational changes in the receptor.
Ethanolamine tail interactions with Asp1133.32 and Asn3127.39 anchor site
The crystal structure of the β2AR-carazolol complex (Cherezov and others 2007) reveals the geometry of the ethanolamine tail anchor site (see Figure 2 and Table 1), which involves both N+ and β-OH groups of the ligand and Asp1133.32 and Asn3127.39 side chains of the β2AR, and also a stabilizing Asp1133.32-Tyr3167.43 intramolecular H-bond. An exceptional strength of this polar interaction network suggests that it may be preserved for agonists as well, and indeed, we consistently found a very similar ethanolamine tail conformation for agonists in all rigid and flexible types of β2AR models (Figures 3AC, 4, 6B,C, 7).
While the salt bridge between Asp1133.32 and N+ is well established as a key anchor for both agonists and antagonists binding (Strader and others 1988), the details of β-OH - Asn3127.39 interaction were poorly understood. Some models suggested that another asparagine side chain, Asn2936.55 forms a hydrogen bond with the β-OH group of agonists, probably resulting in a β-OH “up” orientation towards the extracellular end of the receptor (Hannawacker and others 2002; Wieland and others 1996). The rationale for such models was based on mutation data (Del Carmine and others 2004; Del Carmine and others 2002), which implicated Asn2936.55 in binding affinity of catecholamine agonists.
At the same time, biochemical data from ref (Suryanarayana and Kobilka 1993), support involvement of Asn3127.39 side chain in β2AR H-bonding to both agonists and antagonists, because N312A mutation was found to be responsible for a ∼100-fold (∼2pKd) reduction of the binding affinity for both types of ligands. The β2AR crystal structure with carazolol and our β2AR models with agonists suggest a fully buried β-OH “down” conformation, involved in at least two hydrogen bonds with both Asp1133.32 and Asn3127.39 side chains, but not Asn2936.55 side chain located more than 10Å away. A switch of β-OH from “down” to “up” position for agonist would result in a loss of two strong buried H-bonds in exchange for a single solvent exposed H-bond to Asn2936.55, which is very unfavorable.
The results of agonist-β2AR modeling here support alternative mechanisms of Asn2936.55 side chain contribution to agonist binding affinity and stereospecificity. One of these mechanisms is illustrated by the model in Figure 4, which shows formation of a hydrogen bond between Asn2936.55 and the catechol meta-OH of (-)-isoproterenol. Another indirect contribution to agonist affinity may come from participation of the Asn2936.55 side chain in a stabilizing intramolecular H-bonding network with Ser2045.43 and Tyr3087.35 side chains (see Figure 4 and discussion of TM-V serines below).
Aromatic ring interactions with the hydrophobic patch of the β2AR pocket
Interaction of the ligand aromatic “head” with the hydrophobic patch in the middle part of the β2AR binding pocket is another interaction contributing to β2AR agonists, antagonists and inverse agonist binding. In the β2AR crystal structure with carazolol (Cherezov and others 2007), the hydrophobic contacts include Trp2866.48, Phe2906.52, Val1143.33, Val1173.36 - mostly with the “bottom” ring of the carbazole aromatic system, and also Phe1935.32, Tyr1995.38, Phe2896.51 - mostly with the “upper” ring of the carbazole. In our β2AR models with (-)-isoproterenol and other agonists, the hydrophobic contacts of the catechol ring are similar to those contacts of the “bottom” carbazole ring, with the only exception that a somewhat shifted position of the catechol rings allows an additional Van der Waals contact with Phe2896.51 side chain (Figure 4).
The (-)-isoproterenol molecule can adopt the same axial orientation of its aromatic “head”, as the one found for (-)-carazolol, despite its shorter linker to the ethanolamine “tail”. This makes (-)-isoproterenol sterically compatible with the carazolol-bound pocket in β2AR, and our modeling results confirm the absence of van der Waals clashes or torsional stress in (-)-isoproterenol even when it is docked into β2AR crystallographic coordinates (Figure 3A). A ready accommodation of (-)-isoproterenol within the carazolol-defined β2AR crystal structure is somewhat surprising, because significant rearrangements in the hydrophobic part of the binding pocket were expected between antagonist-bound and agonist-bound states. According to the “rotamer toggle switch” hypothesis (Shi and others 2002), for example agonist binding is expected to trigger rotation in Trp6.48 and Phe2906.52 in the binding pocket. Favorable contacts of these two aromatic side chains with both (-)-carazolol and (-)-isoproterenol in β2AR models do not support a direct mechanistic connection between agonist binding and conformational changes in Trp6.48/Phe2906.52 side chains. On the other hand, our results do not exclude “toggle switch” in these residues as a part of large scale downstream conformational changes in β2AR.
Role of TM-V serines in agonist binding
The most important anchor interaction specific for β2AR agonists is represented by a hydrogen bond network between catechol “head” moiety and serine side chains of the TM-V helical domain (Ambrosio and others 2000; Del Carmine and others 2002; Sato and others 1999; Strader and others 1989). Our modeling consistently predicts specific configurations of this interaction network for several catecholamine agonists (e.g. Figure 4). This extensive network combines not only three H-bonds involving both catechol hydroxyls, but also four intramolecular H-bonds involving Ser2035.42, Ser2045.43 and Ser2075.43 side chains.
Simple distance measurements in ref (Rosenbaum and others 2007) indicate that the “tail” (ethanolamine) and the “head” (catechol) anchor sites are too far apart in the β2AR-carazolol crystal structure to afford simultaneous contacts with isoproterenol. The conformational modeling in this study suggests that a finite tilt of the TM-V extracellular helix (TM-Ve) is required to resolve this discrepancy and bring the anchor sites closer together for optimal binding of isoproterenol and other full agonists. The tilt of TM-Ve helix and engagement of both anchors is associated with a major (∼1000 fold) improvement in the predicted binding affinity for isoproterenol and other full agonists to the β2AR. Such significant energetic coupling between binding of full agonists (but not antagonists) and TM-V tilt suggests important role of this movement in the conformational changes leading to β2AR activation.
A coordinated inward shift of all three TM-Ve serines is also in a good agreement with observed synergistic contribution of individual functional groups into affinity and efficacy of β2AR agonists (Del Carmine and others 2004; Liapakis and others 2000; Liapakis and others 2004). For example, results in ref. (Liapakis and others 2004) show that addition of both hydroxyl groups to the phenol ring of halostachine (HAL) increases its affinity to β2AR about 120-fold, while individual pOH and mOH additions yield only marginal (∼1 fold and ∼3.5 fold) contributions. Our conformational model provides a simple structural basis for such synergy: both catechol hydroxyls together can stabilize an optimal TM-Ve tilt to assure full engagement of corresponding H-bonds, whereas individual hydroxyls are not sufficient to shift TM-V and would make only suboptimal contact with TM-V serines, if any.
Our models consistently reproduce the well characterized H-bonding pattern of the catechol pOH with Ser2075.43 and mOH with Ser2035.42 (Liapakis and others 2000; Strader and others 1989). At the same time Ser2045.43 hydroxyl is predicted to participate in strong intramolecular bonding network in the β2AR but not in direct H-bonding with mOH of agonists, as proposed by early mutagenesis studies (Strader and others 1989). It is possible that the optimized conformation in our models reflects only one static snapshot of the dynamic polar interaction network in the β2AR-agonist complex. However, hydrogen bonding of Ser2045.43 with Asn2936.55 side chain and Ala2005.39 main chain has been also observed in the β2AR crystal structure with antagonist timolol (Hanson and others 2008a). According to our models (Figure 2C and 4) further stabilization of this interaction network, particularly improvement of Ser2045.43 - Asn2936.55 H-bond distance from 3.3 to 2.6 Å, can be achieved upon binding of a full agonist and inward tilt of TM-Ve domain. This stabilizing effect of Ser2045.43 - Asn2936.55 H-bond suggests a significant indirect contribution of Ser2045.43 side chain into agonist binding affinity, and also predicts significant impact of the S204A mutation on the basal activity of β2AR. Indeed, experimental results in ref.(Ambrosio and others 2000) provide some initial evidence in support of this effect by demonstrating decrease in β2AR basal activity by as much as 50%-60% upon the S204A/S207A double mutation. More specific measurements of individual effects of Ser2045.43 mutation on β2AR basal activity and ligand binding will be needed to resolve the direct and/or indirect contributions of Ser2045.43 to agonist binding.
Potential role of TM-V movements in the activation mechanism
Conformational changes required for the optimal accommodation of agonists in the β2AR binding pocket should be considered in the broader context of agonist-induced receptor activation. Previous biophysical studies have pointed to existence of several intermediate steps in the conformational changes associated with binding of full agonists to the β2AR (Del Carmine and others 2004; Liapakis and others 2004; Swaminath and others 2004). The multistep ligand binding hypothesis ((Kobilka and Deupi 2007) and (Kobilka 2007)) describes two fast steps, followed by a distinct slow step in the conformational changes. In the first step, an agonist is expected to fit into the relatively loose binding pocket and engage in polar interactions with either the ethanolamine anchor site (Asp1133.32/ Asn3127.39) or the catechol anchor site (TM-V serines). In the second step, specific for catecholamine-like agonists, conformational changes facilitate full engagement of the β2AR with both “head” and “tail” of the full agonist (Figure 4). Finally, the third, slow step may involve substantial movements/deformations in the TM helices and probably a rotamer “toggle switch” in aromatic side chains, leading to significant changes on the receptor cytoplasmic side and to G-protein binding/activation.
The results presented in the current study suggest that binding affinities of agonists and ligand/receptor interactions can be reliably predicted using an intermediate conformation of the receptor, which corresponds to the first and second (fast) steps described above. This is in line with recent studies from our group (Reynolds and others 2008) and others (de Graaf and Rognan 2008), where models based on minor conformational changes in the TM-V domain of β2AR-carazolol crystal structure were shown to effectively select for agonists in virtual ligand screening (VLS). In the ref (de Graaf and Rognan 2008) though, the adjustments in the agonist-binding model were limited to rotamer changes in Ser2035.42, Ser2045.43 and Ser2075.43 side chains only, similar to those observed in our models with rigid backbone. Screening with this rigid backbone model was not selective for β2AR agonists when used with the standard scoring functions. To gain selectivity, the authors introduced protein-ligand interaction fingerprints (IFP), which explicitly boosts scoring term for user-specified “anchor” hydrogen bonds.
The current study and our VLS model (Reynolds and others 2008) demonstrate that agonist selectivity can be achieved by a minor backbone shift in TM-V helix, without incorporating knowledge-based terms in the scoring function. Interestingly, our model suggests that optimal binding of full agonists requires the largest shift of the TM-Ve anchor site (∼1.6÷2.2 Å), which is also accompanied by a largest gain in binding affinity (∼1000-fold). Partial agonists have weaker interactions with either (or both) anchor sites, which results in a whole spectrum of optimal TM-Ve positions, as shown in Figure 5B and Table 2. On the other end of the spectrum, inverse agonists like carazolol can stabilize the “inactive” receptor conformation by preventing the inward movement of TM-Ve, and thereby suppress spontaneous activation. Such direct ligand control over direction and magnitude of TM-V movement may play an important role in the “rheostat” behavior (Kobilka and Deupi 2007) of the β2AR and some other aminergic GPCRs, where ligands can ‘dial in’ different levels of receptor activity ranging from full activation to full blockage.
The exact mechanism by which the ligand-dependent movements in the extracellular part of TM-V affect signal propagation into the cytoplasmic portion of the β2AR is not clear yet. One plausible explanation would be that the tilting of TM-Ve can result in a “seesaw” movement of the TM-V around Pro2115.50 kink, dislocating the cytoplasmic ends of TM-V and TM-VI away from the receptor axis and changing topology of the G-protein binding site (Schwartz and others 2006). A relatively high flexibility of proline-induced kinks in TM-V and other TM helices though, may hamper such direct seesaw movements. In the recently published ligand-free activated opsin (Ops*) structures {Park, 2008 #374; Scheerer, 2008 #488}, for example, the TM-IV kink angle is sharply increased as compared to dark-state rhodopsin, so that extracellular and cytoplasmic ends of TM-VI are both tilted outward in the Ops*. Therefore, further experimental and theoretical inquiries are needed to grasp a complex interplay between movements of TM helices, their parts and individual side chains that leads to β2AR activation.
Conclusions
A critical role of Ser2035.42/Ser2045.43/Ser2075.43 (“head”) and Asp1133.32/Asn3127.39 (“tail”) anchor sites in agonist binding and receptor activation is well documented for the β2AR (Ambrosio and others 2000; Del Carmine and others 2002; Strader and others 1989) and several other closely related GPCRs in adrenergic, dopamine and serotonin families (Coley and others 2000; Hwa and Perez 1996; Mansour and others 1992; Wang and others 1991). Our study suggests a structural mechanism for direct contribution of these interactions in the conformational changes of the ligand binding pocket, and their role in differentiation between agonistic and antagonistic effect of β2AR ligands. The β2AR models with TM-V hinge flexibility afford prediction of conformational preferences and binding affinities for the whole spectrum of β2AR ligands from full agonists to inverse agonists, which are consistent with existing experimental data. At the same time, note that the polar TM-V anchor site is not universally conserved in GPCRs. Therefore the proposed here role of TM-V in regulation of receptor activation may be specific only to this therapeutically important group of receptors. This suggests that along with some “universal” features similar across different GPCRs classes (Schwartz and others 2006), some family-specific and/or function-specific (Hoffmann and others 2008) mechanisms should be considered in analysis of GPCR activation.
Computational Methods
All-atom molecular models and 3D graphics in this work were generated with the ICM-Pro software package, version 3.5-1 (Molsoft LLC). The molecular objects were described in terms of internal coordinate variables (Abagyan and others 1994), and modified ECEPP/3 potentials(Nemethy and others 1992), as implemented in the ICM program (Abagyan and others 2007). Charges for ligands were taken from the MMFF description (Halgren 1995).
Rigid and flexible models of β2ARprotein
The rigid ICM model of β2AR protein was prepared from the PDB coordinates (PDB ID: 2rh1) (Cherezov and others 2007) using ICM conversion procedure. This includes addition of hydrogen atoms to the receptor structure, selection of the energetically favorable His, Asn and Gln side chains, and local minimization of hydrogens in the internal coordinates space. Water molecules and carazolol ligand were subsequently removed from the model. No coordinates of β2AR heavy atoms were changed from those in the crystal structure.
The β2AR model with flexible side chains was obtained by unfixing specific sets of torsion angles in the rigid model. All χ torsion angles for residues with atoms in 8 Å radius from carazolol atoms were set free, and participate in Monte Carlo optimization.
For the β2AR model with flexible TM-V backbone, in addition to flexible side chains in the binding pocket, all torsion angles (χ ω, ψ and φ) were unfixed in a portion of the extracellular loop EL2 (residues 191-196) and around the proline-induced kink of TM-V (residues 205-210). Free torsion variables in these residues allow for motility of the TM-V extracellular helix (residues 197-204) as a rigid body, while keeping the rest of the protein backbone fixed. Standard disulfide bridging constraints were imposed between thiol groups of Cys191 and Cys106 side chains.
Flexible ligand docking
For all β2AR receptor models, docking was performed by placing ligand in a random position within 10 Å from the binding pocket and global optimization of the complex conformational energy. Stochastic global energy optimization of the complex was performed using the ICM Monte Carlo (MC) procedure with minimization (Abagyan and Totrov 1994; Abagyan and others 1994).
To facilitate side chain rotamer switches in flexible β2AR models, the first 106 steps of the MC procedure used “soft” vdW potentials and high MC temperature, followed by another 106 steps with “exact” vdW method and gradually decreasing temperature. In the models with TM-V backbone flexibility, a minor external force was applied to the top portion of TM5 in the direction of TM3-TM7 helices. The force was adjusted using a ligand-free β2AR model, so that the best conformational energy of the model with TM-Ve helix tilted by 2 Å inward is equal to the best conformational energy of the model with original position of TM-V (no shift), thus providing a mean to evenly sample the whole range of TM-Ve positions in between. Specifically we used an ICM harmonic “distance restraint” between Cα atoms of Ser203 and Gly37 in TM-I, with weight W=0.41 and “upper wall” Du=1.0, as described in ICM manual. The inward shift of the Ser203 Cα atom in angstroms was used as a measure of the tilt for the whole TM-Ve helix backbone.
At least ten independent runs of the docking procedure were performed for each ligand-β2AR complex. The docking results were considered “consistent” when at least 70% of the individual runs resulted in conformations clustered within a root mean square deviation (RMSD) of <0.5Å to the overall best energy pose of the ligand.
Binding affinity predictions
Ligand-receptor binding energy ΔGpred was calculated as the conformational energy of the ligand bound into the β2AR receptor minus optimized conformational energy of the free ligand. Changes in the receptor conformational energy between apo- and ligand-bound forms cannot be predicted with a high accuracy and were not accounted for in ΔGpred calculations. The energy functions included the following ICM terms with the corresponding default weights: van der Waals (“vw” +“14”), hydrogen bonding (“hb”), distant dependent electrostatics (“el”), torsion (“to”) and desolvation term (“sf”, surfaceTension=0.004). The ICM entropy term for ligands was not included in the final affinity calculations, since it did not improve the prediction accuracy in the initial tests. A more accurate treatment of the entropy that accounts for both ligand and receptor changes may be beneficial in the future studies. The predicted binding affinities were calculated by linear transformation of the binding energy (pKdpred = 0.313*ΔGpred - 5.8) to match the scale of the experimentally measured pKd for this set.
Most experimental ligand binding data in Table 2 we obtained from ref. (Del Carmine and others). Though the pKd measurements for carazolol (Manalan and others 1981) and TA-2005 (Kikkawa and others 1998) were performed at different assay conditions, the values of pKd for common ligands in these studies were consistent with those in (Del Carmine and others) with 0.4 pKd, maximum deviation.
Acknowledgements
This work was supported in part by the NIH grants 5-R01-GM071872 and 1-R01-GM074832, and Roadmap Initiative grant P50 GM073197. The authors acknowledge Ray Stevens for assistance with manuscript preparation.
Abbreviations
- β2AR
β2-adrenergic receptor
- GPCR
G protein-coupled receptor
- RMSD
root mean square deviation
- TM
transmembrane
- Maxn
maximum atom deviation
- EL2
extracellular loop 2
Footnotes
Supplementary Materials
Supplementary materials include PDB coordinates of energy optimized β2AR-lignad models, listed in Table 2.
References
- Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235(3):983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Abagyan RA, Totrov MM, Kuznetsov DA. Icm: A New Method For Protein Modeling and Design: Applications To Docking and Structure Prediction From The Distorted Native Conformation. J. Comp. Chem. 1994;15:488–506. [Google Scholar]
- Abagyan RA, Orry A, Raush E, Budagyan L, Totrov M. Version 3.0. MolSoft LLC; La Jolla, CA: 2007. ICM Manual. [Google Scholar]
- Ambrosio C, Molinari P, Cotecchia S, Costa T. Catechol-binding serines of beta(2)-adrenergic receptors control the equilibrium between active and inactive receptor states. Molecular pharmacology. 2000;57(1):198–210. [PubMed] [Google Scholar]
- Baker JG. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. British journal of pharmacology. 2005;144(3):317–22. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Deupi X, Olivella M, Haaksma EE, Pardo L. Serine and threonine residues bend alpha-helices in the chi(1) = g(-) conformation. Biophys J. 2000;79(5):2754–60. doi: 10.1016/S0006-3495(00)76514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Chelikani P, Hornak V, Eilers M, Reeves PJ, Smith SO, RajBhandary UL, Khorana HG. Role of group-conserved residues in the helical core of beta2-adrenergic receptor. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(17):7027–32. doi: 10.1073/pnas.0702024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–65. doi: 10.1126/science.1150577. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C, Woodward R, Johansson AM, Strange PG, Naylor LH. Effect of multiple serine/alanine mutations in the transmembrane spanning region V of the D2 dopamine receptor on ligand binding. Journal of neurochemistry. 2000;74(1):358–66. doi: 10.1046/j.1471-4159.2000.0740358.x. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. Journal of medicinal chemistry. 2008;51(16):4978–85. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- Del Carmine R, Molinari P, Sbraccia M, Ambrosio C, Costa T. “Induced-fit” mechanism for catecholamine binding to the beta2-adrenergic receptor. Molecular pharmacology. 2004;66(2):356–63. doi: 10.1124/mol.66.2.356. [DOI] [PubMed] [Google Scholar]
- Del Carmine R, Ambrosio C, Sbraccia M, Cotecchia S, Ijzerman AP, Costa T. Mutations inducing divergent shifts of constitutive activity reveal different modes of binding among catecholamine analogues to the beta(2)-adrenergic receptor. British journal of pharmacology. 2002;135(7):1715–22. doi: 10.1038/sj.bjp.0704622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Ramakrishnan C. Deterministic features of side-chain main-chain hydrogen bonds in globular protein structures. Protein Eng. 2000;13(4):227–38. doi: 10.1093/protein/13.4.227. [DOI] [PubMed] [Google Scholar]
- Goddard WA, 3rd, Abrol R. 3-Dimensional structures of G protein-coupled receptors and binding sites of agonists and antagonists. J Nutr. 2007;137(6 Suppl 1):1528S–1538S. doi: 10.1093/jn/137.6.1528S. discussion 1548S. [DOI] [PubMed] [Google Scholar]
- Halgren T. Merck molecular force field I-V. J Comp Chem. 1995;17:490–641. [Google Scholar]
- Hannawacker A, Krasel C, Lohse MJ. Mutation of Asn293 to Asp in transmembrane helix VI abolishes agonist-induced but not constitutive activity of the beta(2)-adrenergic receptor. Molecular pharmacology. 2002;62(6):1431–7. doi: 10.1124/mol.62.6.1431. [DOI] [PubMed] [Google Scholar]
- Hanson M, Cherezov V, Roth C, Griffith M, Jaakola V, Chien E, Velasquez J, Kuhn P, Stevens R. A Specific Cholesterol Binding Site Is Established by the 2.8 A° Structure of the Human b2-Adrenergic Receptor. Structure. 2008a doi: 10.1016/j.str.2008.05.001. doi:10.1016/j.str.2008.05.001] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008b;16(6):897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Zurn A, Bunemann M, Lohse MJ. Conformational changes in G-protein-coupled receptors-the quest for functionally selective conformations is open. British journal of pharmacology. 2008;153(Suppl 1):S358–66. doi: 10.1038/sj.bjp.0707615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa J, Perez DM. The unique nature of the serine interactions for alpha 1-adrenergic receptor agonist binding and activation. The Journal of biological chemistry. 1996;271(11):6322–7. doi: 10.1074/jbc.271.11.6322. [DOI] [PubMed] [Google Scholar]
- Javitch JA, Shi L, Liapakis G. Use of the substituted cysteine accessibility method to study the structure and function of G protein-coupled receptors. Methods in enzymology. 2002;343:137–56. doi: 10.1016/s0076-6879(02)43131-x. [DOI] [PubMed] [Google Scholar]
- Javitch JA, Fu D, Chen J. Residues in the fifth membrane-spanning segment of the dopamine D2 receptor exposed in the binding-site crevice. Biochemistry. 1995;34(50):16433–9. doi: 10.1021/bi00050a026. [DOI] [PubMed] [Google Scholar]
- Kikkawa H, Kurose H, Isogaya M, Sato Y, Nagao T. Differential contribution of two serine residues of wild type and constitutively active beta2-adrenoceptors to the interaction with beta2-selective agonists. British journal of pharmacology. 1997;121(6):1059–64. doi: 10.1038/sj.bjp.0701229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa H, Isogaya M, Nagao T, Kurose H. The role of the seventh transmembrane region in high affinity binding of a beta 2-selective agonist TA-2005. Molecular pharmacology. 1998;53(1):128–34. doi: 10.1124/mol.53.1.128. [DOI] [PubMed] [Google Scholar]
- Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends in pharmacological sciences. 2008 doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends in pharmacological sciences. 2007;28(8):397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Kobilka BK. G protein coupled receptor structure and activation. Biochimica et biophysica acta. 2007;1768(4):794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7(4):339–57. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Ballesteros JA, Papachristou S, Chan WC, Chen X, Javitch JA. The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the beta 2-adrenergic receptor. The Journal of biological chemistry. 2000;275(48):37779–88. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Chan WC, Papadokostaki M, Javitch JA. Synergistic contributions of the functional groups of epinephrine to its affinity and efficacy at the beta2 adrenergic receptor. Molecular pharmacology. 2004;65(5):1181–90. doi: 10.1124/mol.65.5.1181. [DOI] [PubMed] [Google Scholar]
- Manalan AS, Besch HR, Jr., Watanabe AM. Characterization of [3H](+/-)carazolol binding to beta-adrenergic receptors. Application to study of beta-adrenergic receptor subtypes in canine ventricular myocardium and lung. Circulation research. 1981;49(2):326–36. doi: 10.1161/01.res.49.2.326. [DOI] [PubMed] [Google Scholar]
- Mansour A, Meng F, Meador-Woodruff JH, Taylor LP, Civelli O, Akil H. Site-directed mutagenesis of the human dopamine D2 receptor. European journal of pharmacology. 1992;227(2):205–14. doi: 10.1016/0922-4106(92)90129-j. [DOI] [PubMed] [Google Scholar]
- Nemethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini MG, Zagari A, Rumsey S, Scheraga HA. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J. Phys. Chem. 1992;96(15):6472–6484. [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–45. doi: 10.1126/science.289.5480.739. others. [DOI] [PubMed] [Google Scholar]
- Reynolds K, Katritch V, Abagyan R. Identifying conformational changes of beta-2 adrenoceptor that enable accurate prediction of ligand/receptor interactions and screening for GPCR modulators. JCAMD, accepted. 2008 doi: 10.1007/s10822-008-9257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318(5854):1266–73. doi: 10.1126/science.1150609. others. [DOI] [PubMed] [Google Scholar]
- Sato T, Kobayashi H, Nagao T, Kurose H. Ser203 as well as Ser204 and Ser207 in fifth transmembrane domain of the human beta2-adrenoceptor contributes to agonist binding and receptor activation. British journal of pharmacology. 1999;128(2):272–4. doi: 10.1038/sj.bjp.0702813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation--a global toggle switch model. Annual review of pharmacology and toxicology. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K, Gether U, Kobilka BK. Functional differences between full and partial agonists: evidence for ligand-specific receptor conformations. The Journal of pharmacology and experimental therapeutics. 2001;297(3):1218–26. [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. The Journal of biological chemistry. 2002;277(43):40989–96. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. The Journal of biological chemistry. 1989;264(23):13572–8. [PubMed] [Google Scholar]
- Strader CD, Sigal IS, Candelore MR, Rands E, Hill WS, Dixon RA. Conserved aspartic acid residues 79 and 113 of the beta-adrenergic receptor have different roles in receptor function. The Journal of biological chemistry. 1988;263(21):10267–71. [PubMed] [Google Scholar]
- Suryanarayana S, Kobilka BK. Amino acid substitutions at position 312 in the seventh hydrophobic segment of the beta 2-adrenergic receptor modify ligand-binding specificity. Molecular pharmacology. 1993;44(1):111–4. [PubMed] [Google Scholar]
- Swaminath G, Deupi X, Lee TW, Zhu W, Thian FS, Kobilka TS, Kobilka B. Probing the beta2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. The Journal of biological chemistry. 2005;280(23):22165–71. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential binding of agonists to the beta2 adrenoceptor. Kinetic evidence for intermediate conformational states. The Journal of biological chemistry. 2004;279(1):686–91. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- Tyndall JD, Sandilya R. GPCR agonists and antagonists in the clinic. Med Chem. 2005;1(4):405–21. doi: 10.2174/1573406054368675. [DOI] [PubMed] [Google Scholar]
- Wang CD, Buck MA, Fraser CM. Site-directed mutagenesis of alpha 2A-adrenergic receptors: identification of amino acids involved in ligand binding and receptor activation by agonists. Molecular pharmacology. 1991;40(2):168–79. [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta(1)-adrenergic G-protein-coupled receptor. Nature. 2008;454(7203):486–91. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland K, Zuurmond HM, Krasel C, Ijzerman AP, Lohse MJ. Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta 2-adrenergic receptor. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(17):9276–81. doi: 10.1073/pnas.93.17.9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xhaard H, Rantanen VV, Nyronen T, Johnson MS. Molecular evolution of adrenoceptors and dopamine receptors: implications for the binding of catecholamines. Journal of medicinal chemistry. 2006;49(5):1706–19. doi: 10.1021/jm0511031. [DOI] [PubMed] [Google Scholar]