

Abstract
The intracellular Ca2+ ([Ca2+]i) level of skeletal muscles must be rapidly regulated during the excitation-contraction-relaxation process 1. However, the signaling components involved in such rapid Ca2+ movement are not fully understood. Here, we report that mice deficient in the novel phosphatidylinositol phosphate (PIP) phosphatase MIP displayed muscle weakness and fatigue. Muscles isolated from MIP−/− mice produced less contractile force, markedly prolonged relaxation, and exhibited exacerbated fatigue. Further analyses revealed that MIP deficiency resulted in spontaneous Ca2+ leak from the internal store — the sarcoplasmic reticulum (SR). This was attributed to the decreased metabolism/dephosphorylation and the subsequent accumulation of MIP substrates, especially PI(3,5)P2 and PI(3,4)P2. Furthermore, we found that PI(3,5)P2 and PI(3,4)P2 bound to and directly activated the Ca2+ release channel/ryanodine receptor (RyR1) of the SR. These studies provide the first evidence that finely controlled PIP levels in muscle cells are essential for maintaining Ca2+ homeostasis and muscle performance.
During our systematic genome-wide survey for tyrosine/dual specificity phosphatases (unpublished work), we discovered a novel phosphatase by hidden Markov database mining using the conserved catalytic motif ([V/I][V/I]HCXXGXXR[T/S]) as the bait sequence. Both human (BC035690) and mouse (BC018294) homologies were identified. They share 90% identity in amino acid sequences (Supplementary Information, Fig. S1). Northern blotting analyses illustrated that this phosphatase was predominantly expressed in skeletal muscle and heart (Fig. 1a). Immunostaining indicates that it is primarily localized in the cytoplasm (data not shown). To verify its phosphatase property, we generated a GST fusion protein and tested its catalytic activity using pNPP (p-Nitrophenyl Phosphate), a widely used non-specific substrate for tyrosine/dual specificity phosphatases. This new phosphatase did not significantly hydrolyze pNPP (Fig. 1b). Instead, it dephosphorylated a variety of PIPs, especially PI(3,5)P2 (Fig. 1c), similar to PTEN and myotubularin and myopathy related (MTMR) phosphatases that also favor PIPs as substrates despite containing tyrosine phosphatase domains 2. As this new phosphatase is mainly expressed in skeletal muscle and heart, we named it MIP (muscle-specific inositol phosphatase). While our gene knockout work on MIP was ongoing, the Mustalin group also identified this phosphatase (FLJ20133) in their comprehensive collection of tyrosine phosphatases from the human genome and listed it as the 14th member of the MTMR family (MTMR14) based on the homology of its catalytic motif to myotubularin 3. More recently, inactivating mutations in this phosphatase were identified in two cases of human autosomal centronuclear myopathy 4. However, whether these mutations play a causal role in this disease has not yet been determined.
Figure 1.



Characterization of human MIP. (a) The human tissue RNA blot (Clontech, Mountain View, CA) was hybridized with α-32P-deoxycytidine triphosphate (dCTP)-labeled human MIP cDNA probe following a standard protocol. The blot was stripped and re-probed with the β-actin positive control probe to monitor RNA loading. Full scans of the images are shown in Fig. S6. (b) Purified GST-MIP fusion protein and GST alone protein (1 μM) were incubated with pNPP as described in Supplementary Information, Methods. The reaction was stopped by the addition of 0.1 N NaOH, and the pNPP hydrolysis was measured by absorbance at 410 nm. Dual specificity phosphatase 23 was included as the positive control. (c) GST-MIP fusion protein (1μg) was tested for its dephosphorylating activity using the indicated PIPs (Echelon Biosciences Inc., Salt Lake City, UT) as substrates. Three independent experiments were performed and similar results were obtained in each (b and c). Results shown are the mean ± s.d. of triplicates from one experiment.
To characterize the physiological function of MIP, we generated MIP deficient mice through gene targeting (Supplementary Information, Fig. S2). MIP−/− mice were born at the Mendelian ratio and showed no obvious deficiencies during the first 12 months of life. It appears that heart development and basal functions of MIP−/− mice under resting conditions were not significantly affected (Supplementary Information, Table S1 and S2). Interestingly, animal behavior tests revealed a decreased motor function in MIP−/− mice (Supplementary Information, Fig. S3). We then reasoned that a muscle specific phenotype might manifest itself more evidently during or after a stress response. Thus, untrained 16 to 24-week old female wildtype (WT) and MIP−/− mice were run in a rodent treadmill until exhausted following the protocol previously reported 5. The running time of the knockout mice was dramatically decreased (Fig. 2a). These strenuous exercise results indicate that MIP−/− mice are prone to greater skeletal muscle fatigue. However, treadmill performance does not necessarily reflect intrinsic changes within the essential contractile features of intact skeletal muscles and may be confounded with other systemic changes. To directly measure muscle function, extensor digitorum longus (EDL), a fast-twitch muscle, was isolated from the animals used in the treadmill experiments 1–2 weeks after the fatigue tests and assessed for contractility and fatigability characteristics. As illustrated in Fig. 2b, the force-generating capacity (magnitude of the maximal fused isometric tetanic contraction, Tmax) of EDL muscles from the MIP−/− mice was reduced by ~ 60% compared to the control muscles. In addition, the relaxation profile of MIP−/− muscles was substantially prolonged. To further determine the performance of isolated muscles, in vitro intermittent fatigue of the muscles 6,7 was studied. Compared to WT controls, upon exposure of MIP mutant muscles to repeated electrical stimulations, resting baseline force increased, while maximum tetanic force quickly decreased and muscles became non-responsive to subsequent stimulations (Fig. 2c).
Figure 2.
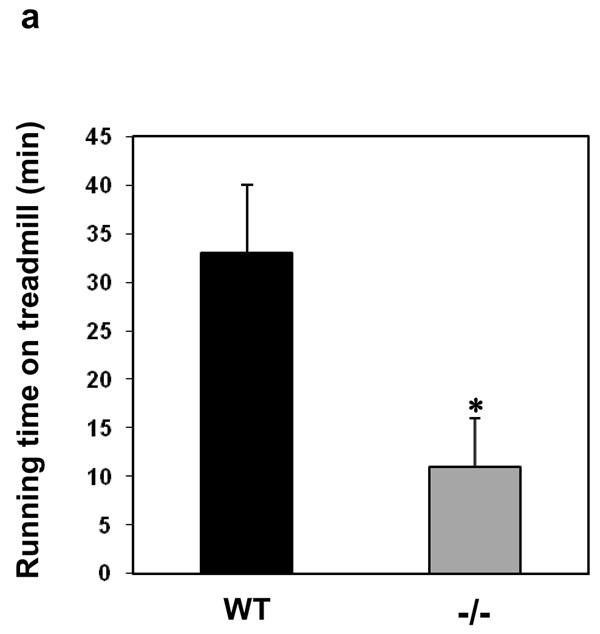




Decreased force production, prolonged relaxation, and exacerbated fatigue in MIP−/− muscles. (a) 16–24-week old female WT and MIP−/− mice were run in a rodent treadmill until exhausted (see Supplementary Information). Three trials were conducted on each animal and the average running times were obtained. Results shown are mean ± s.e. (n=7) (* p < 0.003 by one-way ANOVA followed by Tukey’s post hoc test). (b) One-two weeks following the treadmill tests, EDL muscles isolated from WT and MIP−/− mice were subjected to contractility measurement. Cross sectional area-normalized Tmax (mean ± s.e.) of MIP−/− (n=8) and WT (n=12) EDL muscles were 130 ± 28 kPa and 280 ± 32 kPa, respectively; p < 0.001 by Kruskal-Wallis one-way ANOVA on ranks test. Representative results are shown. (c) EDL muscles isolated from WT and MIP−/− mice were subjected to in vitro muscle fatigue tests using repeated electrical stimulations (n=7–8). (d) Soleus muscles dissected from WT and MIP−/− mice were subjected to 5 min of fatiguing stimulation (80 Hz, 500 msec) followed by 10 min of recovery from fatigue. Contractile forces were normalized to Tmax (100%). (e) Summary data (mean ± s.e.) (n=8) of the response of soleus muscles to fatiguing stimulation and recovery from fatigue (* p < 0.001 by one-way ANOVA followed by Tukey’s post hoc test). (f) Kinetic profile of a single tetanic contraction in soleus muscles following stimulation with a tetanic stimulation train (80 Hz, 1000 msec). Horizontal scale bar shows time in msec and vertical bar shows absolute force in kPa. (g) WT and MIP−/− soleus muscles were subjected to the force vs. frequency relationship tests using frequencies of stimulation in the range of 1–180 Hz. Data shown are mean ± s.e. from 8 preparations (p < 0.02 by one-way ANOVA followed by Tukey’s post hoc test). The force-generating capacity (Tmax) of MIP−/− soleus was reduced by ~15% (180 ± 22 kPa in WT vs. 153 ± 18 kPa in MIP−/− soleus muscles). (h) Thirty percent of aged MIP−/− mice display accelerated muscle wasting/atrophy (n=10). Shown are a 18-month-old WT mouse and a MIP−/− mouse with muscle wasting.
To systematically compare fatigability and recovery from fatigue in WT and MIP−/− muscles, we tested the effects of in vitro fatiguing stimulation and recovery from fatigue in the slow-twitch, fatigue-resistant soleus muscle. We found that soleus muscles from MIP−/− mice fatigued to a greater extent and recovered significantly less than soleus from WT animals (Fig. 2d and 2e). In addition, the kinetics of single tetanic contractions were altered in soleus muscles from MIP−/− mice as seen in Fig. 2f. The plateau phase of the contraction was not well maintained in MIP−/− muscles. We next determined the force-generating capacity (Tmax) of soleus muscles from MIP−/− mice and found that Tmax of MIP−/− soleus was reduced by ~15% (Fig. 2g). Finally, the normalized force vs. frequency relationship was shifted to the right in MIP−/− muscles (Fig. 2g), i.e. at any given frequency of stimulation less force was generated. These results obtained from isolated intact muscles reaffirm that the contractility and relaxation defects of MIP−/− muscles are muscle cell autonomous. Remarkably, the muscle phenotypes appear to be exacerbated in aged mutant mice. About 30% of MIP−/− mice older than 18 months displayed accelerated muscle wasting/atrophy compared to WT littermates (Fig. 2h).
The poor performance of MIP−/− muscles may indicate a defect in the regulation of [Ca2+]i since rapid cycling of [Ca2+]i is critical for force production, relaxation, and recovery from fatigue 1. As Ca2+ handling in muscle cells primarily takes place in a highly specialized junctional structure known as the triad junction that is formed by the transverse tubule invagination of the plasma membrane and the terminal cisternae of SR 8, we surveyed morphology of this anatomic structure in mutant muscle cells. Electron microscopic examination revealed that the terminal cisternae of SR were slightly swollen (Fig. 3a). Ultrastructures of mutant cells otherwise appeared normal.
Figure 3.




Compromised store-operated Ca2+ signaling in MIP−/− muscle cells. (a) Soleus muscles dissected from 4-week old WT and MIP−/− mice were processed for transmission electron microscopic examination. Myofibers of the longitudinal ultrathin sections were photographed at the magnification of × 20,000. Arrows indicate transverse tubules while arrow heads indicate the terminal cisternae of SR. Scale bars, 200 nm. (b) Myotubes were prepared as described in Methods and subjected to ratiometric Ca2+ analyses. Myotubes were loaded with 5 μM Fura-2-AM in the presence of 1.5 mM CaCl2. The ECB was replaced sequentially with Ca2+-free ECB containing caffeine and ryanodine (b) or TG (c), Ca2+ (2 mM)-containing ECB, and Ca2+ (2 mM)-containing ECB supplemented with SKF-96365 at 50 μM (b) or 100 μM (c). The kinetic changes of [Ca2+]i concentration were continuously monitored based on the average ratio of F340/F380 readings of all cells (4–5 cells) in the field. Scale bars, 3 min. Three independent experiments were performed and similar results were obtained in each. Note that basal levels of [Ca2+]i in MIP−/− cells were significantly increased. F340/F380 ratios (mean ± s.d.) of WT and MIP−/− cells were 0.848±0.007 and 0.825±0.009, respectively (n=26 preparations. p <0.001 by Student’s t-test). (d) Intact FDB muscle fibers from WT and MIP−/− mice were prepared as detailed in Methods. The muscle fibers were loaded with Fura 2 and then treated with ionomycin (5 μM). The intracellular Ca2+ was monitored. (e) Summary data of [Ca2+]i response to ionomycin treatment (* p < 0.03 by one-way ANOVA followed by Tukey’s post hoc test) (f) Summary data of the basal levels of [Ca2+]i in WT and MIP−/− FDB fibers (* p < 0.03 by one-way ANOVA followed by Tukey’s post hoc test). Data shown in (e) and (f) are mean ± s.e. from 12–13 preparations. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test.
To functionally test whether MIP−/− muscle cells might have an impaired Ca2+ signaling, we prepared primary myotubes and assessed [Ca2+]i by the ratiometric fluorescence technique. As shown in Fig. 3b and 3c, the basal F340/F380 ratio in MIP−/− myotubes was higher than that in WT cells, indicating elevated resting [Ca2+]i levels in MIP−/− myotubes. Changing the extracellular buffer (ECB) from Ca2+-containing (1.5 mM) to Ca2+-free ECB in tandem with the addition of caffeine and ryanodine (which are known to induce sustained opening of the RyR channel 9,10) (Fig. 3b) or thapsigargin (TG), a potent inhibitor of the SR Ca2+ pump (Sarco/Endoplasmic Reticulum Ca2+-ATPase, SERCA) that inhibits the reuptake of Ca2+ into the SR 11 (Fig. 3c), resulted in a rapid and transient elevation of [Ca2+]i followed by loss of Ca2+ from the myotubes. Under these conditions, the magnitude of the initial Ca2+ transient was reduced in MIP−/− cells as compared to WT myotubes (Fig. 3b and 3c), suggesting decreased Ca2+ content of the mutant SR. Moreover, the duration of the decaying phase was markedly prolonged in the mutant cells, indicating a prolonged release or a defective clearance of [Ca2+]i in the mutant cells.
Store operated Ca2+ entry (SOCE), the capacitative Ca2+ entry through store-operated Ca2+ channels on the plasma membrane following the depletion or the decrease of Ca2+ from internal stores, is a universal mechanism that provides a direct way of refilling intracellular Ca2+ stores 12. Since MIP−/− muscles had lower levels of SR Ca2+, we reasoned that a compromised SOCE could contribute to the reduced Ca2+ storage capacity in MIP−/− muscles. In fact, our data showed that SOCE was decreased in the mutant myotubes, as evidenced by the reduced [Ca2+]i response to the restoration of 2 mM extracellular Ca2+ in the ECB medium (Fig. 3b and 3c). Subsequent treatment with SKF-96365, a known blocker of the store-operated Ca2+ channel 13 in the plasma membrane, quickly decreases [Ca2+]i concentration. By comparing [Ca2+]i traces of WT and MIP−/− cells (Fig. 3b and 3c), it is clear that while SOCE is still functional in MIP−/− myotubes, this response is significantly blunted.
To further determine if SR Ca2+ storage is reduced in MIP−/− adult muscles, we prepared single intact flexor digitorum brevis (FDB) fibers and exposed them to ionomycin (Ca2+ ionophore), an experimental condition designed to assess total Ca2+ storage within the SR 14,15. As shown in Fig. 3d and 3e, ionomycin-mobilized Ca2+ was greatly decreased in MIP−/− muscle fibers. In addition, we found that resting levels of [Ca2+]i were increased in MIP−/− fibers (Fig. 3d and 3f), consistent with earlier observations in myotubes (Fig. 3b and 3c). Thus, ablation of MIP causes diminished Ca2+ storage within the SR and an elevated [Ca2+]i, findings that are fully consistent with and supported by the phenotypic changes of muscle weakness and impairment of muscle relaxation in MIP−/− mice (Fig. 2b, 2c, 2d, and 2f).
MIP dephosphorylates multiple PIPs (Fig. 1d), especially PI(3,5)P2, the most recently identified phosphatidylinositol-bisphosphate (PIP2) isomer whose subcellular localization and function in mammalian cells have not been characterized 16,17. To test whether the dysfunction of store-operated Ca2+ signaling in MIP deficient cells is associated with decreased dephosphorylation/metabolism of PI(3,5)P2, we first determined subcellular localization of PI(3,5)P2 in myotubes. Confocal microscopic examination following immunostaining showed that it resided in the SR, as evidenced by co-localization with the endoplasmic reticulum specific protein disulfide isomerase (PDI) (Fig. 4a). We then assessed PI(3,5)P2 levels in MIP−/− SR by immunostaining of myotubes with anti-PI(3,5)P2 and anti-PDI antibodies followed by laser scanning cytometric analyses 18,19. As shown in Fig. 4b, immunofluorescence intensity of PI(3,5)P2 in MIP−/− SR defined by PDI positive areas is doubled compared to that in WT counterparts, confirming excessive accumulation of PI(3,5)P2 in the mutant SR. We next dialyzed WT and MIP−/− myotubes with 200 nM PI(3,5)P2 or PI(5)P, the hydrolyzing product of PI(3,5)P2, and monitored Ca2+ signaling. As illustrated in Fig. 4c, perfusion of PI(3,5)P2 resulted in dysfunction of the SR in WT myotubes, recapitulating the defects of Ca2+ signaling in MIP−/− cells (Fig. 3b and 3c). The decaying phase of the Ca2+ transient triggered by TG was substantially prolonged by PI(3,5)P2 overloading. By contrast, addition of PI(5)P did not rescue Ca2+ signaling in MIP−/− cells, nor were the Ca2+ profiles in WT cells changed (Fig. 4d). Furthermore, we tested other PIPs that can be dephosphorylated by MIP. As shown in Fig. 4e, overloading of PI(3,4)P2 and PI(3)P (to a less extent), but not PI(4,5)P2 and PI(3,4,5)P3, also prolonged the decaying phase of TG-triggered Ca2+ transients in WT myotubes. Overloading of these PIPs into MIP−/− cells either did not or only slightly increased Ca2+ signaling defects (Fig. 4f). Together, these PIP perfusion results support the concept that dysfunction of the Ca2+ store (SR) in MIP deficient muscle cells is attributed to the increased levels of PIP substrates, especially PI(3,5)P2 and PI(3,4)P2.
Figure 4.




Perfusion of PI(3,5)P2, PI(3,4)P2, and PI(3)P (to a less extent) into WT myotubes results in aberrant Ca2+ signaling, recapitulating the defects of MIP−/− muscle cells. (a) Differentiated myotubes were double immunostained with anti-PI(3,5)P2 (Echelon Biosciences Inc.) and anti-PDI (Stressgen, Ann Arbor, MI)) antibodies. PI(3,5)P2 and PDI were visualized using FITC-conjugated anti-mouse 2nd antibody and Cy5-conjugated anti-rabbit 2nd antibody (Molecular Probes, Eugene, OR), respectively. Images were analyzed with the Zeiss laser-scanning microscope LSM510 confocal imaging system. Scale bar, 18 μm. (b) Primary WT and MIP−/− myotubes were immunostained with anti-PI(3,5)P2 and anti-PDI antibodies followed by laser scanning cytometric analyses as described in Supplementary Information, Methods. Immunofluorescence intensity of PI(3,5)P2 in PDI positive areas (SR) in MIP−/− cells was quantified and normalized against the WT control. Representative images are shown in the left panel and statistical results (mean ± s.d.) from three independent experiments are shown in the right panel (* p < 0.05 by Student’s t-test). Scale bars, 20 μM. PI(3,5)P2 (di-C16) (c), PI(5)P (di-C16) (d), and other PIPs (di-C16) as indicated (e and f) were delivered (shuttled) into the myotubes at the concentration of 200 nM for 20 min using Shuttle PIP kits following the protocol provided by the manufacturer (Echelon Biosciences Inc.). Carrier 2 was used to deliver PI(3,4)P2, PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3. Carrier 3 was used to deliver PI(3)P and PI(5)P. Ca2+ signaling was then analyzed as described in Fig. 3c. Representative results of three experiments are shown. Scale bars, 3 min.
To define the molecular mechanisms underlying the disrupted Ca2+ homeostasis in MIP−/− cells, we assessed SERCA ATPase activity that is responsible for reuptake of Ca2+ into the SR, as reported 20,21. We also tested for the contribution of a potential functional change in Na+/Ca2+ exchangers in the plasma membrane to the phenotypes by treating myotubes with KB-R9743, a selective inhibitor for the reverse mode of the Na+/Ca2+ exchanger 22. Neither SERCA ATPase nor the Na+/Ca2+ exchanger appeared to be affected in MIP−/− cells (Supplementary Information, Fig. S4). These data thus led us to the hypothesis that Ca2+ stores in MIP−/− cells were spontaneously leaky. To test this hypothesis, we focused on potential functional changes in RyR1, the skeletal muscle Ca2+ release channel on the SR 23,24. First, we tested potential physical interactions of MIP PIP substrates with the RyR1 protein by PIP array analyses. As shown in Fig. 5a, PI(3)P, PI(5)P, and PI(3,5)P2 clearly bound to purified RyR1. In addition, PI(4)P and PI(3,4,5)P2 showed weak binding to RyR1. The direct physical interaction between PI(3,5)P2 and RyR1 was further verified by the PI(3,5)P2-agarose beads pull-down assay and anti- PI(3,5)P2 co-immunoprecipitation analyses (Fig. 5b). We then determined whether the interactions of PIPs with RyR1 on the purified SR microsomes produced functional effects. We used the [3H]ryanodine binding assay to assess RyR1 channel activity because [3H]ryanodine binds with high affinity to the open conformational state of the channel 25,26. The dose-response curve conducted at near-optimal (pCa 5) free Ca2+ (Fig. 5c) showed that among all PIPs tested, PI(3,5)P2 had the most effective activation, even though its physical interaction with the RyR1 protein was weaker than PI(3)P and PI(5)P (Fig. 5a). In addition, PI(3,4)P2 also activated RyR1 despite very weak binding to RyR1 protein. The observation that the RyR1 binding affinity of PIPs does not directly correlate with their effects on activation of the RyR1 channel suggests that some RyR1-PIP interactions are functionally more productive than others and that each PIP elicits a distinct re-arrangement of the channel protein that has different repercussions on channel gating. The complexity of PIP function in regulating ion channels and transporters is exemplified by PI(4,5)P2, the best characterized PIP. The effects of PI(4,5)P2 on various channels and transporters include activation, inhibition, and insensitivity 27. Clearly, further systemic studies are needed to determine the structure-activity relationship of the PIPs on the RyR1 channel function.
Figure 5.




Activation of the skeletal muscle RyR1 Ca2+ channel by PIPs. (a) PIP array blots (Echelon Biosciences Inc.) were blocked with 3% fatty acid-free BSA followed by incubation with RyR1 (2 μg/ml) purified from rabbit skeletal muscle SR microsomes. RyR1 bound to the blots was then detected with anti-RyR1 antibody following standard immunoblotting procedures. The right panel shows statistical results (mean ± s.d.) from three independent experiments. The fluorescence intensity of each spot of 100 and 50 pmole groups was quantified using a PhosphoImager (Amersham Biosciences) and normalized against the intensity of PI spot of the same group. (b) PI(3,5)P2-agarose beads (lane 2), control agarose beads (lane 1), anti- PI(3,5)P2 antibody (lane 4) and IgG control (lane 3) were incubated with lysates prepared from 426 μg of purified rabbit SR vesicles. Proteins bound to the beads or the antibody were detected using immunoblotting with anti-RyR1 antibody. (c) [3H]ryanodine binding to muscle RyR1 in the presence of 10 μM free Ca2+ and PI(3,5)P2 (di-C8) or other PIPs (di-C8) at the indicated concentrations was determined as described in Methods. (d) Effects of PI(3,5)P2 (150 μM) on Ca2+-dependent binding of [3H]ryanodine to RyR1 at various concentrations of free Ca2+ were determined. Nonspecific binding (~20% of total binding) was determined with 20 μM ryanodine and has been subtracted. (e) and (f) Effects of PI(3,5)P2 on Ca2+-release activity of single RyR1 channel reconstituted in planar lipid bilayer were determined in the presence of nominally (5 μM) free Ca2+ (e) or pCa 7 (10−7 M) (f). Activity of a reconstituted RyR1 channel was continuously recorded at -30 mV holding potential. Channel openings are presented as downward deflections. Results shown were obtained from the same channels, ~ 1 min after addition of 30 μM or 60 μM of PI(3,5)P2 to the cis side. c indicates closed and o indicates opened channel. Results displayed in panels b to f are representative results of two to four experiments.
We next conducted Ca2+-dependence of [3H]ryanodine binding curves to determine whether PI(3,5)P2 activates RyR1 at free Ca2+ concentrations compatible to those found in myofibers at rest (pCa 7) or during contraction (pCa 6-5). The Ca2+-dependence of [3H]ryanodine binding to RyR1 has been well established. In the range of pCa 9 (10−9 M) to pCa 5 (10−5 M), Ca2+ has an activating effect and increases [3H]ryanodine binding to RyR1, whereas in the range of pCa 4 to pCa 2, Ca2+ has an inactivating effect. This dual effect of Ca2+ gives rise to a characteristic bell-shaped curve (Fig. 5d), which is similar to the Ca2+-Po (open probability) relationship of single RyR1 channels 25,26. In the presence of PI(3,5)P2, the binding curve maintained the same bell shape; however, the absolute binding values were dramatically increased, especially between pCa 7 and pCa 4. These results strongly suggest that the partial Ca2+ depletion of SR in MIP−/− muscle cells may be caused by direct activation of the RyR1 Ca2+ release channel by accumulated MIP PIP substrates.
Finally, we directly tested the functional significance of the interaction between PI(3,5)P2 and RyR1. PI(3,5)P2 was added to RyR1 reconstituted lipid bilayers. A nominally-free Ca2+ solution (~5 μM contaminant Ca2+) in the cis (cytosolic) side served to activate RyR1 (Fig. 5e). Before addition of PI(3,5)P2, NPo, the probability of the RyR1 channel being open (P0) times the number of channels in the recording, was low (1.15±0.3, n=3). Addition of 30 and 60 μM PI(3,5)P2 induced a significant increase in open events, increasing NP0 to 1.92±0.4 and 2.6±0.6 (n=3), respectively (Fig. 5e, and Supplementary Information, Fig. S5a). The effect of PI(3,5)P2 on the RyR1 channel was even more dramatic at a low concentration of free Ca2+ (pCa7) (Fig. 5f), suggesting an enhancement of Ca2+ leak and Ca2+-induced Ca2+ release (CICR) activities by PI(3,5)P2. The basal activity of the RyR1 channel was minimal under this condition (Po = 0.08±0.03, n=3) (Fig. 5f and Supplementary Information, Fig. S5b), but increased to Po = 0.28±0.04 and 0.51±0.11 (n=3) immediately after addition of 30 and 60 μM PI(3,5)P2, respectively (n=3). Unitary conductance (~700 pS) remained unchanged after addition of PI(3,5)P2. These electrophysiological analyses demonstrate that PI(3,5)P2 binds to RyR1 and directly modifies channel activity at contracting (pCa 5) and resting (pCa 7) levels of Ca2+, suggesting that the enhanced CICR and increased Ca2+ leak observed in MIP−/− cells may be due, at least in part, to the increased PI(3,5)P2 levels resulting from MIP absence.
In summary, our studies provide the first evidence that finely controlled PIP levels in the SR membrane and possibly the sarcolemmal membrane are critical for the proper function of the SR as the Ca2+ storage compartment and in rapidly regulating intracellular Ca2+. However, the mechanisms that regulate MIP activity and PIP levels in normal skeletal muscle physiology remain to be addressed. Since PIPs, especially PI(3,5)P2, are also the major substrates of other phosphatases of the MTMR family 2, dysregulated metabolism/dephosphorylation of these PIPs may be responsible for MTMR phosphatase-associated muscle disorders in general. Further exploration of the PIP pathway involved in intracellular Ca2+ homeostasis may provide new therapeutic targets for these muscle diseases.
Methods
Generation of MIP−/− mice
The MIP allele was targeted by homologous recombination. Exon 3 containing the start codon ATG to Exon 5 was replaced with the Neo cassette. The targeting vector was constructed using the ‘recombineering’ technique. It was then electroporated into D1 mouse embryonic stem (ES) cells with the 129S6 × C57BL/6J hybrid background. G418 resistant ES clones were isolated and screened by PCR genotyping using the primers as shown in Supplementary Information, Fig. S2a. Two ES cell clones containing a correctly targeted MIP allele were used to generate chimeric mice. Three germline transmitted chimeric mice were obtained and used to cross C57BL6J mice to produce heterozygous mice. MIP+/− mice were backcrossed with C57BL6J mice for 3 generations and F4 generation of mice were used for the studies. No differences in the two lines of mutant mice derived from the original two ES cell clones were detected.
Contractile properties of skeletal muscle
We tested the in vitro contractility properties of intact, anatomical muscles from the animals as previously described 5,28. WT and mutant EDL and soleus muscles were dissected intact and placed into 2.5 mM Ca2+ modified Ringer solution (142 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4) bubbled with 100% O2. We also compared our results using a bicarbonate buffer system and did not observe significant differences. After careful dissection of the intact muscles, a pair of control and mutant intact muscles were mounted vertically onto 20-ml Radnotti glass chambers with built-in platinum stimulating electrodes (Radnoti Products, Monrovia, CA). One tendon of the muscle was attached to a force transducer and the other one to a stationary arm in preparation for in vitro fatiguing experiments. A Powerlab computer-interface program (AD Instruments, Colorado Springs, CO) was used to control the electrical stimulation protocols and to record, digitize, and store force output data. Field stimulation (squared-waves electrical currents of 500-msec duration, 300 mA using frequencies of stimulation in the range of 1–180 Hz) was accomplished with platinum electrodes running on both sides of intact muscles. After mounting, WT and MIP−/− muscles were mounted in parallel, and their resting lengths were adjusted to produce maximal isometric twitch force. The muscles were then subjected to the force vs. frequency relationship, and the stimulating frequencies that produced maximal isometric tetanic force (Tmax) were determined. Frequencies that produced Tmax were then employed for the remaining of the protocols. After 20 min of equilibration (1-min interval, 0.83% duty cycle) at a high frequency, muscles were exposed to a fatiguing stimulation protocol (1-sec interval, 50% duty cycle) using the same frequency of stimulation for 5 min. The fatiguing protocol used in this study was modified from our previous protocol 6,7. Force output data during fatigue were normalized to Tmax before the onset of fatigue and analyzed by Origin software (OriginLab Corp. Northampton, MA). Tmax was normalized to the cross sectional area of each muscle and is reported in kPa. All experiments were conducted at room temperature (23 ± 2°C).
Ca2+ fluorometry
Primary myoblasts were isolated from hind-limb muscles of WT and MIP−/− neonates, grown and differentiated into myotubes as described previously 29. Three independent isolates of each genotype were analyzed. Spatial and temporal distribution of intracellular Ca2+ concentration in individual myotubes was determined as previously reported 10,30. Myotubes derived from primary myoblasts after 4–5 days of differentiation were loaded with 5 μM Fura-2-AM for 45 min at 25°C in ECB (130 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 25 mM HEPES, pH7.5, 1 mg/ml BSA, and 5 mM glucose). The buffer was replaced with fresh ECB and the incubation was continued for 45 min at 25°C to permit deesterification. A dual-wavelength spectrofluorometer (excitation at 340 nm and 380 nm) was used to determine the kinetic changes of [Ca2+]i concentration, which is reflected by the average ratio of F340/F380 of all cells (4–5 cells) in the field. Changes in intracellular Ca2+ were measured for 8 min following exposure to 10 μM TG or 10 mM caffeine plus 1 μM ryanodine in the Ca2+-free balanced salt solution (ECB was deficient in Ca2+ and supplemented with 0.5 mM EGTA). The extracellular bath solution was then changed with ECB with 2 mM Ca2+ and the average ratio of F340/F380 was continuously monitored. In some cases, after 3 min, the extracellular buffer was changed to ECB containing 50 μM or 100 μM SKF-96365. The [Ca2+]i concentration was monitored for additional 8 min.
For determination of resting cytosolic Ca2+ and total SR Ca2+ content in intact muscle fibers directly dissected from adult mice, FDB fibers were used for the experiments. In brief, FDB fibers were isolated by enzymatic disassociation in 0.2% type I collagenase (Sigma-Aldrich, St. Louis, MO). Mean FDB fiber size was 1 mm × 20 μM. FDB fibers were loaded with 10 μM Fura-2 AM for 45 min at room temperature in Tyrode solution. N-benzyl-p-toluene sulphonamide (20 μM), a myosin II inhibitor, was applied for 15 min to prevent motion artifact from muscle contraction 31. Fibers were also embedded into silicone grease to maintain their position in the culture dish 32. The ratio of Fura-2 fluorescence at excitation wavelength of 350 and 380 nm was measured on a PTI spectrofluorometer (Photon Technology International) to assess the [Ca2+]i level. Total SR Ca2+ storage was measured by addition of 5 μM ionomycin in the presence of 2 mM CaCl2 in the bath solution.
[3H]ryanodine binding assay
[3H]ryanodine binding to purified rabbit skeletal muscle SR was carried out as previously described 25,26. The incubation medium contained 0.2 M KCl, 40 mM Na-HEPES (pH 7.2), 7 nM [3H]ryanodine, 1 mM EGTA and various CaCl2 concentrations necessary to set free Ca2+ [Ca2+] from 10 nM to 10 mM. Sixty μg of mouse skeletal muscle SR-enriched microsomes and PI(3,5)P2 were added into the incubation medium to the indicated concentrations. Samples (0.1 ml) were incubated at 37 °C for 90 min. Mathematical fitting of data was accomplished with the computer program Origin (version 7.0, Micro-cal Inc., Northampton, MA).
Planar lipid bilayer technique
Purified rabbit skeletal muscle RyR1 protein was reconstituted into Muller-Rudin planar lipid bilayers as described previously 25,26. Single channel data were collected at steady voltages (+35 mV, cis chamber grounded) for 2 min in symmetrical 300 mM cesium methane-sulfonate and 10 mM Na-HEPES (pH 7.2). The recording solution contained ~5 μM free Ca2+ as assessed by a calibration curve, which was sufficient to activate the RyR1 channel. Different concentrations of PI(3,5)P2 were added to the cis chamber, which corresponded to the cytosolic side of the channel. Signals were digitized at 4 kHz and analyzed after filtering with a low-pass 8-pole Bessel filter at a sampling frequency of 1.5 kHz. Data acquisition and analyses were performed with Axon Instruments software and hardware (pClamp version 8.03, Digidata 200 AD/DA interface). The full and non-conducting current values were obtained from Gaussian fits to the amplitude histograms, as previously described 24,25.
Supplementary Material
Acknowledgments
We are very grateful to Mr. David Chess, Dr. Margaret Chandler, Ms. Tammy Stefan, Dr. James Jacobberger, Dr. Leticia Brotto, and Dr. Jianjie Ma for technical assistance and helpful discussions. Motor function tests were performed by the Case Western Reserve University Rodent Behavior Core. This work was supported by The National Institutes of Health grants HL068212 and HL082670 (to C.K.Q.), HL55438 (to H.H.V.), The American Heart Association grant 0535555N (to M.B.), and a pilot grant from Case Center for Transdisciplinary Research on Energetics and Cancer (to T.M.N., C.K.Q., and M.B.).
Footnotes
AUTHOR CONTRIBUTIONS
J.S., W.M. Y., M.B., J.A.S., and C.S. conducted the research and summarized the data. C.K.Q., M.B., H.H.V., T.M.N., and C.G. designed the experiments and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.MacLennan DH. Ca2+ signalling and muscle disease. Eur J Biochem. 2000;267:5291–7. doi: 10.1046/j.1432-1327.2000.01566.x. [DOI] [PubMed] [Google Scholar]
- 2.Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. 2002;12:579–85. doi: 10.1016/s0962-8924(02)02412-1. [DOI] [PubMed] [Google Scholar]
- 3.Alonso A, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 4.Tosch V, et al. A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum Mol Genet. 2006;15:3098–106. doi: 10.1093/hmg/ddl250. [DOI] [PubMed] [Google Scholar]
- 5.Zhao X, et al. Enhanced resistance to fatigue and altered calcium handling properties of sarcalumenin knockout mice. Physiol Genomics. 2005;23:72–8. doi: 10.1152/physiolgenomics.00020.2005. [DOI] [PubMed] [Google Scholar]
- 6.Nagaraj RY, et al. Increased susceptibility to fatigue of slow- and fast-twitch muscles from mice lacking the MG29 gene. Physiol Genomics. 2000;4:43–9. doi: 10.1152/physiolgenomics.2000.4.1.43. [DOI] [PubMed] [Google Scholar]
- 7.Nosek TM, et al. Functional properties of skeletal muscle from transgenic animals with upregulated heat shock protein 70. Physiol Genomics. 2000;4:25–33. doi: 10.1152/physiolgenomics.2000.4.1.25. [DOI] [PubMed] [Google Scholar]
- 8.Takeshima H, et al. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 1994;369:556–9. doi: 10.1038/369556a0. [DOI] [PubMed] [Google Scholar]
- 9.Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1988;331:315–9. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- 10.Pan Z, et al. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat Cell Biol. 2002;4:379–83. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- 11.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–7. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 13.Merritt JE, et al. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J. 1990;271:515–22. doi: 10.1042/bj2710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhogal MS, Colyer J. Depletion of sarcoplasmic reticulum calcium prompts phosphorylation of phospholamban to stimulate store refilling. Ann N Y Acad Sci. 1998;853:260–3. doi: 10.1111/j.1749-6632.1998.tb08274.x. [DOI] [PubMed] [Google Scholar]
- 15.Toth A, et al. Quantitative assessment of [Ca2+]i levels in rat skeletal muscle in vivo. Am J Physiol. 1998;275:H1652–62. doi: 10.1152/ajpheart.1998.275.5.H1652. [DOI] [PubMed] [Google Scholar]
- 16.Michell RH, Heath VL, Lemmon MA, Dove SK. Phosphatidylinositol 3,5-bisphosphate: metabolism and cellular functions. Trends Biochem Sci. 2006;31:52–63. doi: 10.1016/j.tibs.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–7. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 18.Min J, et al. Forward chemical genetic approach identifies new role for GAPDH in insulin signaling. Nat Chem Biol. 2007;3:55–9. doi: 10.1038/nchembio833. [DOI] [PubMed] [Google Scholar]
- 19.Niswender KD, et al. Immunocytochemical detection of phosphatidylinositol 3-kinase activation by insulin and leptin. J Histochem Cytochem. 2003;51:275–83. doi: 10.1177/002215540305100302. [DOI] [PubMed] [Google Scholar]
- 20.Ishii T, Lemas MV, Takeyasu K. Na(+)-, ouabain-, Ca(2+)-, and thapsigargin-sensitive ATPase activity expressed in chimeras between the calcium and the sodium pump alpha subunits. Proc Natl Acad Sci U S A. 1994;91:6103–7. doi: 10.1073/pnas.91.13.6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartolommei G, et al. Clotrimazole inhibits the Ca2+-ATPase (SERCA) by interfering with Ca2+ binding and favoring the E2 conformation. J Biol Chem. 2006;281:9547–51. doi: 10.1074/jbc.M510550200. [DOI] [PubMed] [Google Scholar]
- 22.Amran MS, Homma N, Hashimoto K. Pharmacology of KB-R7943: a Na+-Ca2+ exchange inhibitor. Cardiovasc Drug Rev. 2003;21:255–76. doi: 10.1111/j.1527-3466.2003.tb00121.x. [DOI] [PubMed] [Google Scholar]
- 23.Treves S, Jungbluth H, Muntoni F, Zorzato F. Congenital muscle disorders with cores: the ryanodine receptor calcium channel paradigm. Curr Opin Pharmacol. 2008;8:319–26. doi: 10.1016/j.coph.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Benkusky NA, Farrell EF, Valdivia HH. Ryanodine receptor channelopathies. Biochem Biophys Res Commun. 2004;322:1280–5. doi: 10.1016/j.bbrc.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, Zamudio FZ, Olbinski BA, Possani LD, Valdivia HH. Activation of skeletal ryanodine receptors by two novel scorpion toxins from Buthotus judaicus. J Biol Chem. 2004;279:26588–96. doi: 10.1074/jbc.M403284200. [DOI] [PubMed] [Google Scholar]
- 26.Zhu X, Ghanta J, Walker JW, Allen PD, Valdivia HH. The calmodulin binding region of the skeletal ryanodine receptor acts as a self-modulatory domain. Cell Calcium. 2004;35:165–77. doi: 10.1016/j.ceca.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001:RE19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 28.Brotto MA, Nosek TM, Kolbeck RC. Influence of ageing on the fatigability of isolated mouse skeletal muscles from mature and aged mice. Exp Physiol. 2002;87:77–82. doi: 10.1113/eph8702224. [DOI] [PubMed] [Google Scholar]
- 29.Frock RL, et al. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20:486–500. doi: 10.1101/gad.1364906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin DW, et al. A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J Biol Chem. 2003;278:3286–92. doi: 10.1074/jbc.M209045200. [DOI] [PubMed] [Google Scholar]
- 31.Cheung A, et al. A small-molecule inhibitor of skeletal muscle myosin II. Nat Cell Biol. 2002;4:83–8. doi: 10.1038/ncb734. [DOI] [PubMed] [Google Scholar]
- 32.Jacquemond V. Indo-1 fluorescence signals elicited by membrane depolarization in enzymatically isolated mouse skeletal muscle fibers. Biophys J. 1997;73:920–8. doi: 10.1016/S0006-3495(97)78124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.