

Abstract
In this study, we performed the first systematic survey of DNA methylation status of the CpG islands of the PEG3 (Paternally expressed gene 3) imprinted domain in the mouse, cow, and human genomes. Previous studies have shown that the region surrounding the first exon of PEG3 contains a differentially methylated CpG island. In addition, we have discovered two previously unreported differentially methylated regions (DMR): one in the promoter region of mouse Zim3 and another in the promoter region of human USP29. In the cow, the Peg3-CpG island was the only area that showed DMR status. We have also examined the methylation status of several CpG islands in this region using human tumor derived DNA. The CpG islands near PEG3 and USP29 both showed hypermethylation in DNA derived from breast and ovarian tumors. Overall, this study shows that the PEG3 imprinted domain of humans, cows, and mice contains differing numbers of DMRs, but the PEG3-CpG island is the only DMR that is conserved among these three species.
Keywords: CpG island, imprinting, differentially methylated region, PEG3
1. Introduction
The majority of autosomal genes are expressed equally from two parental alleles. However, in fewer than 200 mammalian genes, transcription from one allele is epigenetically repressed based on its parent of origin due to a process called genomic imprinting (Wagschal and Feil, 2006). Genomic imprinting is found only in marsupials and placental mammals that utilize a unique reproductive strategy in which young offspring develop inside females' wombs. Most imprinted genes are also involved in controlling fetal growth rates and nurturing behaviors (Tilghman, 1999). The proper dosage of imprinted genes is very critical for the survival of these mammals, and abnormal dosages quite often manifest as genetic diseases in humans. The imprinting-related diseases include Beckwith-Wiedemann, Prader/Willi, Angelman, and Silver-Russell syndromes as well as autistic spectrum disorders (Ferguson-Smith et al., 2004; Ideraabdullah et al., 2008). In addition, several imprinted genes are involved in many types of cancers (Jirtle and Skinner, 2007). Thus, genomic imprinting is regarded as a gene dosage control mechanism for this subset of genes critical for mammalian-specific reproductive scheme (John and Surani, 2000).
The promoter regions of imprinted genes are usually associated with CpG-rich regions, termed CpG islands. Some of these CpG islands are differentially methylated in an allele-specific manner, and thus called Differentially Methylated Regions (DMRs). Besides monoallelic expression, the allele-specific methylation pattern of these CpG islands is another molecular signature that is used to define imprinted genes (Ferguson-Smith et al., 2004). Some of these DMRs also inherit their methylation as a gametic signal from the previous generations, and these DMRs play critical roles for maintaining the imprinting and transcription of a given domain (Edwards and Ferguson-Smith, 2007). Abnormal methylation levels of these DMRs, either hyper or hypomethylation, are also often associated with many types of human diseases as ‘epimutations’ (Hatchwell and Greally, 2007).
An evolutionarily conserved imprinted region, the PEG3 domain, is found on human chromosome 19q13.4/proximal mouse chromosome 7/cow chromosome 18. In the mouse, this 500-kb genomic region contains 6 additional imprinted genes, including Usp29/Mim1 (Mer-repeat containing imprinted transcript 1), Zim1 (Zinc finger gene imprinted 1), Zim2, Zim3, Zfp264 (Zinc finger protein gene 264), and APeg3 (Antisense to Paternally expressed gene 3) (Kim and Stubbs, 2005). Several members of this domain are also imprinted in human and cow (Kim et al., 2007). According to recent studies, human PEG3 expression is often missing in several types of cancers and DNA hypermethylation on the promoter region appears to be a prime cause for this loss of PEG3 expression (Maegawa et al., 2001). Other studies suggest that the epigenetic abnormalities of this domain may be associated with other human diseases (Van den Veyver et al., 2001). Despite this close linkage to human diseases, this domain has not been systematically studied so far in humans and other mammals. Thus, the current study sought to analyze the genome sequence and DNA methylation status of this evolutionarily conserved imprinted domain. This study revealed that the PEG3 domain of humans and mice contains at least two DMRs, but contains only one DMR in cows. In addition, the methylation status of the two human DMRs is often affected in the ovary and breast tumor DNA.
2. Methods
2.1 CpG island prediction and sequence analysis
A Perl script was used to analyze the genomic sequences surrounding the PEG3 imprinted domain (Chr 19: 61750000-62500000, 750 kb for human; Chr. 7: 6293901-7043900, 750 kb for mouse; Chr. 18: 6398699-6473700, 750 kb for cow) and a nonimprinted region containing similar types of genes that was used to provide a basis for comparison for sequence analysis (Chr 1: 244543476-246543476 for human; Chr. 11: 58303940-60303939 for mouse; Chr. 7: 38923539-40923539 for cow). For the cow and human sequences, this Perl script was set to recognize a sequence as a CpG island only if three conditions were met: length greater than 500 bp, C+G content greater than 55%, and observed/expected CpG ratio at least 0.65 (Takai and Jones, 2002). An initial CpG island prediction using these criteria resulted in very few predicted islands in mice, so the minimum length parameter was reduced to 200 bp for this species only. To test evolutionary conservation, the sequence of each CpG island was analyzed using BLAST (Altschul et al., 1990) and the ECR browser of the dcode website (http://www.dcode.org/) (Ovcharenko et al., 2004). The CpG islands predicted by this program were also analyzed for the presence of repetitive elements using RepeatMasker and Tandem Repeat Finder (Smit 1996-2004, Benson 1999). The default parameters and appropriate species were used for RepeatMasker, and the parameters for Tandem Repeat Finder were adjusted as follows (Hutter et al., 2006): match score 2, mismatch score 5, indel score 7, match probability 80, indel probability 10, minscore to report 100, maxperiod 2000. The sequence of each CpG island and related information regarding repeat contents and evolutionary conservation are available upon request.
2.2 COBRA (COmbined Bisulfite Restriction Analysis) and bisulfite sequencing
Mouse genomic DNA was isolated from the liver tissues of the F1 (3 months old) and F2 (2 weeks old) offspring of interspecific crossing of Mus musculus and M. spretus (Kim et al., 2001). Mouse placentas were isolated from 17 day embryos. Mouse sperm DNA was isolated from the epididymis of 3 month old male mice according to a previously established protocol (Bunch and Saling, 1991). Briefly, the epididymides were incubated in sperm elution buffer (130 mM NaCl, 20 mM Tris, 2 mM EDTA pH 7.4) for ten minutes at 37°C. The epididymides were then removed and the solution was centrifuged for 30 seconds at 800 rpm. Then, the sperm were washed twice more with the sperm elution buffer. The isolated sperm were examined under a microscope, and only samples that did not display somatic cell contamination were used for the methylation analyses. The sperm from a single mouse (∼105-106 sperm) was pooled and subjected to bisulfite conversion. In preparation for isolation of blastocyst-stage embryos, female mice were superovulated (Eppig and Telfer, 1993; Horgan et al., 1994). First, 5 IU of Pregnant Mare Serum (PMS) (Cat. G4877, Sigma) was injected subcutaneously. Then, the same mice were injected with 5 IU of human Chorionic Gonadotropin (hCG) hormone (Cat. C1063, Sigma) 48 hours after the PMS injection. The treated mice were mated with male littermates, and sacrificed 3.5 days later. The embryos were flushed from each uterus, and the isolated blastocysts were examined under the microscope to assess their developmental stages and purity. Only embryos at the blastocyst stage were used for methylation analysis. Seven blastocysts were pooled for bisulfite treatment of the DNA. To isolate oocytes, female mice were superovulated as described above, but the mice were sacrificed 12 hours after the second injection. Mature eggs were isolated from the swollen ampulla of the oviducts, incubated in hyaluronidase solution (Cat. H3506, Sigma) for several minutes to separate the cumulus cells, and subsequently washed three additional times to remove potential somatic tissue contamination. DNA isolated from approximately 400 oocytes obtained from ten females (8 weeks old) was pooled and used for bisulfite conversion.
Cow genomic DNA was also isolated from the liver of the hybrid offspring of interspecific crossing of Bos taurus and B. indicus. These hybrid animals have been previously used to test imprinting of several genes in the PEG3 domain (Kim et al., 2001). Human genomic DNAs derived from normal and tumor tissues were obtained from a commercial firm (Biochain).
Each DNA (2 μg) was modified with the bisulfite conversion reaction according to the manufacturer protocol (EZ DNA methylation kit, Zymo Research). The converted DNA was eluted with 15 μl of TE. Each converted DNA (1 μl) was used as a template for PCR with primers designed using the MethPrimer program (Li and Dahiya, 2002). The PCR amplification was performed using the Maxime PCR premix kit (Intron Biotech, South Korea). Information regarding the primer sequences and detailed PCR conditions for each tested region are available in Supplemental Material 1.
The amplified PCR products were analyzed using restriction enzyme digestion (COBRA) (Xiong and Laird, 1997). Each PCR product was analyzed with two sets of restriction enzymes. First, the efficiency of the bisulfite conversion reaction was monitored with a set of enzymes that contain non-CpG cytosines in their recognition sites (DdeI, HpaII; data not shown). Any digestion by these enzymes indicates that the conversion reaction was incomplete. A second set of enzymes that distinguish between unmethylated and methylated DNA were chosen to analyze the degree of methylation in each tested region. The recognition site of each of these enzymes contains a CpG site (TaqI, BstUI, HhaI and HpyCH4IV) or a TpG site (HphI). Since methylation inhibits the conversion of cytosines into thymidines during the bisulfite conversion, bisulfite-treated DNA will be digested by the first group of enzymes if the DNA is methylated. The recognition site for HphI is GGTGA(N)8, so it will only digest DNA in which the CpG site is unmethylated in vivo. Each of these restriction digestion reactions was repeated at least three times.
Some of the PCR products amplified from the bisulfite-treated DNA were further analyzed through cloning and sequencing. Each of the selected PCR products was purified using the MEGA-Spin agarose gel purification kit (Intron), and then individually cloned into the pGEM-tEasy vector (Promega). At least 10 different clones were randomly selected for DNA sequencing for each PCR product. Due to a cloning bias toward methylated fragments in the mouse Zim2 and Zim3 regions, methylated and unmethylated fragments were separated using COBRA, gel purified, and the unmethylated fragment was ligated into the pGEM-tEasy vector. The purified plasmid DNA was sequenced using BigDye v3.1 (Applied Biosystems). Unincorporated primers and dye terminators were removed via ethanol precipitation, and an ABI 3130 XL was used to analyze the results. To determine methylation status, the resulting electropherograms were visually inspected in BioEdit (Hall, 1999).
3. Results
3.1 Characterization of the CpG islands located in the PEG3 domain
The overall organization of the 500-kb genomic region of the PEG3 domain is well conserved among mammals, but the neighboring regions show large-scale genomic changes, such as the loss of an Olfactory Receptor (OLFR) cluster as well as a Vomeronasal Organ Receptor (VNO) cluster in the human genome (Fig. 1). Since the exact boundaries of this imprinted domain are unknown, we analyzed the 750 kb region surrounding PEG3 from mouse chromosome 7, cow chromosome 18, and human chromosome 19. We identified 50, 34, and 19 CpG islands in the mouse, cow, and human PEG3 regions, respectively (Table 1). A 2 Mb nonimprinted region that is syntenic between human, mouse, and cow and contains a cluster of zinc finger genes was used as a basis for analysis of the sequence characteristics of this region. The PEG3 region and its associated CpG islands show several interesting differences when compared to the reference region. First, although the overall GC content of the PEG3 region and the reference region are similar, the PEG3 region contains more CpG islands than expected. Second, both the PEG3 region and the CpG islands contain more tandem repeats than expected. These tandem repeats were detected using the Tandem Repeat Finder program, and consist of simple sequence repeats ranging in size from approximately 15 bp to several hundred bp. Third, the repeat content of the PEG3 domain deviates greatly from the reference region. Lower levels of SINEs, LINEs, and LTRs were found in the mouse, and reduced levels of LINEs and LTRs were found in both the human and cow sequences. Fourth, the CpG islands in this region are mainly found in regions near genes: 50% (mouse), 58% (cow), and 84% (human) of the CpG islands are within 10 kb of an annotated gene. In addition, 60% (mouse), 45% (cow), and 63% (human) of the above CpG islands overlap at least one exon. Although most of the CpG islands are not conserved among the three species in terms of sequence and position, one CpG island surrounding the first exon of PEG3 appears to be well conserved.
Figure 1. Organization of the PEG3 genomic region.
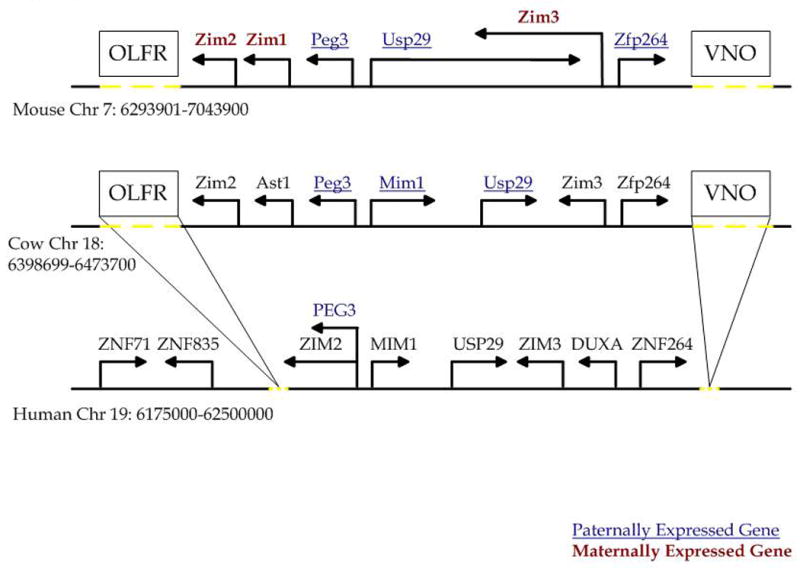
Outline of the 750 kb genomic region surrounding PEG3 in the mouse, cow, and human. Directions of arrows indicate the direction of transcription. Maternally expressed genes are indicated by bold red text; paternally expressed genes are indicated by underlined blue text. The positions of the olfactory receptor gene cluster (OLFR) and the vomeronasal gene cluster (VNO) are indicated by boxes. Dotted yellow lines indicate the approximate regions in the mouse and cow genomes that contain the OLFR and VNO clusters as well as the approximate region from which these clusters were lost from the human genome.
Table 1. Analysis of CpG islands and repeat content in the PEG3 imprinted region.
| Mouse | Cow | Human | ||||
|---|---|---|---|---|---|---|
| Peg3 Region | Control Region | Peg3 Region | Control Region | Peg3 Region | Control Region | |
| Sequence size | 750 kb | 2 Mb | 750 kb | 2 Mb | 750 kb | 2 Mb |
| GC content | 43.66% | 47.31% | 47.85% | 39.86% | 44.36% | 40.86% |
| CpG islands | 36 (15)* | 41 | 34 (13) | 16 | 19 (9) | 26 |
| Tandem Repeats in CpG islands | 9 (3.75) | 10 | 1 (0.75) | 2 | 10 (0) | 0 |
| Tandem Repeats in entire region | 173 (104) | 279 | 223 (129) | 343 | 49 (31.75) | 85 |
| Total masked bases | 19.79% | 31.46% | 35.49% | 49.42% | 58.05% | 55.78% |
| Total interspersed repeats | 14.13% | 28.89% | 34.36% | 48.39% | 55.68% | 54.53% |
| SINEs | 3.04% | 10.93% | 15.34% | 15.17% | 21.57% | 14.45 |
| LINEs | 6.75% | 7.89% | 14.60% | 28.32% | 18.87% | 25.50% |
| LTR | 3.70% | 8.99% | 2.65% | 3.78% | 10.67% | 11.71% |
number in parentheses indicates expected number, based on number in control region
3.2 DNA methylation analysis of the CpG islands from the mouse Peg3 domain
We analyzed the methylation status of the computationally predicted CpG islands along with several other regions associated with genes in the Peg3 imprinted domain using genomic DNA from the mouse, cow, and human. Briefly, this genomic DNA was treated via bisulfite conversion, and then the bisulfite-converted DNA was amplified using PCR. The resulting bisulfite PCR products were analyzed by COBRA (COmbined Bisulfite Restriction Analysis) (Xiong and Laird, 1997) and/or subcloning and sequencing (Fig. 2A).
Figure 2. Bisulfite sequencing and COBRA analyses of selected CpG islands in the mouse Peg3 domain.
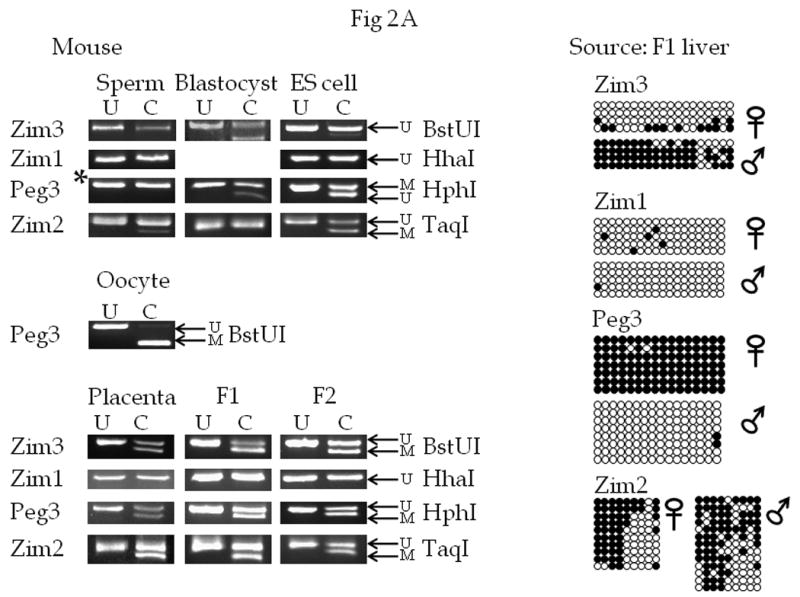
Mouse genomic DNA was obtained from Mus musculus sperm, oocytes, blastocysts, embryonic stem cells, placenta, and from the liver tissues of the F1 (3 months old) and F2 (2 weeks old) offspring of interspecific crossing of M. musculus and M. spretus.
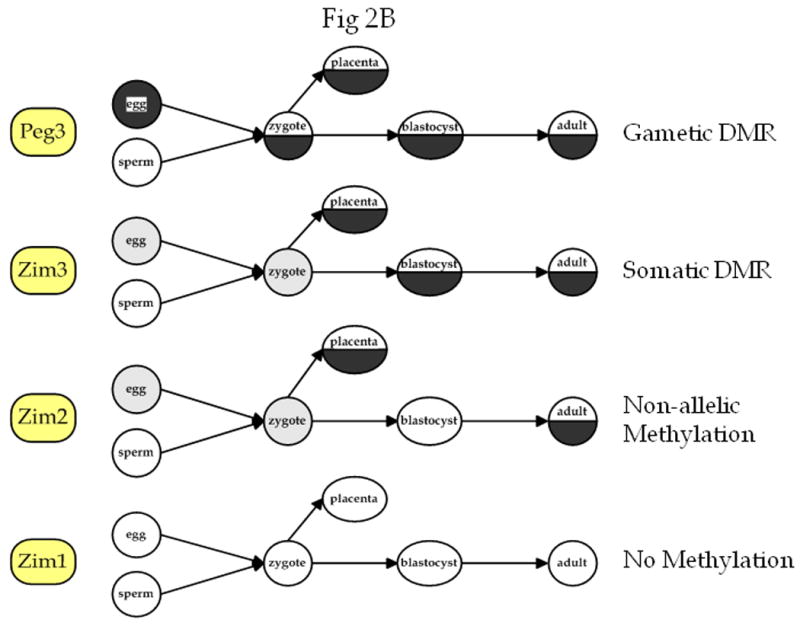
A) COBRA and bisulfite sequencing analysis. The gene associated with each PCR product is shown to the left of the figure. Each DNA sample was treated with sodium bisulfite and used in the PCR reaction. Next, each PCR product was cloned and sequenced and/or analyzed by the COBRA analysis in which each PCR product was digested with the enzyme shown to the right of each picture to assess the methylation level of the region (*: the Peg3-CpG island was digested with BstUI in the sperm DNA sample). Total digestion by this enzyme indicates methylation of the region and a lack of digestion indicates the absence of methylation in the region. For each COBRA panel, column U contains DNA that was not exposed to the selected restriction enzyme, and column C contains DNA that was exposed to the restriction enzyme. The arrow labelled U indicates the position of undigested, unmethylated DNA, and the arrow labelled M indicates the position of digested, methylated DNA. Repeated analyses (at least three times) yielded similar results, so a representative picture is shown for each COBRA. For the bisulfite sequencing results, each row represents a different clone, and each column represents a different CpG site. The name of the gene associated with each CpG island is indicated above each set of results. Filled circles indicate methylated cytosines and open circles indicate unmethylated cytosines. B) Summary of methylation at different developmental time points in each of the four loci tested. Open circles indicate unmethylated regions, completely filled black circles indicate methylated regions, half-filled black regions indicate probable DMRs, and gray circles indicate time points for which there is no data available.
Within the mouse Peg3 domain, we targeted four CpG islands (Zim1, Zim2, Zim3, and Peg3) using genomic DNA isolated from various Mus musculus samples as well as from the livers of F1 and F2 offspring of interspecific crosses between M. musculus and M. spretus. The use of DNA from both F1 and F2 animals allowed us to exclude the possibility that the genetic background of the mice affected our methylation analysis. In addition, since the F1 and F2 samples are from mice of different ages (three months and two weeks old, respectively), we are able to determine if age has an effect on the methylation status of the analyzed regions. Analysis of DNA from placenta, germ cells, blastocyst stage embryos, ES cells, and adult mice gave us a developmental stage-specific view of the methylation status of this domain. Sequence polymorphisms between the two parental species were first identified within the studied CpG regions, and later used to differentiate the parental alleles in the F1 and F2 samples. The Zim1-CpG island was not digested by the enzyme HhaI in any of the tested samples, showing a lack of methylation in the region at all of these stages, although we were unsuccessful in obtaining data on this region in the blastocyst stage embryo. Bisulfite sequence analysis of this region confirmed this result. As demonstrated in earlier studies (Li et al., 2000), the CpG island near the promoter of Peg3 displayed a maternal allele-specific methylation pattern based on results from both COBRA and bisulfite sequencing. The Peg3-CpG island was almost completely methylated in oocyte DNA and lacked methylation in the sperm DNA, as expected for a maternally methylated DMR, while ES cell, placenta, and both F1 and F2 adult tissues derived DNA showed the expected DMR pattern. At the Zim2-CpG island, a small amount of PCR product was digested after incubation with the enzyme TaqI in the sperm-derived DNA. This pattern could either be a result of somatic contamination of the sperm-derived DNA or could indicate that there are low levels of methylation in sperm DNA. In contrast, the placenta, ES cell, F1, and F2 samples all show digestion of approximately 50% of the PCR products. This pattern indicates about half of these PCR products are methylated. Bisulfite sequencing of the PCR products confirmed this result, but the methylation was not allele-specific. The first four CpG sites were all methylated in most clones, while the next four CpG sites were unmethylated in most clones, and the ninth CpG site (which is part of the TaqI enzyme site used for COBRA) was methylated in approximately half of the clones. The PCR product from the Zim3-CpG island showed approximately 50% digestion with the enzyme BstUI in DNA derived from blastocyst, placenta, F1, and F2 mice, indicating that half of the PCR products were methylated (Fig. 2A). Sequencing revealed that the unmethylated DNA was mostly derived from the maternal allele (Fig. 2A). This is consistent with the maternal allele-specific expression of Zim3 (Kim et al., 2001). However, the Zim3-CpG island is unmethylated in sperm DNA and ES cell DNA, suggesting that the methylation on this CpG island is not inherited as a gametic signal, but that allele-specific methylation at this CpG island may be a somatic signal established during early development of the mouse.
Overall, the Zim3-CpG island appears to be the second DMR (Differentially Methylated Region) discovered in the mouse Peg3 domain. In addition, we show that the CpG island near Zim1 is unmethylated, based on the lack of digestion of bisulfite PCR products by HhaI and analysis of the sequence of individual Zim1 PCR products. The Zim2-CpG island showed a pattern of DNA methylation that is not allele-specific, but rather stage- or tissue-specific (Fig. 2B).
3.3 DNA methylation analysis of the CpG islands from the cow Peg3 domain
We performed a similar series of analyses using DNA prepared from the liver of F1 hybrid offspring of Bos taurus and B. indicus. We analyzed CpG islands associated with Peg3, Usp29, Ast1, Zim2, and Zim3. The COBRA results from the CpG islands near Zim2 and Zim3 showed digestion of nearly all of the PCR products, and bisulfite sequencing confirmed that most of the CpG sites are methylated. The CpG island associated with Zim2 showed a similar pattern of total digestion indicating complete methylation. We also analyzed the CpG island located close to the promoter of cow Usp29 since this has been shown to be an imprinted gene with paternal allele specific expression (Kim et al., 2007). However, the results from the DNA methylation analyses revealed that the CpG island near this gene is almost completely methylated in the cow (Fig. 3). It is important to note that although cow Usp29 is imprinted, this gene has lost its ORF (Open Reading Frame) capability in recent evolutionary time, suggesting some changes in the bovine lineage (Kim et al., 2007). Thus, the hypermethylation of the Usp29-CpG island of cow might be related to the loss of its ORF during evolution. In contrast, lack of digestion by HhaI shows that the CpG island near Ast1 is not methylated at all. As predicted, the CpG island close to the Peg3 promoter is methylated in an allele-specific manner (Fig. 3). Bisulfite sequencing results further confirmed maternal allele-specific methylation at this CpG island (Fig. 3), consistent with the paternal allele-specific expression of cow Peg3. In summary, this survey indicated that the Peg3-CpG island is the only DMR known at this time to exist in the cow Peg3 region.
Figure 3. Bisulfite sequencing and COBRA analyses of the cow Peg3 domain.
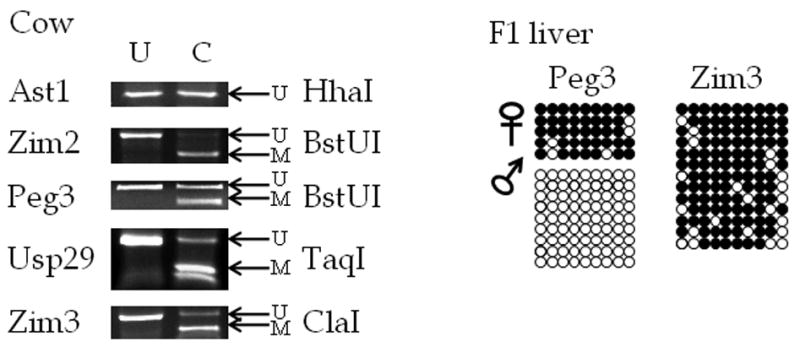
Cow DNA was obtained from the liver of offspring of interspecific crossing of Bos taurus and Bos indicus. The gene associated with each PCR product is shown to the left of the figure. Each DNA sample was treated with sodium bisulfite, used in the PCR reaction, and then cloned and sequenced and/or analyzed by the COBRA analysis. For each COBRA panel, column U contains DNA that was not exposed to the selected restriction enzyme, and column C contains DNA that was exposed to the restriction enzyme listed on the right side. On the right side of the COBRA panel, the arrow labelled U indicates the position of undigested, unmethylated DNA, and the arrow labelled M indicates the position of digested, methylated DNA. Repeated analyses (at least three times) yielded similar results, and a representative picture is shown for each COBRA. For the bisulfite sequencing results, each row represents a different clone, and each column shows a different CpG site. Filled circles indicate methylated cytosines and open circles indicate unmethylated cytosines.
3.4 DNA methylation analysis of the CpG islands from the human PEG3 domain
We also performed a series of DNA methylation analyses of the CpG islands identified from the human PEG3 domain (Fig. 4). These include the CpG islands located close to ZNF71 (Zinc finger protein71), ZNF835 (Zinc finger protein 835), PEG3, USP29, DUXA (double homeobox A), and ZNF264 (Zinc finger protein 264). In addition, we analyzed the promoter region of ZIM3 to allow for comparison of the methylation status of this region between the cow, mouse and human, and also a CpG island that was predicted by MethPrimer to exist in the first intron of DUXA (DUXA-5/6). We used human genomic DNA isolated from the brain, testis, lung and liver of 4 normal individuals. Representative results from the adult brain and testis of different individuals are shown in Fig. 4.
Figure 4. COBRA analysis of the CpG islands located in the human PEG3 imprinted region.
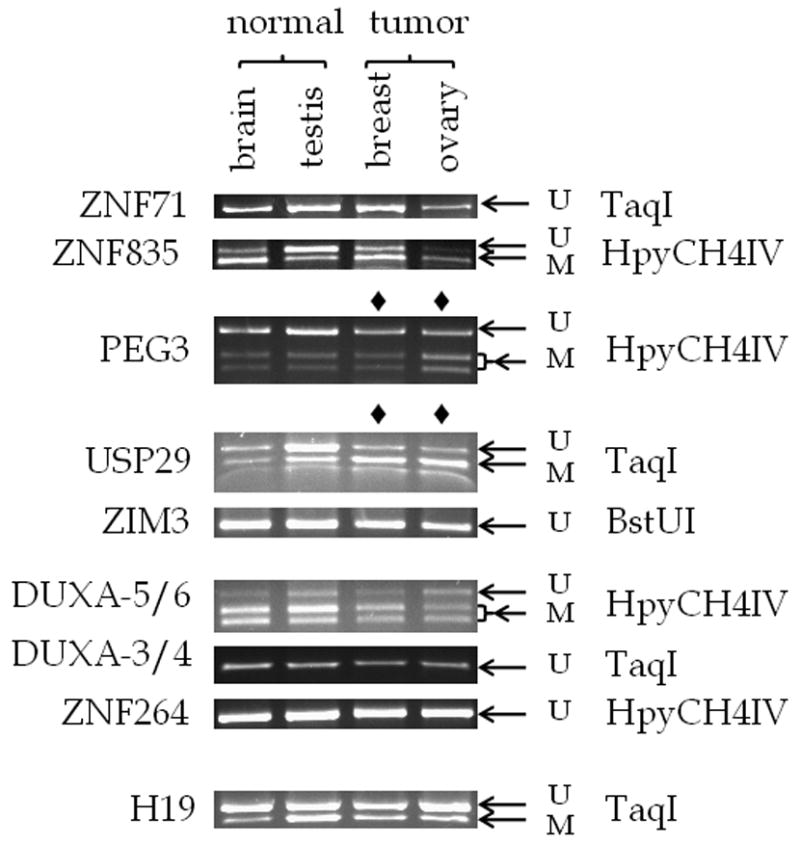
Human DNA was derived from normal brain, normal testis, breast tumor, and ovary tumor. Each DNA was converted using sodium bisulfite and used in the PCR product. Each PCR product was incubated with the enzyme indicated to the right of the figure. Repeated analyses (at least three times) yielded similar results, so a representative picture is shown for each COBRA. The gene associated with each PCR product is shown to the left of the figure. Each lane contains DNA that was incubated with the appropriate restriction enzyme. The arrow labelled U indicates the position of undigested, unmethylated DNA, and the arrow labelled M indicates the position of digested, methylated DNA.
The methylation status of the CpG islands in the human PEG3 domain can be summarized as follows. The PCR products derived from the CpG islands of ZNF71, ZIM3, ZNF264, and DUXA (DUXA-3/4) were not digested by their respective restriction enzymes, indicating that these CpG islands were unmethylated in the tested DNA from adult brain, testis, liver and lung. In contrast, the PCR products from the CpG island in the first intron of DUXA (DUXA-5/6) showed the opposite pattern in the COBRA analysis: the majority of this region was digested, indicating methylation. Digestion of the ZNF835 PCR product by HpyCH4IV revealed a tissue-specific methylation pattern: less DNA was digested in the sample derived from testis, indicating hypomethylation compared to the brain sample. Finally, the CpG islands of PEG3 and USP29 showed a digestion pattern characteristic of a differentially methylated region: only half of the PCR products were digested while the other half remained undigested (Fig. 4). We could not determine allele-specific methylation in either locus since the tested regions of both PEG3 and USP29-CpG islands lack sequence polymorphisms. However, the results from bisulfite sequencing of the PEG3-CpG island clearly indicated that half of the clones were methylated while the other half were unmethylated, the typical pattern of a DMR (Fig. 5). In contrast, the bisulfite sequencing results of the USP29-CpG island showed a less clear pattern than the PEG3-CpG island. About half of the clones from testis DNA (4 out of 10 clones, 26.3% of the CpGs) showed methylation whereas a much greater number of the clones from brain DNA showed methylation but with high levels of mosaicism (54.5% of the CpGs were methylated). It is important note that human USP29 is expressed only in testis (Kim et al., 2000). Thus, it is possible that the observed higher levels of methylation at brain might reflect the transcriptional activity of human USP29, but this remains to be tested in the near future.
Figure 5. Bisulfite sequencing of the CpG islands located in the human USP29 and PEG3 promoter regions.
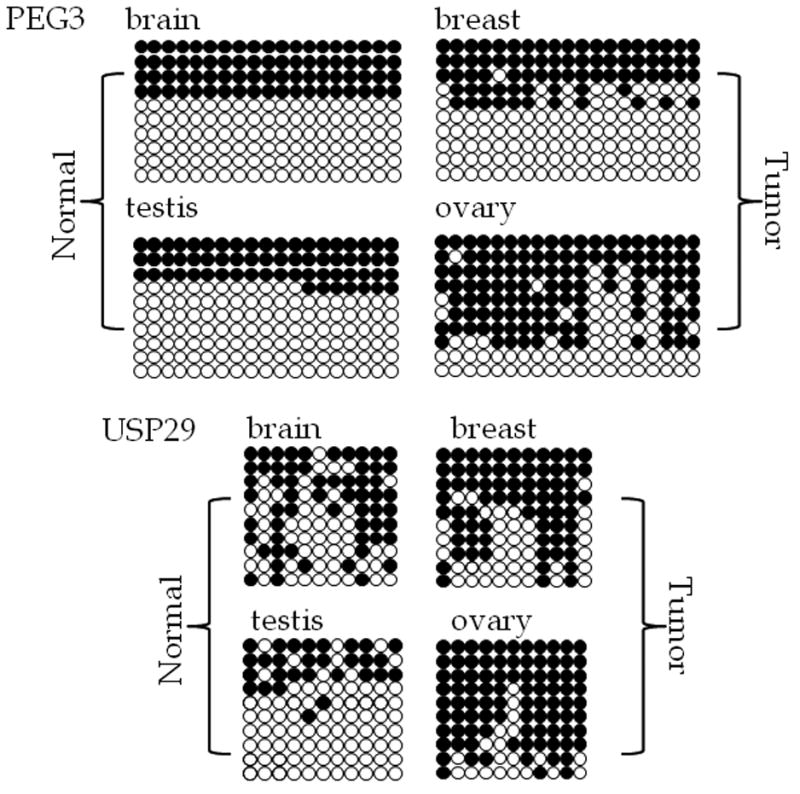
Results obtained from analysis of normal brain and testis derived DNA are shown along with breast and ovary tumor-derived DNA. Each row indicates a different clone, and each column indicates a different CpG site. Filled circles indicate methylated cytosine and open circles indicate unmethylated cytosine. The analyzed CpG islands are indicated with the name of the gene with which they are associated.
Since several independent reports previously indicated the hypermethylation of human PEG3-DMR in cancers (Maegawa et al., 2001), we also performed a similar series of DNA methylation analyses using 4 representative cancer DNAs of different tissue origin, including ovary, breast, lung and liver. According to the results from this survey (Fig. 4), the two DMRs of the human PEG3 domain were affected in two cancer types, ovary and breast, but not in lung and liver (Supplemental Material 2). In the case of the PEG3-DMR, half of the PCR products from breast cancer DNA were digested by HpyCH4IV, which is similar to the pattern observed from normal brain and testis DNA. However, the same analysis revealed that more than half of the PCR products from ovarian cancer DNA were digested by HpyCH4IV, suggesting hypermethylation in this cancer DNA (Fig. 4, lanes marked by ◆). About 80% of the ovarian cancer DNA showed methylation at the PEG3-DMR although three other samples show much lower levels of methylation. This result was further confirmed through sequencing the PCR products as shown in Fig. 5. These results are consistent with those seen in a large scale study that included over forty samples of ovarian tumor DNA (Feng et al., 2008). In addition, the previously undiscovered USP29-DMR showed a similar pattern-the majority of the PCR products from both breast and ovarian cancer DNA were digested by TaqI, suggesting that this CpG island is also hypermethylated in these two types of cancer DNA. Our sequencing analyses indeed confirmed the hypermethylation of USP29-DMR in breast and ovarian cancer DNAs (Fig. 5). The hypermethylation levels in the ovarian cancer DNA appears to be much greater (81.8% of CpGs are methylated) than those in the breast cancer DNA (65.5% of CpGs are methylated). Also, it is interesting to note that the methylation pattern at the breast cancer DNA is somewhat mosaic, which is similar to the pattern seen in normal brain DNA but with much more methylation. For this series of analyses, we have also included the human H19-DMR as a control. As shown in Fig. 4, this region showed a DMR pattern in the normal DNA as well as two types of cancer DNA. This also suggests that the observed DNA hypermethylation may be specific to the two DMRs of the human PEG3 domain. In sum, the data presented above is consistent with the previous observations revealing the hypermethylation of human PEG3 in cancer (Maegawa et al., 2001), and further indicates that this hypermethylation probably occurs on the DMRs of both PEG3 and USP29 in cancer DNA.
4. Discussion
In this study, we surveyed methylation status of the CpG islands of the PEG3 imprinted domain in the mouse, cow, and human genomes. This survey led to the discovery of two previously unreported differentially methylated regions: mouse Zim3 and human USP29. In addition, we examined the methylation status of the CpG islands in this region using human tumor derived DNA. The CpG islands near PEG3 and USP29 both showed hypermethylation in DNA derived from breast and ovarian tumors.
We performed a comprehensive analysis of the sequence in the 750 kb region containing the PEG3 imprinted domain. This analysis showed that the sequence structure in this region was different from a non-imprinted region containing similar types of genes (Table 1). Both tandem repeats and CpG islands were over-represented in both the entire sequence and the CpG island sequence of the PEG3 region. Tandem repeats have been reported to be associated with imprinted genes, and may play a role in setting up DNA methylation for the CpG islands of imprinted genes during gametogenesis (Hutter et al., 2006). It is well known that tandem repeat sequences can attract DNA methylation although it is still unclear how only one allele of tandem repeats become methylated in the case of imprinted genes, such as the DMRs of imprinted genes. At the same time, the tandem repeats may also play a role in the genesis and maintenance of CpG islands during evolution. Since methylated cytosines are prone to mutation, CpG islands (which contain high numbers of cytosines) must be protected from this mutation by some mechanism. Tandem duplication of CpG islands, which would increase the overall size of the island, could be one way to prevent attrition of cytosines. These two conflicting needs might have contributed to increasing the number of tandem repeats in the CpG islands of mammalian imprinted genes.
In the mouse, we analyzed four CpG islands associated with Zim1, Peg3, Zim3, and Zim2, respectively. We assessed methylation status of each CpG island in DNA obtained from sperm, blastocyst, embryonic stem cell (ES cell), placenta, and in somatic tissue from mice of two different ages (two weeks and three months) (Fig. 2). Methylation status of other loci is known to show differences at different stages of development, and also with age, but a broad survey such as this has not been previously performed in the PEG3 imprinted region. The Zim2- and Zim3-CpG islands showed different patterns between blastocysts and ES cells, which are at a similar stage of development: Zim2 was unmethylated in the blastocyst and showed a DMR-type digestion pattern in the COBRA results from the ES cell, and Zim3 had a DMR pattern in the blastocyst and was unmethylated in the ES cell. While the Peg3-DMR has been shown to be stable in ES cells, other regions have not, so this change in methylation at these regions can probably be attributed to the effects of ES cell culture (Chang et al., 2009). Peg3, Zim2, and Zim3 all show approximately 50% digestion of DNA derived from the placenta. This may indicate that the methylation pattern of these regions in the placenta is similar to that found in the adult somatic tissue, which could indicate that these genes are regulated similarly in the placenta and somatic tissues, although it is also possible that this pattern is due to the fact that the placenta is derived from both maternal and fetal cells.
The current study identified two additional DMRs, Zim3-DMR in the mouse and USP29-DMR in the human (Fig. 6). However, the DMR status of these CpG islands appears to be lineage-specific. First, although the Zim3-CpG island is methylated in an allele-specific manner in the mouse, both alleles of the human ZIM3-CpG island are unmethylated, and both alleles of the cow Zim3-CpG island are methylated (Fig. 6). Second, the human USP29-CpG island appears to be a DMR in a tissue-specific manner in the testis, but the homologous region in cow showed hypermethylation, indicating that both alleles may be methylated. These differences might be an indication of the presence of some species-specific changes in the imprinting status of the surrounding genes. In this regard, it is interesting to point out the presence of a genomic rearrangement in the rodent lineage: the mouse genome does not contain the Duxa gene between Zim3 and Zfp264 although two other lineages, human and cow, have this gene, suggesting that a lineage-specific deletion event occurred during rodent evolution. Also, the mouse genome contains a very long (over 300 kb) Usp29 transcript while this long transcript has been truncated in the human and cow lineages (Fig. 6). This transcript could be one of the mechanisms that maintains the imprinted status of the surrounding genes, similar to the functions of Air and Kcnq1ot1 in their respective imprinted domains (Ideraabdullah et al., 2008). Once we obtain the imprinting status of human and cow ZIM3, it is possible that the relationship between this genomic deletion and the imprinting status of the surrounding genes will be clarified.
Figure 6. Summary of the methylation status of CpG islands in the PEG3 region.
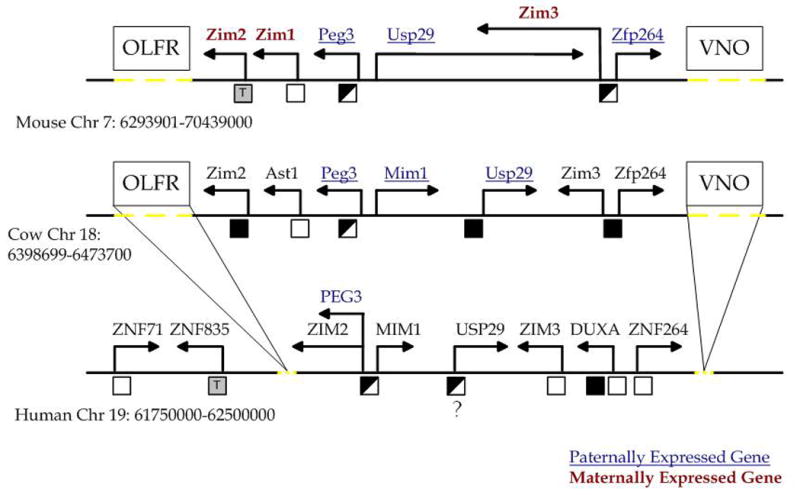
Outline of the 750 kb genomic region surrounding PEG3 in the mouse, cow, and human. Directions of arrows indicate the direction of transcription. Maternally expressed genes are indicated by bold red text; paternally expressed genes are indicated by underlined blue text. The boxes show the approximate position of the olfactory (OLFR) and vomeronasal (VNO) gene clusters. Dotted yellow lines indicate the approximate regions in the mouse and cow genomes that contain the OLFR and VNO clusters as well as the approximate region from which these clusters were lost from the human genome. Below each chromosome, black boxes indicate methylated regions, empty boxes indicate unmethylated regions, half-filled boxes indicate differentially methylated regions, and grey boxes containing the letter T indicate potential tissue-specific methylation patterns.
PEG3 expression is silenced in various tumor types, including gliomas, choriocarcinomas, and ovarian tumors (Maegawa et al., 2001; Van den Veyver et al., 2001). This silencing of human PEG3 was found to be a result of DNA methylation (Murphy et al., 2001). Our results also confirm the hypermethylation of cytosines at the PEG3-DMR in ovarian tumor-derived DNA (Fig. 4&5). Although some regions show tissue-specific methylation patterns, the PEG3-DMR is methylated on the maternal allele only over a range of normal tissues. In each normal tissue, we expect to see a pattern in which approximately half of the bisulfite clones are unmethylated and half are methylated. Since this CpG pattern has such a consistent methylation pattern, any deviation from this in abnormal tissues is probably meaningful. This study only analyzed a limited number of samples, but other studies of human cancers have shown hypermethylation of this region and reduced PEG3 expression (Maegawa et al., 2001; Feng et al., 2008). Our study showing hypermethylation in the PEG3-CpG island in ovarian cancer DNA adds to the evidence supporting the hypothesis that PEG3 functions as a tumor suppressor. We also found an increase in the DNA methylation level at the USP29-DMR in breast and ovarian tumor DNA. Since this CpG island tends to show mosaicism in its DNA methylation pattern, it is unclear whether the observed increase in the DNA methylation levels in the two types of cancer DNA truly represents abnormal DNA methylation or is simply a tissue-specific methylation pattern. However, it is relevant to note that human USP29 was previously discovered through a screen of a tumor-derived expression library (Tureci et al., 2002). Although human USP29 is known to be expressed only in the testis, the expression of this gene has been seen in several types of cancers. This further suggests a potential role of human USP29 in human cancer. Also, it is well known that several genes in a given imprinted domain tend to be co-regulated in terms of its expression and epigenetic modifications. Thus, it is likely that the observed DNA methylation changes in the USP29-DMR along with PEG3-DMR might reflect together the abnormal status of DNA methylation in the two tissues tested in this study. This could suggest a link between abnormal expression of USP29 and PEG3 in human cancers.
Supplementary Material
Acknowledgments
We would like to thank Michelle Aucoin, Uduak Udoh, Letitia Lacour, Dr. Jeong Do Kim, Keunsoo Kang, and Anne Bergmann (Lawrence Livermore National Laboratory) for their technical assistance. We also thank Christopher Faulk and Deepa Srikanta for critical reading of this manuscript. This work was supported by NIH (R01-GM66225).
Abbreviations
- CpG
cytosine followed immediately by guanine
- DMR
differentially methylated region
- LTR
long terminal repeat
- SINE
small interspersed nuclear element
- LINE
long intersperse nuclear element
- COBRA
combined bisulfite restriction analysis
- ES cell
embryonic stem cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder. a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch D, Saling P. Generation of a mouse sperm membrane fraction with zona receptor activity. Biol Reprod. 1991;44:672–680. doi: 10.1095/biolreprod44.4.672. [DOI] [PubMed] [Google Scholar]
- Chang G, Liu S, Wang F, Zhang Y, Kou Z, Chen D, Gao S. Differential methylation status of imprinted genes in nuclear transfer derived ES (NT-ES) cells. Genomics. 2009;93:112–119. doi: 10.1016/j.ygeno.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Edwards CA, Ferguson-Smith AC. Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol. 2007;19:281–289. doi: 10.1016/j.ceb.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Eppig JJ, Telfer EE. Isolation and culture of mouse oocytes. Methods Enzymol. 1993;225:77–84. doi: 10.1016/0076-6879(93)25008-p. [DOI] [PubMed] [Google Scholar]
- Feng WW, Marquez RT, Lu Z, Liu J, Lu KH, Issa JP, Fishman DM, Yu Y, Bast RC. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer. 2008;112:1489–1502. doi: 10.1002/cncr.23323. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Lin SP, Youngson N. Regulation of gene activity and repression: a consideration of unifying themes. Curr Top Dev Biol. 2004;60:197–213. doi: 10.1016/S0070-2153(04)60006-8. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hatchwell E, Greally JM. The potential role of epigenomic dysregulation in complex human disease. Trends Genet. 2007;23:588–595. doi: 10.1016/j.tig.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: a laboratory model. second. Cold Spring Harbor Laboratory Press; New York: 1994. [Google Scholar]
- Hutter B, Helms V, Paulsen M. Tandem repeats in the CpG islands of imprinted genes. Genomics. 2006;88:323–332. doi: 10.1016/j.ygeno.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Ideraabdullah FY, Vigneau S, Bartolomei MS. Genomic imprinting mechanisms in mammals. Mutat Res. 2008;647:77–85. doi: 10.1016/j.mrfmmm.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John RM, Surani MA. Genomic imprinting, mammalian evolution, and the mystery of egg-laying mammals. Cell. 2000;101:585–588. doi: 10.1016/s0092-8674(00)80870-3. [DOI] [PubMed] [Google Scholar]
- Kim J, Noskov VN, Lu X, Bergmann A, Ren X, Warth T, Richardson P, Kouprina N, Stubbs L. Discovery of a novel, paternally expressed ubiquitin-specific processing protease gene through comparative analysis of an imprinted region of mouse chromosome 7 and human chromosome 19q13.4. Genome Res. 2000;10:1138–1147. doi: 10.1101/gr.10.8.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Bergmann A, Wehri E, Lu X, Stubbs L. Imprinting and evolution of two kruppel-type zinc-finger genes, ZIM3 and ZNF264, located in the PEG3/USP29 imprinted domain. Genomics. 2001;77:91–98. doi: 10.1006/geno.2001.6621. [DOI] [PubMed] [Google Scholar]
- Kim J, Stubbs L. Rapidly evolving imprinted loci. In: Jorde L, Little P, Dunn M, Subramaniam S, editors. Encyclopedia of Genetics, Genomics, Proteomics, and Bioinformatics. John Wiley Publishers; West Sussex, UK: 2005. pp. 147–158. [Google Scholar]
- Kim J, Bergmann A, Choo JH, Stubbs L. Genomic organization and imprinting of the PEG3 domain in bovine. Genomics. 2007;90:85–92. doi: 10.1016/j.ygeno.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Li LL, Szeto IY, Cattanach BM, Ishino F, Surani MA. Organization and parent-of-origin-specific methylation of imprinted Peg3 gene on mouse proximal chromosome 7. Genomics. 2000;63:333–340. doi: 10.1006/geno.1999.6103. [DOI] [PubMed] [Google Scholar]
- Maegawa S, Yoshioka H, Itaba N, Kubota N, Nishihara S, Shirayoshi Y, Nanba E, Oshimura M. Epigenetic silencing of PEG3 gene expression in human glioma cell lines. Mol Carcinog. 2001;31:1–9. doi: 10.1002/mc.1034. [DOI] [PubMed] [Google Scholar]
- Murphy SK, Wylie AA, Jirtle RL. Imprinting of PEG3, the human homologue of a mouse gene involved in nurturing behavior. Genomics. 2001;71:110–117. doi: 10.1006/geno.2000.6419. [DOI] [PubMed] [Google Scholar]
- Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32(Web Server issue):W280–286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AH, Green R. RepeatMasker Open-3.0. 19962004 http://www.repeatmasker.org.
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman SM. The sins of the fathers and mothers: genomic imprinting in mammalian development. Cell. 1999;96:185–193. doi: 10.1016/s0092-8674(00)80559-0. [DOI] [PubMed] [Google Scholar]
- Türeci O, Sahin U, Koslowski M, Buss B, Bell C, Ballweber P, Zwick C, Eberle T, Zuber M, Villena-Heinsen C, Seitz G, Pfreundschuh M. A novel tumour associated leucine zipper protein targeting to sites of gene transcription and splicing. Oncogene. 2002;21:3879–3888. doi: 10.1038/sj.onc.1205481. [DOI] [PubMed] [Google Scholar]
- Van den Veyver IB, Norman B, Tran CQ, Bourjac J, Slim R. The human homologue (PEG3) of the mouse paternally expressed gene 3 (Peg3) is maternally imprinted but not mutated in women with familial recurrent hydatidiform molar pregnancies. J Soc Gynecol Investig. 2001;8:305–313. doi: 10.1016/s1071-5576(01)00129-0. [DOI] [PubMed] [Google Scholar]
- Wagschal A, Feil R. Genomic imprinting in the placenta. Cytogenet Genome Res. 2006;113:90–98. doi: 10.1159/000090819. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.