

SUMMARY
In near all animals, sleep is consistently concentrated to a specific time of the day. The timing and consolidation of sleep and wake bouts depend on the interplay between a homeostatic and a circadian processes of sleep regulation [1–3]. Sleep propensity rises as a homeostatic response to increasing wake time while a circadian clock determines the specific time when sleep will likely occur. This two-process regulation of sleep also determines which specific sleep stage will be manifested and specifically, the circadian process governs tightly the manifestation of rapid eye movement sleep (REMS) [1, 4]. The role of the hypothalamic suprachiasmatic nucleus (SCN) in the circadian gating of sleep and wakefulness has been unequivocally established by lesion studies [5] but its role in the timing of specific sleep stages has remained unknown. Using a forced desynchrony paradigm that induces the stable uncoupling of the ventrolateral (vl) and dorsomedial (dm) SCN, and a jetlag paradigm that induces a transient desynchronization between these SCN subregions we provide evidence that the SCN can time the occurrence of specific sleep stages. Specifically, the circadian regulation of REMS is associated with rhythmic clock gene expression within the dmSCN. Our results also provide the first neurophysiological model for the disruptions of sleep architecture that may result from temporal challenges such as rotational shift-work and transmeridional flights.
RESULTS
REMS propensity is associated with clock gene expression within the dorsomedial SCN
The role of the SCN in the circadian regulation of sleep has been widely supported by a variety of experimental approaches including neuroanatomical lesions and tract tracing involving SCN efferent projections to sleep centers [5–7]. The circadian regulation of specific sleep stages, in particular the remarkable faithfulness of REMS propensity to the circadian nadir of core body temperature (CBT), has been widely supported by studies in humans in which the circadian and homeostatic regulation of sleep are experimentally desynchronized from each other by exposing subjects to rest-activity cycles different from 24 hours [4, 8]. Under this forced desynchrony protocol, slow-wave sleep (SWS) shows a tight synchronization with the scheduled sleep episodes, whereas REMS propensity free-runs with the endogenous circadian period. Although these studies clearly implicate a circadian oscillator in the regulation of REMS, the role of the master circadian clock within the SCN on the circadian gating of REMS or other sleep stages has been confounded by the fact that SCN lesions completely abolish the sleep-wake circadian rhythm.
We have recently developed a rat model of circadian forced desynchrony that displays the key features of forced desynchronized human subjects, including the desynchronization of sleep stages [9, 10]. Rats exposed to a 22-h light-dark (LD) cycle exhibit two rhythms of locomotor activity: one rhythm is entrained to the LD cycle while the other rhythm free-runs with a period longer than 24 h. In these animals, NREMS and SWS show a dual periodicity in synchrony with the entrained and free-running activity bouts; however, REMS only exhibits a single, free-running rhythm phase-locked to the nadir of the free-running CBT rhythm [9]. In the forced desynchronized rat (FDR), the entrained locomotor activity rhythm is associated with the rhythmic expression of the clock genes Per1, Per2 and Bmal1 within neurons in the retinorecipient vlSCN. In contrast, the free-running locomotor activity rhythm is associated with the rhythmic expression of these genes within neurons in the dmSCN [10]. Based on these results we hypothesized that the desynchronization of sleep stages could emerge from the desynchronized activity of these anatomically and functionally distinct SCN subregions [11, 12]. To test this possibility, we implanted 22-h FDRs with electroencephalographic (EEG) electrodes. Locomotor activity was continuously monitored through infrared detectors and after desynchronization was confirmed, animals were sacrificed during one of two conflicting phases of the locomotor activity rhythms. One of these phases occurs when the subjective day of the free-running rhythm coincided with the night of the entrained rhythm; the other phase occurs when the subjective night of the free-running rhythm coincided with the day of the entrained rhythm. Brains were removed, frozen and processed for in situ hybridization for the clock gene Per1. As we have previously shown [13], the RNA levels for this gene showed high (daytime) values during the 22-h light phase only within the vlSCN (Figure 1a) and during the free-running subjective day only within the dmSCN (Figure 1b). Scoring of the EEG recordings during the last hour before sacrifice showed a statistically positive correlation between REMS intensity and the level of expression within the dmSCN (linear regression analysis, n = 6, R2 = 0.6885, p = 0.0413, Figure 1c). The occurrence of REMS was not associated with daytime clock gene expression within the vlSCN, as REMS intensity was not associated with vlSCN Per1 expression (linear regression analysis, n = 6, R2 = 0.5043, p = 0.1138, Figure 1c).
Figure 1.
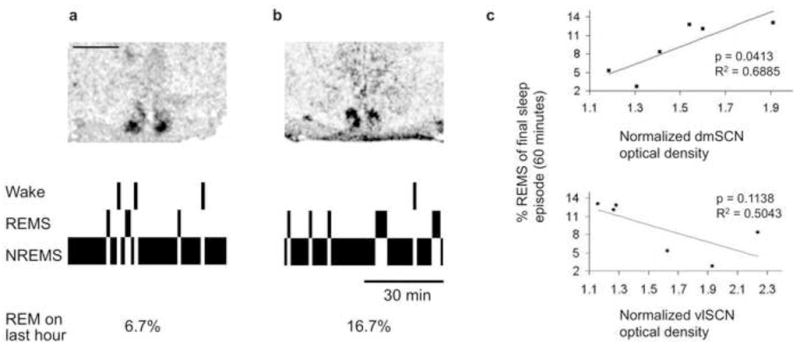
Circadian REMS propensity is associated with clock gene expression within the dmSCN. a and b: Autoradiographic films of SCN sections hybridized with a riboprobe against the clock gene Per1 (top) and hypnograms of the last hour before the animals were sacrificed (bottom). Tissue sections from an animal sacrificed during 22-h light and rest phase of the entrained rhythm coincided with the subjective night of the free-running rhythm (a) and an animal sacrificed during the 22-h dark and active phase for the entrained rhythm coincided with the subjective day of the free-running rhythm (b). c: Linear regression analysis between the intensity of Per1 mRNA expression in different SCN subregions, as normalized optical intensity, and the percent of time spent in REMS in the last hour before sacrifice in six animals. Animals were sacrificed in the phases shown in a and b (n = 6). Scale bar for a and b represents 1 mm.
Transient circadian desynchronization of the ventrolateral and dorsomedial SCN leads to desynchronization between REMS and SWS
Our hypothesis of specific sleep stage circadian control by SCN subregions predicts that desynchronization of circadian oscillators in the dm- and vlSCN should lead to desynchrony of SWS and REMS. Besides forced desynchronization, abrupt phase shifts of the LD cycle similar to those that result from jetlag can induce transient desynchronization between these SCN subregions. In Wistar rats, a 6-h delay in the LD cycle mimicking a westward transmeridional flight induces a quick phase delay of the vlSCN characterized by rapid entrainment to the new phase. This has been determined by measuring both the rhythm in action potential frequency [14] and in Per1 expression as reported by luciferase bioluminescence [15]. In contrast, the dmSCN requires 3–6 cycles, depending on the delaying protocol, to reach the new phase. We tested the prediction that transient desynchronization between the vl- and dmSCN induced by a 6-h delay would lead to desynchrony between SWS and REMS. Rats implanted with EEG electrodes and intraperitoneal temperature sensors were exposed to a 24-h LD cycle. Entrainment was confirmed by monitoring continuous locomotor activity, and the rats were then exposed to a 6-h delay of the LD cycle achieved by extending the light phase by 6 h (day 0), followed by 12 h of darkness and then 12 h of light (day 1). After the end of this delayed light phase, animals were released into constant darkness (day 2). This protocol has been shown to induce the immediate (day 0) delay of the ventral SCN and a slower delay of the dorsal SCN, which does not reach the new phase until 6 days after the shift [14]. As predicted by our hypothesis, SWS showed an immediate adjustment after the phase delay, whereas REMS required several days to adjust to the delay (Figure 2a). The phase transitions are more clearly seen in Figure 2b, where the evolution of delta power and REMS acrophases on the days before and after the delay of the LD cycle are depicted. Before the phase delay, the phase angle difference between the REMS and delta power acrophases was approximately 2 hours. Immediately after the phase shift, this phase angle difference was reduced to less than an hour. The normal phase relationship was not reestablished until at least 3–5 days after the phase delay. This is confirmed by the analysis of the difference between these acrophases throughout the days, which shows a statistically significant reduction on days 1–2 compared to the days before the shift and days 3–5 after the shift (one-way repeated-measures ANOVA with time as a single factor, n = 5, F = 4.47, p = 0.007, and Tukey contrasts, Supplemental Figure 1). Interestingly, total sleep appeared to undergo a quick delay as that seen for SWS, a phenomenon that is consistent with the rapid delay of the locomotor activity rhythm (data not shown).
Figure 2.
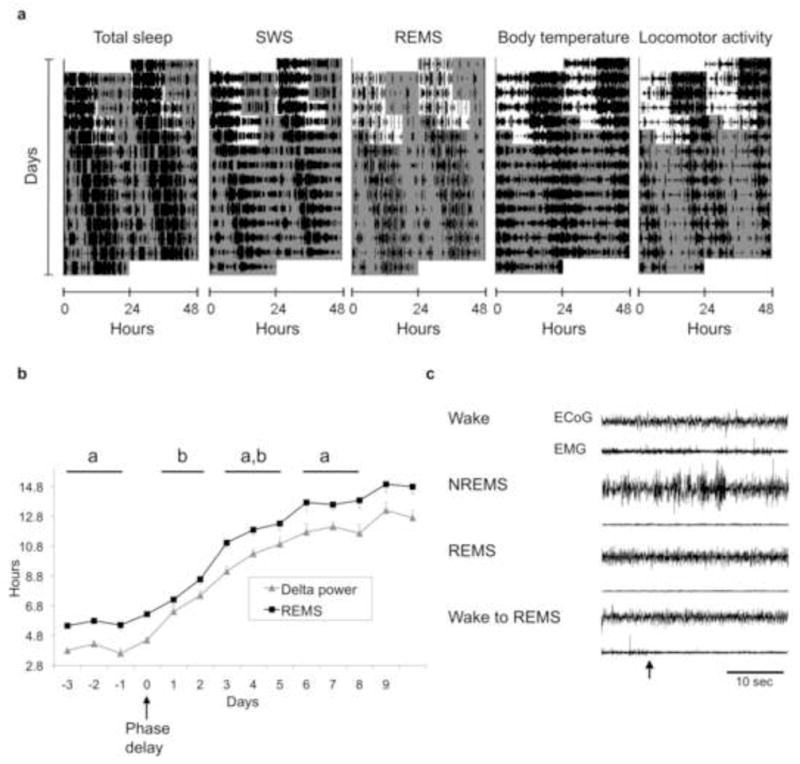
An abrupt phase delay of the LD cycle, similar to jetlag, induces transient desynchrony of sleep stages in the rat. a: actograms representing the daily course of total sleep, REMS, delta power, CBT and locomotor activity of an animal exposed to an abrupt 6-h phase delay of its 12:12 LD cycle. Lights-OFF time was delayed by 6 h on day 0, followed by one 12:12 cycle of the new LD schedule (day 1) before release into constant darkness (day 2). White and gray areas represent the times of lights ON and OFF, respectively. b: The time course change of the acrophase of REM and delta power before and after the 6-h delay in the LD cycle (day 0). Points represent means + or − the standard error (n = 5). Blocks of days marked with different letters denote statistically significant differences (one-way ANOVA) in the acrophase difference between the two stages. c: An example of a spontaneous transition from wake to REMS in an animal that underwent a phase delay under the conditions shown in a. Representative EEG and EMG recordings are shown for each vigilance state; the wake-to-REMS transition (arrow) is shown in the bottom traces.
The slower phase delay of REMS relative to SWS and total sleep produces a transient abnormal circadian sequence of sleep stages that increases REMS propensity at early stages of the subjective day. Consistent with this idea, we detected instances in which REMS occurs directly after a wake state without the normal transition through NREMS (Figure 2c). Although rare (about 10 total observations out of 7 phase delayed animals) these wake-REMS transitions were never observed in the same animals before the delay of the LD cycle or after the delay was completed, day 6 and beyond.
CBT is under tight circadian regulation and its circadian nadir is typically linked to high REMS propensity, even under forced desynchrony [8, 9] or spontaneous internal desynchronization [16, 17]. Previous data from our laboratory indicated that in the FDR, CBT shows a strong free-running component coupled to the free-running rhythm of REMS [9]. Because locomotor activity-induced heat production can mask the CBT rhythm, we “demasked” the temperature data by subtracting the temperature increment due to the activity just before the intraperitoneal temperature was recorded. The nadir of this activity-independent CBT remained in phase with the acrophase of REMS throughout the delay protocol (data not shown; one-way repeated-measures ANOVA with time as a single factor, n = 5, F = 1.68, p = 0.22). This was also confirmed by the fact that the 95% confidence interval for the phase difference between the CBT nadir and the REMS acrophase always included the zero value, even on the first and second day after the phase delay, indicating that the circadian phase of these two events was statistically undistinguishable throughout the jetlag paradigm.
DISCUSSION
Here we show that the activity of the dmSCN, as determined by the expression of the clock gene Per1, is associated with REMS intensity. Although the role of the SCN as a master circadian pacemaker gating the occurrence of sleep and wakefulness has been recognized for decades [5–7, 18], our study suggests that the SCN can also control sleep architecture by regulating the circadian sequence of specific sleep stages. Furthermore, the present study shows that the manifestation of specific sleep stages throughout the circadian cycle is associated with the activity of anatomically identifiable neuronal oscillators within the SCN. In the rat, the vl- and dmSCN, also known as the core and shell respectively, are recognized as functionally distinct subregions, showing differences in histochemistry, cytoarchitecture and afferent and efferent projections [11, 19, 20]. Our study exploits both a forced desynchrony paradigm and jetlag-like protocol to demonstrate a link between this subregional organization and the regulation of sleep architecture. This is consistent with the notion that the vl- and dmSCN are associated with the circadian regulation of sleep and SWS, whereas the circadian regulation of REMS relies solely on the activity of the dmSCN.
Early studies in humans that dissociated the circadian and homeostatic regulation of sleep using spontaneous internal desynchronization in constant environmental conditions showed a striking dependence of REMS propensity on the phase of the circadian pacemaker [16]. This circadian REMS propensity was also tightly associated with the rising phase of the CBT circadian nadir [17]. The use of forced desynchrony protocols in humans, in which the circadian and homeostatic regulation of sleep can be dissociated in a stable and predictable manner, confirmed this relationship between body temperature and REMS and provided further evidence for a stronger homeostatic control of SWS ([17] and reviewed in [1, 4]). Results in the rat from our laboratory later confirmed this differential regulation of sleep stages as well as the tight association between the CBT nadir and the presence of REMS [9]. The present study offers a neuroanatomical basis for the circadian gating of CBT and REMS, as our CBT rhythm remained tightly associated to the REMS propensity rhythm.
Previous to our study, Deboer et al [21] showed that the electrical activity of the SCN in vivo, as recorded by multiple unit activity, is associated with specific sleep stages. The authors show that the action potential frequency of SCN neurons is positively correlated with the occurrence of REMS and negatively correlated with slow-wave activity, and that this correlation is present irrespective of the circadian time at which the recording is performed. These results are interpreted as evidence for afferent input from sleep centers to the SCN. Although it is not known whether these changes in electrical activity are correlated with changes in the expression of clock genes within the SCN, our results suggests that these sleep-stage-associated changes in electrical activity are transient changes that do not reflect the circadian pattern of gene expression. Nevertheless, it is important to note that whereas our study is focused on subregional gene expression, multiple unit activity recordings in vivo do not discriminate between vl- and dmSCN neurons.
In view of previous studies showing transient uncoupling between the ventral and the dorsal SCN after abrupt phase shifts of the LD cycle [14, 15], the transient desynchronization of sleep stages after an abrupt phase delay of the LD cycle is consistent with our hypothesis of SCN subregional circadian gating of sleep stages. Furthermore, our results point to the dramatic effects that transmeridional travel may have on sleep quality. Immediately after the delay of the LD cycle, the normal phase relationship between SWS and REMS is disrupted so that REMS is relatively phase advanced to SWS and sleep onset. This result is indeed expected by the well-known regulation of REMS by a circadian oscillator that is relatively resilient to phase shifting stimuli. Nevertheless, thus far the important prediction that SWS should phase shift more rapidly than REMS after abrupt phase shifts has only been tested once (E. Vivaldi, Universidad de Chile, personal communication). The abnormal circadian sequencing that results from our jetlag protocol leads to spontaneous wake-to-REMS transitions. This short latency to REMS is typical of narcoleptic patients and animal models of narcolepsy [22]. Given the known role of sleep on learning processes, the transient disruption of the normal circadian sequence of sleep stages we observe may account for the known learning deficits that result from phase shifting circadian rhythms in animals [23–25].
Direct and indirect SCN efferent projections reach both sleep-promoting regions including the ventrolateral preoptic area and median preoptic nucleus, as well as wake-promoting regions such as orexinergic network of the lateral and posterior hypothalamus and the noradrenergic locus coeruleus [5–7]. This strategic neuroanatomical position is coherent with a preeminent role of the SCN in the master circadian control of the sleep-wake cycle. Furthermore, SCN input to both wake- and sleep-promoting regions is consistent with a bidirectional role of the rat master circadian pacemaker, in which the SCN promotes sleep during the subjective day and wake during the subjective night [5]. Our study not only supports a sleep-promoting role for the SCN but also suggests that this sleep-promotion is stage specific, so that not only the occurrence of sleep but also of specific sleep stages is gated by the SCN circadian pacemaker.
In summary, our results support a preeminent role of the master circadian pacemaker within the SCN in the circadian regulation of not only the sleep-wake cycle but of specific sleep states as well. The desynchronization of subregional neuronal oscillators within the SCN both by forced desynchrony and by a jetlag-like protocol are consistent with a subregion-specific regulation of sleep stages, in which the circadian gating of REMS relies on the activity dmSCN. Our studies highlight the importance of neuronal coherence within the SCN’s master circadian clock. They also constitute direct evidence that circadian internal desynchronization associated with rotational shift-work or with jetlag can induce internal desynchrony of sleep stages, and provide the first model for the neuroanatomical bases of this disruption of sleep architecture.
EXPERIMENTAL PROCEDURES
Animals
All experiments were approved by the Animal Care and Use Committee of the University of Washington. Male Wistar rats, two months old on arrival were purchased from Charles River (Raleigh, NC) and used for all the experiments.
Sleep stages and clock gene expression in the forced desynchronized rat
Animals were housed individually in transparent polycarbonate cages (20×25×22 cm) were maintained under a symmetrical LD cycle of 11 h of light and 11 h of dark. Light consisted of cool white light (100–150 lux) and darkness of dim red light (less than 1 lux). Locomotor activity was continuously monitored by a system consisting of two crossed infrared beams and binned every 15 min. After 10 to 15 days under 22-hr LD cycle desynchronization was confirmed by the appearance of two statistically significant rhythmic components of locomotor activity as determined by periodogram analysis. Non-desynchronized rats were removed from the study and desynchronized animals were anesthetized during the light phase of the LD cycle and implanted with low impedance cortical electrodes for EEG recordings and electromyographic (EMG) electrodes as previously described [9]. After recovery, cortical electrical activity, EMG activity and locomotor activity was recorded for at least 15 days before the animals were sacrificed. Based on the locomotor activity recordings, animals were sacrificed in one of two phases of the desynchrony protocol. The first group of animals was sacrificed during the phase in which the 22-h light and rest phase of the entrained rhythm coincided with the subjective night of the free-running rhythm. The second group of animals was sacrificed during a phase in which the 22-h dark and active phase for the entrained rhythm coincided with the subjective day of the free-running rhythm. These two phases were chosen because clock gene expression in the SCN shows high vlSCN and low dmSCN Per1 expression or vice versa [10] and because REMS propensity is expected to be low and high in these two phases respectively [9]. Animals were sacrificed by decapitation and the brains were quickly removed and frozen until they were processed using radioactive in situ hybridization as previously described [26]. Hybridized brain sections were exposed to autoradiographic film and the images were scanned and quantified with the ImageJ software. Templates for the dm- and vlSCN were used for sections from all animals and the optical density of each SCN subregion was normalized to the background optical density within the hypothalamus. The normalized optical density values were correlated with the percentage of REMS within the last hour before sacrifice using least-squares linear regression analysis (n = 6).
Transient desynchronization by abrupt phase shifts of the LD cycle
Animals were housed under the same conditions as above but under a 24-h symmetrical LD cycle. Animals were implanted with EEG and EMG electrodes and with intraperitoneal temperature sensors (Thermochrone iButtons, Dallas Semiconductor, Dallas, TX). After recovery, cortical electrical activity, EMG activity and locomotor activity was recorded for at least 11 days before the animals were subjected to an abrupt delay of the LD cycle. This delay was achieved by extending the light phase by 6 h on one cycle (day 0), followed by 12 h of darkness and then 12 h of light (day 1). After the end of this delayed light phase, animals were released into constant darkness. Locomotor activity, CBT, and EEG and EMG activity were recorded for 9 more days. At the conclusion of the experiment, animals were sacrificed by CO2 anesthesia and temperature sensors were recovered and the data (sampled every 15 minutes) downloaded onto a PC computer for analysis.
Analysis of sleep stages
The vigilance states of wakefulness, NREMS and REMS were determined off-line in 10-s epochs by an experimenter masked to the circadian phase of the animal. Wakefulness is characterized by fast, low-amplitude EEG waves in coincidence with high EMG activity. NREMS is associated with slow high-amplitude EEG waves. REMS is characterized by fast, low-amplitude EEG waves, appearance of theta waves (visualized through a fast Fourier transform) and lack of EMG activity. SWS (EEG frequencies of 0.5–4.0 Hz) was identified through fast Fourier transform, normalizing to the power calculated for NREMS episodes. The percentage of time spent in each state was calculated for every 10 min. The percentage data, normalized to the top 85th percentile found in a 10-min interval, was plotted as raster plots or actograms in which the daily profile for each variable is plotted on a horizontal axis and successive 24-h data blocks are stacked vertically. Delta power was calculated as the product of percent of delta activity (0.5–4 Hz) and duration of NREMS within a 10 minute bin. Sleep stage acrophases were calculated with the El Temps® software (http://www.el-temps.com/) through cosinor analysis, using a least squares method to fit a cosine curve to the time series of each variable. This results in a single time point indicating the time of the daily peak of the specific sleep stage.
Analysis of core body temperature and locomotor activity
CBT and locomotor activity temperature data points were expressed as deviation from each animal’s daily mean. Activity data were normalized to the 85th percentile of each animal’s daily maximum. To account for the effect of locomotor activity on CBT it is necessary to eliminate the activity-dependent increase of CBT. We performed a least-squares linear regression analysis with total locomotor activity within 15 min as independent variable and the CBT recording at the end of that 15-min interval as dependent variable, as previously described [9]. This analysis encompassed over 4 days of recordings in animals subjected to a 12:12 LD before the abrupt phase shift. Because all time points across a day are considered, this regression analysis is independent of circadian temperature control and describes the overall relationship between activity and temperature. Based on the best-fit linear equation, each 15 minute CBT value was demasked by subtracting the temperature increase that was accounted for by the locomotor activity. The difference between the measured CBT and activity-dependent CBT results in an activity-independent temperature. Changes in this latter value reflect the circadian modulation of CBT irrespective of the levels of locomotor activity. The nadir of circadian temperature was calculated by inverting normalized activity-independent temperature and calculating the acrophase of this function.
Experiment 2: Phase shifting of sleep stages after an abrupt phase delay
The average difference between SWS and REMS was calculated by taking the daily acrophase difference between delta power and REMS in an individual animal. The acrophase differences among 5 animals (n = 5) were averaged and binned into 2–3-day windows. The first window includes 1 to 3 day before phase delay (days -3 to -1). The second window includes the first and second days (days 1 and 2) after the day of the phase delay. The third and fourth windows include days 3–5 and 6–8 respectively. A one-way repeated measures ANOVA with time window as the factor was used to analyzed the progression of the delta power-REMS phase differences. The same type of analysis was performed for the phase differences between REMS acrophase and the nadir of activity-independent CBT.
Acknowledgments
We would like to thank Dr. Mike Schwartz for comments on the manuscript and advise with statistical analysis. Supported by National Institutes of Health Grant R01 MH075016 to H.O.d. and by the Mary Gates Endowment for Students to B.E.S..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borbely AA, Dijk DJ, Achermann P, Tobler I. Processes underlying the regulation of sleep-wake cycle. In: Takahashi JS, Turek FW, Moore RY, editors. Handbook of Behavioral Neurobiology: Circadian Clocks. Vol. 12. New York: Kluwer Academic/Plenum Publishers; 2001. pp. 457–479. [Google Scholar]
- 2.Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- 3.Daan S, Beersma DG, Borbely AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–183. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- 4.Czeisler CA, Dijk DJ. Human circadian physiology and sleep-wake regulation. In: Takahashi JS, Turek FW, Moore RY, editors. Handbook of Behavioral Neurobiology: Circadian Clocks. Vol. 12. New York: Kluwer Academic/Plenum Publishers; 2001. pp. 531–569. [Google Scholar]
- 5.Mistlberger RE. Circadian regulation of sleep in mammals: Role of the suprachiasmatic nucleus. Brain Research Reviews. 2005;49:429–454. doi: 10.1016/j.brainresrev.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 7.Fuller PM, Gooley JJ, Saper CB. Neurobiology of the sleep-wake cycle: sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythms. 2006;21:482–493. doi: 10.1177/0748730406294627. [DOI] [PubMed] [Google Scholar]
- 8.Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15:3526–3538. doi: 10.1523/JNEUROSCI.15-05-03526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cambras T, Weller JR, Angles-Pujoras M, Lee ML, Christopher A, Diez-Noguera A, Krueger JM, de la Iglesia HO. Circadian desynchronization of core body temperature and sleep stages in the rat. Proc Natl Acad Sci U S A. 2007;104:7634–7639. doi: 10.1073/pnas.0702424104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de la Iglesia HO, Cambras T, Schwartz WJ, Diez-Noguera A. Forced Desynchronization of Dual Circadian Oscillators within the Rat Suprachiasmatic Nucleus. Curr Biol. 2004;14:796–800. doi: 10.1016/j.cub.2004.04.034. [DOI] [PubMed] [Google Scholar]
- 11.Moore RY, Speh JC, Leak RK. Suprachiasmatic nucleus organization. Cell Tissue Res. 2002;309:89–98. doi: 10.1007/s00441-002-0575-2. [DOI] [PubMed] [Google Scholar]
- 12.Silver R, Schwartz WJ. Circadian Rhythms. Vol. 393. 2005. The suprachiasmatic nucleus is a functionally heterogeneous timekeeping organ; pp. 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cambras T, Chiesa J, Araujo J, Diez-Noguera A. Effects of photoperiod on rat motor activity rhythm at the lower limit of entrainment. J Biol Rhythms. 2004;19:216–225. doi: 10.1177/0748730404264201. [DOI] [PubMed] [Google Scholar]
- 14.Albus H, Vansteensel MJ, Michel S, Block GD, Meijer JH. A GABAergic mechanism is necessary for coupling dissociable ventral and dorsal regional oscillators within the circadian clock. Curr Biol. 2005;15:886–893. doi: 10.1016/j.cub.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura W, Yamazaki S, Takasu NN, Mishima K, Block GD. Differential response of Period 1 expression within the suprachiasmatic nucleus. Journal of Neuroscience. 2005;25:5481–5487. doi: 10.1523/JNEUROSCI.0889-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czeisler CA, Weitzman E, Moore-Ede MC, Zimmerman JC, Knauer RS. Human sleep: its duration and organization depend on its circadian phase. Science. 1980;210:1264–1267. doi: 10.1126/science.7434029. [DOI] [PubMed] [Google Scholar]
- 17.Czeisler CA, Zimmerman JC, Ronda JM, Moore-Ede MC, Weitzman ED. Timing of REM sleep is coupled to the circadian rhythm of body temperature in man. Sleep. 1980;2:329–346. [PubMed] [Google Scholar]
- 18.Eastman CI, Mistlberger RE, Rechtschaffen A. Suprachiasmatic nuclei lesions eliminate circadian temperature and sleep rhythms in the rat. Physiol Behav. 1984;32:357–368. doi: 10.1016/0031-9384(84)90248-8. [DOI] [PubMed] [Google Scholar]
- 19.Leak RK, Moore RY. Topographic organization of suprachiasmatic nucleus projection neurons. J Comp Neurol. 2001;433:312–334. doi: 10.1002/cne.1142. [DOI] [PubMed] [Google Scholar]
- 20.Silver R, Schwartz WJ. The suprachiasmatic nucleus is a functionally heterogeneous timekeeping organ. Methods Enzymol. 2005;393:451–465. doi: 10.1016/S0076-6879(05)93022-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deboer T, Vansteensel MJ, Detari L, Meijer JH. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat Neurosci. 2003;6:1086–1090. doi: 10.1038/nn1122. [DOI] [PubMed] [Google Scholar]
- 22.Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–181. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- 23.Devan BD, Goad EH, Petri HL, Antoniadis EA, Hong NS, Ko CH, Leblanc L, Lebovic SS, Lo Q, Ralph MR, et al. Circadian phase-shifted rats show normal acquisition but impaired long-term retention of place information in the water task. Neurobiol Learn Mem. 2001;75:51–62. doi: 10.1006/nlme.1999.3957. [DOI] [PubMed] [Google Scholar]
- 24.Fekete M, van Ree JM, Niesink RJ, de Wied D. Disrupting circadian rhythms in rats induces retrograde amnesia. Physiol Behav. 1985;34:883–887. doi: 10.1016/0031-9384(85)90008-3. [DOI] [PubMed] [Google Scholar]
- 25.Tapp WN, Holloway FA. Phase shifting circadian rhythms produces retrograde amnesia. Science. 1981;211:1056–1058. doi: 10.1126/science.7193351. [DOI] [PubMed] [Google Scholar]
- 26.de la Iglesia HO. In situ hybridization of suprachiasmatic nucleus slices. In: Rosato E, editor. Methods Mol Biol. Vol. 362. Humana Press; 2007. pp. 513–531. [DOI] [PubMed] [Google Scholar]