

Abstract
Using a novel loading technique, IL-12 is reported here to be efficiently encapsulated within large multilamellar liposomes. The preclinical efficacy of the cytokine loaded liposomes to deliver IL-12 into human tumors and to reactive tumor-associated T cells in situ is tested using a human tumor xenograft model. IL-12 is released in vivo from these liposomes in a biologically active form when injected into tumor xenografts that are established by the subcutaneous implantation of non-disrupted pieces of human lung, breast or ovarian tumors into immunodeficient mice. The histological architecture of the original tumor tissue, including tumor-associated leukocytes, tumor cells and stromal cells is preserved anatomically and the cells remain functionally responsive to cytokines in these xenografts. The local and sustained release of IL-12 into the tumor microenvironment reactivates tumor-associated quiescent effector memory T cells to proliferate, produce and release IFN-γ resulting in the killing of tumor cells in situ. Very little IL-12 is detected in the serum of mice for up to 5 days after an intratumoral injection of the IL-12 liposomes. We conclude that IL-12 loaded large multilamellar liposomes provide a safe method for the local and sustained delivery of IL-12 to tumors and a therapeutically effective way of reactivating existing tumor-associated T cells in human solid tumor microenvironments. The potential of this local in situ T cell re-stimulation to induce a systemic anti-tumor immunity is discussed.
Keywords: Tumor Immunotherapy, T Cells, Interleukin-12, Liposomes, SCID mice
INTRODUCTION
The majority of T cells within the microenvironment of a variety of different murine and human tumors express a phenotype of CD4+ and CD8+ effector memory cells (1–3). However, tumors often progress and spread inspite of the presence of these memory T cells, and a broad array of other inflammatory leukocytes that are directly associated with the tumor. Multiple and diverse explanations have been offered to indicate why the cognitive and innate immunocompetent cells in the tumor microenvironment fail to control tumor growth (4,5). T cells isolated from human renal cell carcinomas (6), non-Hodgkin lymphoma (1) and non-small cell lung carcinomas (7), were found to be hyporesponsive to activation via the T cell receptor. We reported that the hyporesponsiveness of the human lung tumor-associated memory T cells was due to a TCR signaling arrest that was at least partially dependent upon membrane-associated TGF-β1 present in the tumor microenvironment (8). The hyporesponsiveness of these T cells to activation signals (immobilized anti-CD3 and anti-CD28) was found to be reversible by a brief ex vivo exposure to IL-12 (8).
While the molecular mechanisms by which IL-12 reverses the TCR signaling blockade in tumor-associated memory T cells have not yet been completely defined, this cytokine is known to exert numerous regulatory effects on effector, helper, and memory T lymphocytes and natural killer (NK) cells (9). IL-12 facilitates CD8+ T cell responses, promotes the generation of CD4+ TH1 helper T cells (10–12), enhances the lytic activity of NK cells and induces the secretion of IFN-γ in both T cells and NK cells (13–15). Because of its recognized potent immunostimulatory effects, IL-12 was extensively evaluated for its potential to enhance anti-tumor immune responses. The systemic administration of IL-12 established its ability to augment immunity and to significantly retard the growth of a variety of different murine tumors and prolong the survival of the tumor bearing mice (16–20). Based upon these early successes with IL-12 in murine tumor models, clinical trials with cancer patients were initiated. Unfortunately the repeated systemic administration of IL-12 resulted in some dose and schedule-dependent serious toxicities, including death in these early clinical trials (13, 21–24). Recognition of the need to avoid the toxicities associated with the systemic bolus injections of IL-12 has spawned the development of a number of different approaches to deliver IL-12 locally into tumors. One paracrine delivery system that has achieved some success is based upon viral vector delivery methodologies. However, these virus based approaches are logistically daunting, expensive and patients’ existing or developing immune responses to potent viral antigens may lead in vivo to the premature clearance of these viral vectors (25, 26). Clearly there is a need to develop and test simpler and more cost effective novel non-viral IL-12 delivery approaches that will reduce or eliminate the cytokine toxicity and enhance the immunostimulatory activity of the cytokine locally, i.e. within the tumor microenvironment. Currently there is considerable interest and activity in the generation of systems that are designed for the non-viral delivery of IL-12 that include both IL-12 protein or IL-12 genes that are delivered by employing alum, liposomes and polymer-based methods. A variety of different routes of injection of these different IL-12 delivery systems have been tested in mice including subcutaneous, intraperitoneal, intratumoral, and peritumoral inoculations, and have shown promising therapeutic effects in murine tumor models (Reviewed in 27). However, little is known about the safety or efficacy of any of these IL-12 non-viral delivery systems in preclinical or clinical settings with cancer patients.
It is now possible to evaluate cytokine delivery systems in vivo with human tumors and human tumor-associated lymphocytes in situ by employing tumor xenograft models in which non-disrupted tumor tissues are engrafted into severely immunodeficient mice (28). In this xenograft model the histological architecture of the original tumor microenvironment is maintained, and the human cells within these xenografts (including inflammatory leukocytes, fibroblasts, endothelial cells as well as tumor cells) remain viable and predictably responsive to selected biologically active molecules. This model has been used successfully by us to evaluate a polymer-based IL-12 protein delivery system (29). A single intratumoral injection of IL-12 loaded biodegradable poly-lactic-acid microspheres led to the reactivation of CD4+ and CD8+ effector memory T cells within the microenvironment of human non-small cell lung tumor xenografts. The T cells proliferated, and secreted IFN-γ, which led to the suppression or complete arrest of the tumor (29, 30).
In the present report we have designed a novel protocol that results in the efficient loading of IL-12 into large multilamellar vesicles (MLV) for the local and sustained delivery of IL-12 into the tumor microenvironment. Liposomes have many recognized advantages for the therapeutic delivery of a wide variety of biologically active molecules. Liposomes are easy to prepare, cost effective, and are loaded without exposure to organic solvents. In addition, liposomes can be lyophilized and stored for prolonged periods. The ability of the local and sustained release of IL-12 from the MLV to reactivate effector memory T cells in human tumors is tested here using our tumor xenograft model. We report here that a single intratumoral injection of IL-12 loaded MLV activates T cells to proliferate, secrete IFN-γ and kill tumor cells in the microenvironment of human lung, breast and ovarian carcinomas.
MATERIALS AND METHODS
Materials
Recombinant human interleukin-12 (IL-12) was obtained as a gift from Wyeth Research Genetics Institute (Andover, MA). Distearoyl phosphatidylcholine (DSPC) and dimyristoyl phosphatidylglycerol (DMPG) were obtained from Avanti Polar Lipids (Alabaster, AL) and were used without further purification. Cholesterol (CHOL) was obtained from Sigma (St. Louis, MO). All other buffer salts and solvents used in the study were obtained from Fisher Scientific (Fairlawn, NJ) and were used without further purification.
Circular Dichroism Studies
CD spectra were acquired on a JASCO-715 spectropolarimeter calibrated with d-10 camphor sulfonic acid. Samples in phosphate buffer (10 mM Na2HPO4 and 145 mM NaCl, pH - 7) containing 50 μg/ml of rhIL-12 in a 1 mm quartz cuvette were scanned from 195 nm to 255nm for secondary structural analysis. To improve signal quality multiple scans were acquired and averaged. CD spectra of the protein were corrected by subtracting the spectrum of the buffer baseline. Thermal denaturation studies were conducted to monitor the unfolding of IL-12 under thermal stress. Samples in MOPS buffered saline (10 mM MOPS and 155 mM NaCl, pH - 7) containing 20μg/ml of IL-12 were used for thermal denaturation studies. MOPS buffer was chosen mainly because of its low temperature coefficient (ΔpKa/°C = −0.006) which ensures minimal pH change at elevated temperatures (31). Hence, observed changes in protein structure in thermal denaturation studies are mainly due to the influence of temperature. The unfolding profile was acquired at a controlled heating rate of 60°C/hr over the temperature range of 20–90°C. The transition temperature (Tm) for the unfolding profile was obtained by fitting the data to a sigmoid function (Equation 1) using WinNonlin (Pharsight Corporation, Mountainview, CA):
| Equation 1 |
where, Yobserved is the ellipticity at 230 nm at a given temperature, Ynative is ellipticity of the native protein, Yunfolded is the ellipticity of the unfolded protein, Tm is the transition temperature, and γ is the fitting function.
Preparation of Liposomal IL-12
A thin film was prepared by mixing appropriate amounts of lipid solutions in chloroform in a kimax tube and evaporating the chloroform in a Rota-evaporator (Buchi R-200, Fischer Scientific, NJ). Large multilamellar liposomes were prepared by rehydrating the lipid film of appropriate molar ratios of DSPC, DMPG and CHOL (DSPC:DMPG:CHOL; 80:20:25) with phosphate buffer containing IL-12 at 45°C. The lipid film was dispersed by gentle swirling and incubated at 45°C for 10 min. This was followed by incubation at 4°C for 15 min. This cycle of alternate incubation at 45°C and 4°C was repeated two additional times to ensure proper hydration of the liposomes. The molar ratio between the lipid and the protein was maintained at 2000:1 (0.534μmoles:0.267 nmoles) for all experiments. The large multilamellar liposome was separated from free protein by centrifugation at 15000×g for 20 minutes at room temperature. The liposome associated protein was obtained as a pellet which was reconstituted in an appropriate volume of buffer. Association efficiency was determined by fluorescence spectroscopy utilizing the intrinsic fluorescence of IL-12 and by an IL-12 bioassay (32). Briefly, the fluorescence intensity (LT) of the liposomal preparation prior to separation of free and associated IL-12 was measured. After centrifugation the fluorescence intensities of the supernatant (LS) and the reconstituted pellet (LP) were obtained. The amount of IL-12 associated with the liposomes was calculated as:
| Equation 2 |
The relationship LT = LS + LP, was found to be true in all the experiments. The liposomal IL-12 was stored at 4°C and utilized within 24 hours for all experiments.
The particle size of the multi-lamellar liposomes was determined by dynamic light scattering using a Nicomp Model CW 380 particle size analyzer (Particle Sizing Systems, Santa Barbara, CA). All measurements were carried out at a temperature of 23°C with viscosity and refraction index set at 0.933 cP and 1.333 respectively. Data were fitted to an intensity weighted-Nicomp (non-Gaussian) distribution.
Fluorescence Spectroscopy and Acrylamide Quenching
Emission spectra of IL-12 and liposomal IL-12 at a protein concentration of 2μg/ml in phosphate buffer were obtained using a PTI fluorometer (QuantaMaster, Photon Technology International, Lawrenceville, NJ). The samples were excited at 280 nm and the emission spectrum was monitored from 300–400 nm. A slit width of 4 nm was used on both the excitation and emission paths. A peak observed at 310 nm was due to Raman scattering. This was confirmed by exciting the sample at 260 nm which resulted in disappearance of the peak.
For fluorescence quenching experiments, the samples were excited at 280 nm and the emission was monitored at 335 nm for both IL-12 and liposomal IL-12. Studies were carried out at 20°C using a slit width of 4 nm on the excitation and emission paths. In order to minimize the contribution due to inner filter effect for samples containing acrylamide, an “I-shaped” cuvette with two different path lengths was used to acquire the fluorescence emission spectra. Quenching was monitored following successive addition of aliquots of 5 M acrylamide stock solution. The data were analyzed according to the classical Stern-Volmer relationship (33).
| Equation 3 |
Plots of FO/F vs. Q were generated, where Fo and F are the unquenched and quenched fluorescence intensities respectively and Q is the concentration of acrylamide (quencher). KD is the Stern Volmer constant. Fluorescence quenching experiments were conducted at protein:lipid ratios of 1:2000, 10,000 and 20,000.
Tumor Samples
Primary non-small cell lung tumor and ovarian serous carcinoma biopsy specimens were received from Roswell Park Cancer Institute Tissue Procurement Facility. Breast carcinomas were received from Sisters of Charity Hospital of Buffalo. Specimens were obtained under sterile conditions and using Institutional Review Board approved protocols. Histological diagnosis for each specimen was obtained anonymously at a later date.
Tumor Implantation
As previously described, necrotic tissue was removed from the tumor specimens and sections (3.0–5.0 mm on a side) were cut resulting in cubic pieces of non-disrupted tumor (3). CB17-scid mice (SCID) were obtained from the breeding colony at the State University of New York (SUNY) at Buffalo. All mice were housed in specific pathogen-free conditions at SUNY at Buffalo. Prior to implantation, mice were anesthetized with avertin (10mg/mouse; Sigma-Aldrich) and treated with one intraperitoneal injection of TMβ-1 (mAB to murine IL-2R β-chain). A small ventral midline incision was made just caudal to the xiphoid process and a subcutaneous pocket was created. One piece of tumor was implanted into the pocket and the incision closed using Nexaband Liquid (Burns Veterinary Supply). All animal experiments were approved by the Institutional Animal Care and Use Committee at SUNY at Buffalo.
Treatment of Mice with Recombinant Human IL-12
Human IL-12 (Wyeth Research) was incorporated into liposomes as indicated above. Mice bearing xenografts were randomly divided into control and treatment groups 7 days after surgical implantation of fresh tumor tissue. All mice were treated with a single injection of (a) IL-12 loaded liposomes (10μg/mouse), (b) empty liposomes, or (c) left untreated. The dose of 10μg/mouse was determined based upon the calculated loading efficiency for each liposome preparation (see the calculation above). The amount of cytokine delivered to each mouse was a total of 10μg. All of the cytokine was associated with the liposomes as the free IL-12 was removed from the liposome preparations prior to injection.
Mouse Serum or Plasma Collection
Blood samples were collected via a tail clipping or retro-orbital sinus bleed. Sera or plasma were collected by microcentrifugation at 14,000 rpm, for 10 minutes and stored at −20°C until use.
Human Interferon Gamma ELISA
A sandwich ELISA for the detection of human IFN-γ in mouse serum was performed as previously described (34). Briefly, 96-well plates were coated using a monoclonal antibody to IFN-γ (M-700-A; Endogen). Mouse sera were added to the plate with a biotinylated monoclonal anti-human IFN-γ (M-701-B; Endogen). Positive binding was detected using streptavidin-conjugated HRP (Sigma-Aldrich), peroxide and 3,3′,5,5′-tetramethylbenzidine (Kirkegaard & Perry Laboratories). Results were measured on a Bio-Tek Instruments automated microplate reader at OD450–540 and analyzed by comparison with a recombinant IFN-γ standard using SigmaPlot software.
Histology and Immunohistochemistry
H&E staining was performed by the SUNY at Buffalo Histology Service Laboratory where fresh surgical sections of the original tumor, xenografts, and mouse tissues were fixed in 10% neutral-buffered formalin and processed for paraffin embedding. Anti-human specific antibodies to the following markers were used for immunohistochemical staining of xenografts and mouse tissues: anti-CD3, anti-CD68, anti-CD45RO, anti-CD138 (DAKO), and anti-mouse CD20 (Abcam). Staining was performed as previously reported (35). The AE1/AE3 cytokeratin (Thermo Shandon) and Ki67 (NeoMarker) staining was performed by the pathology core facility at Roswell Park Cancer Institute. H&E and immunohistochemistry images were taken with a Zeiss Axioimager using Axiovision Rel 4.6 Software.
Results
Formulation, Generation and IL-12 Loading of Large Multilamellar Liposomes
To avoid the toxic effects of IL-12 delivered systemically, liposomes were designed that would deliver biologically active IL-12 in vivo in a local and sustained fashion into the microenvironment of tumors with little or no escape into the circulation. Previous studies have established that the subcutaneous injection of large (>400 nM) multilamellar liposomes remain at the site of administration (36), and provide optimal sustained release (37). Therefore, large multilamellar liposomes were prepared by rehydrating a lipid film of appropriate molar ratios of DSPC:DMPG:CHOL in phosphate buffer as described in the Methods section. The formulation of these liposomes was selected for the following reasons: a) The high phase transition temperature of DSPC, offers rigidity and stability in vivo (38); b) DMPG which carries a negative charge, increases the electrostatic interaction between positive residues of the protein and liposomes; c) CHOL enhances the in vivo stability and rigidity of the liposomes. The alkyl chain mismatch between DSPC and DMPG was intentional as it can lead to interdigitation of the bilayers (39), thereby increasing the encapsulated aqueous volume for higher protein encapsulation.
The spontaneous loading of large complex protein molecules such as IL-12 into the lipid bilayer or lumen of liposomes is hampered by molecular architecture. This obstacle has been successfully overcome previously using a novel approach for the loading of other proteins, such as Factor VIII (40), IFN-γ and erythropoietin (EPO). This technique called “triggered loading”, relies upon mild denaturing conditions to partially and reversibly unfold the protein to expose hydrophobic domains resulting in the intercalation of the protein within or between the lipid bilayers of the liposomes.
To apply the concept of triggered loading to IL-12, it was first necessary to determine the optimal denaturing conditions for achieving liposomal association of IL-12 with minimal or no loss of its biological activity. Thermal stress is commonly used to unfold proteins (40,41). By determining the intrinsic stability of IL-12 to heat, the optimal conditions for the loading of this cytokine could be more rationally determined. Far-UV circular dichroism spectroscopy was used to monitor the effects of increasing temperature upon the secondary structure of IL-12. The far UV-CD spectrum for IL-12 at 20°C shows negative bands at ~208 nm and 215nm, and a positive band below 200nm (Fig. 1A). The spectral features are characteristic for proteins containing both α-helical and β sheet elements consistent with the X-ray crystal structure for IL-12 that shows the p40 subunit of IL-12 exists predominantly as a β-sheet and the p35 subunit as an α-helix (42). The ellipticity values of IL-12 at 230 nm were monitored over a temperature range of 20–90°C. No significant changes were observed in the ellipticity values below 50°C suggesting that IL-12 undergoes minimal secondary structural changes up to 50°C (Fig. 1B). In the same temperature range, fluorescence spectra showed changes in the tertiary structure and exposure of hydrophobic domains (data not shown). These spectral properties observed at 45°C are ideal characteristics for the triggered loading of proteins. At temperatures greater than 50°C the negative ellipticity at 230 nm increased progressively due to the unfolding of the protein and the midpoint of the transition Tm was approximately 67°C (Fig. 1B). Based upon these thermal stability studies, IL-12 loading into the liposomes was conducted at an initial temperature of 45°C where minimal inactivation of the protein due to unfolding was expected. The multilamellar vesicles containing IL-12 were prepared as described in the Methods section. Using both fluorescence spectroscopy and an IL-12 bioactivity assay to monitor IL-12, we determined that at a protein to lipid ratio of 1:2000 a consistent IL-12 loading efficiency of 52±4% (n=12) was achieved (data not shown). Further, dynamic light scattering studies showed that liposomes had a particle size greater than 1 micron, which is suitable for a local and sustained delivery in the tumor microenvironment.
Figure 1. IL-12 liposomal loading is maximal at 45°C with minimal protein denaturation.

A, Far U.V circular dichroism spectroscopy for IL-12 at 20°C. IL-12 consists of both α helical and β helical elements. B, Temperature dependent elipciticy changes of IL-12 monitored at 230nm. To determine the Tm (68°C), the changes in ellipticiy were fit (solid line) using equation 1 described in the Materials and Methods. The secondary structure of IL-12 is stable below temperatures of 50°C
Intercalation of IL-12 into the Lipid Bilayers of the Liposomes
The direct association of IL-12 with the lipid bilayer is essential for the optimal loading and slow release of this cytokine from the liposomes. Two independent studies have provided evidence for this association. In the first study, fluorescence spectroscopy utilizing the intrinsic fluorescence of IL-12 was utilized to determine the nature of the IL-12 association with the liposomes. The emission maximum for IL-12 in the liposome is significantly blue shifted (<325 nm) compared to free IL-12 (Fig. 2A) which is consistent with the a subunit of IL-12 being intercalated into the lipid bilayers of the liposomes. The change in the emission maximum for liposomal IL-12 was also accompanied by a significant increase in the fluorescence intensity (data not shown). This is likely due to the incorporation of fluorescent residues (10 tryptophan residues in the p40 subunit of IL-12) within the hydrophobic environment.
Figure 2. IL-12 is directly intercalated into the lipid bilayer of the liposome.
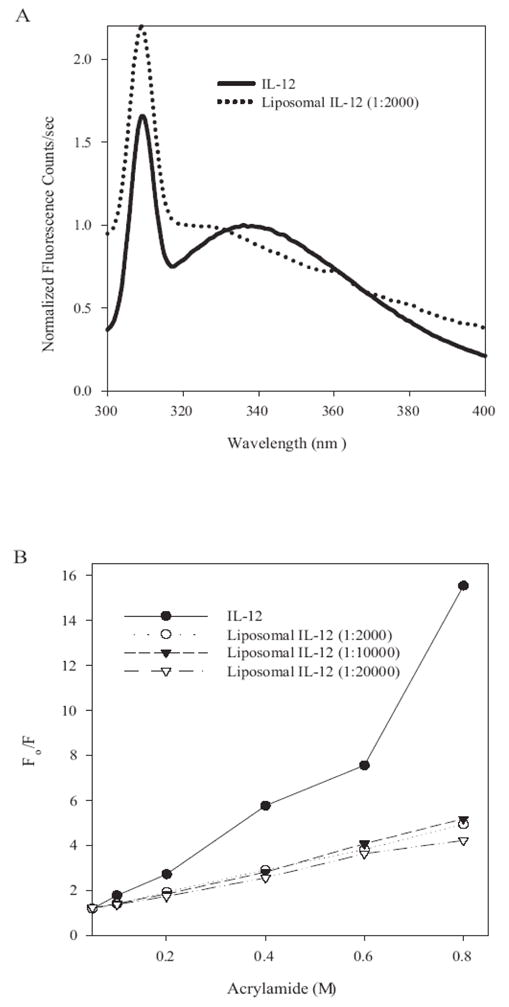
A, Fluorescence emission spectrum of IL-12(solid line) and lipososomal IL-12 (dotted line). The emission maximum for liposomal IL-12 was significantly greater compared to free IL-12. B, Acrylamide quenching of IL-12 and liposomal IL-12. Samples were excited at 280nm and emission monitored at 335nm. The increasing concentrations of acrylamide quenched the fluorescence of free IL-12 to a greater extent than liposomal IL-12.
The above observations suggest that a substantial fraction of the IL-12 associated with the liposomes maybe sequestered away from the aqueous medium with the liposomes shielding the fluorescent tryptophan residues. If this were accurate, the ability of collisional quenchers to quench the fluorescence of liposome associated IL-12 would be hindered relative to free IL-12. To investigate the accessibility of fluorescent tryptophan residues to the aqueous medium the collisional quencher acrylamide was added to the liposomal IL-12 (Figure 2B). As observed, the addition of increasing concentrations of acrylamide quenched the fluorescence of free IL-12 (KD = 18.5) to a greater extent relative to liposomal IL-12 (protein:lipid ratio, 1:2000, KD = 4.8). This demonstrates that the liposomes shield a large fraction of the IL-12 molecule which is inaccessible to quenchers. In addition, concentric bilayer structure of MLVs ensures shielding of large number of IL-12 molecules inside the vesicles.
IL-12 Liposomes Activate Tumor-Associated Lymphocytes in the Microenvironment of Human Tumor Xenografts
To assess the ability of the IL-12 liposomes to deliver biologically active IL-12 into human tumor microenvironments a human tumor xenograft model was utilized. In this model fresh non-disrupted pieces of human tumor tissues are engrafted into SCID mice. The resultant xenografts retain the histological architecture of the original tumor tissues including the tumor resident inflammatory leukocytes that remain predictably responsive to cytokine stimulation (29, 30). Previous studies using this model have established that a local and sustained release of IL-12 from biodegradable microspheres into the microenvironment of human lung tumor xenografts results in the reactivation of resident effector memory T cells (29, 30). The activated CD4+ and CD8+ T cells proliferate and secrete IFN-γ that reaches a peak five days after IL-12 treatment.
This xenograft model is used here to evaluate, for the first time, the ability of the IL-12 loaded liposomes to deliver IL-12 to human tumor xenografts and activate tumor-resident T cells. Xenografts of human breast, ovarian and lung carcinomas were established in SCID mice and one week later were injected intratumorally with the IL-12 loaded liposomes (10μg/mouse), control unloaded liposomes or untreated. The dose of 10μg/mouse was determined based upon the calculated loading efficiency for each liposome preparation (see materials and methods). Five days after treatment, mice were bled and the sera assayed for human IFN-γ. Both the amount of IFN-γ in the sera and the kinetics of its occurrence in xenograft bearing mice following the injection of IL-12 microspheres or IL-12 liposomes into tumor xenografts varies from tumor to tumor. However, we consistently see a significant increase in the IFN-γ levels of IL-12 treated mice compared to the control. This increase is first observed at Day 1 and has usually peaked between day 5 and day 7 post IL-12 treatment (data not shown). Therefore, we typically assay for IFN-γ (as we have done in these experiments) 5 days after treatment.
IFN-γ was undetectable (<1 pg/ml) in all of the control mice while significant levels (~100 – 800 pg/ml) of IFN-γ were present in the sera of mice bearing breast, ovarian and lung tumor xenografts (Fig. 3). We conclude from these studies that IL-12 is delivered to the xenograft microenvironment and that the tumor-associated lymphocytes are activated in response to the biologically active IL-12 released from the liposomes.
Figure 3. Treatment of human tumor xenografts with IL-12 liposomes induces the production of human IFN-γ, which is detected in the sera of SCID mice.

Fresh patient tumor xenografts from breast, ovarian and lung carcinoma were engrafted into SCID mice, as described in the Materials and methods and treated 1 week later with empty or IL-12 loaded liposomes. Mice were bled 5 days post treatment and the IFN-γ levels in the sera monitored by ELISA. Each data plot represents the value of a single mouse. All xenograft bearing mice treated with the IL-12 liposomes had detectable levels of IFN-γ.
Elimination of Tumor and Proliferation of CD45RO+ T cells in Xenografts Treated with IL-12 Liposomes
Xenografts were established in twenty mice by the subcutaneous implantation of human lung tumor tissues. One week after engraftment 10 xenografts were injected with IL-12 loaded liposomes (10μg of IL-12/mouse) and 10 xenografts injected with empty control liposomes. Prior to engraftment tumor cells and inflammatory leukocytes were found to be present throughout the biopsy tissue obtained from a patient with an invasive moderately differentiated squamous cell carcinoma (Fig. 4A). While tumor growth and survival are valid endpoints with mouse tumor models we are unable to correlate human tumor xenograft size and growth to tumor burden. The human tumor xenografts themselves contain various cells of different amounts. We have found that human tumor xenografts of the same approximate volume may contain differences with regard to the number of tumor cells, inflammatory cells, fibroblasts and necrotic tissue.. Therefore, we have determined that the most reliable way to monitor tumor progression of human tumor xenografts is by histology and immunohistochemistry. Three weeks after engraftment and two weeks after treatment with control empty liposome, healthy looking cytokeratin positive, and Ki67 positive proliferating tumor cells are present in the tumor xenografts (Fig. 4C1, D1 respectively). In sharp contrast tumor cells are either completely absent or significantly decreased in all of the IL-12 liposome treated xenografts (Fig. 4B2, C2 and D2). Tumor cells are replaced by a diffuse and florid accumulation of lymphocytes some of which are Ki67 positive proliferating cells in the IL-12 treated xenografts (Fig. 4D2). These data suggest that the local and sustained release of IL-12 from the liposomes is responsible for at least a partial eradication of the xenografts (as defined by an absence of cytokertain staining).
Figure 4. H&E and immunohistochemistry sections of a non-small lung tumor and corresponding xenografts in SCID mice 2 weeks post treatment.
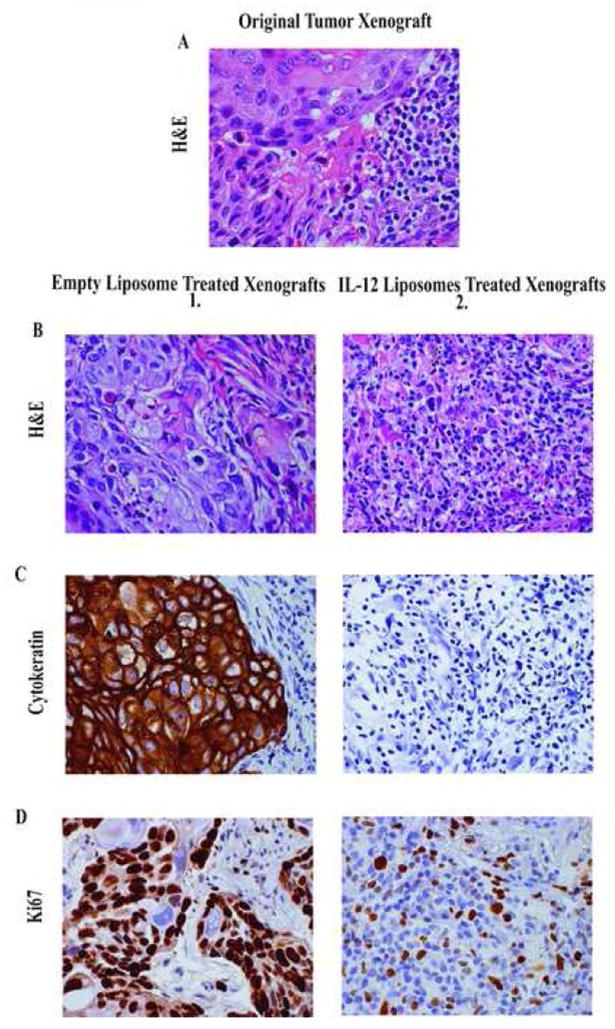
A, H&E stained formalin fixed specimens of the original tumor prior to engraftment. B, Sections of the corresponding empty liposome treated (1) and liposomal IL-12 treated xenografts (2) were also stained with H&E. C, The empty liposome treated xenografts (1) demonstrated retention of the cytokertain staining which was absent in the IL-12 treated xenografts (2). D, The tumor cells in the empty liposome treated xenografts (1) were positive for Ki67 staining. The cells staining positively for Ki67 in the IL-12 liposome treated xenografts (2) are proliferating lymphocytes. All images were taken at 400X magnification.
To identify the populations of human inflammatory cells responding to IL-12, the control treated and Il-12 treated xenografts were subjected to various immunhistochemical stains. The lymphocytes in the cytokine treated xenografts are largely CD3+, CD45RO+ T cells (Fig. 5B2 and 5C2). In contrast to the diffuse accumulation of T cells throughout the IL-12 treated xenograft, fewer foci of CD20+ B cells (Fig. 5D2), CD138+ plasma cells (Fig. 5E2) and CD68+ macrophages (Fig. 5F2) are present in the cytokine treated xenograft. Very few T cells (Fig 5B1), B cells (Fig 5D1), plasma cells (Fig 5E1) or macrophages (Fig 5F1) are observed in any of the xenografts treated with control liposomes. The few CD3+ T cells that are present in the control xenografts are located at tumor margins and are not seen infiltrating into the tumor parenchyma (Fig. 5B1). Note the antibody to CD138 (a molecule called syndecan 1) that stains the plasma cells in the xenograft treated with IL-12 liposomes (Fig. 5E2) also stains tumor cells in the xenograft treated with the control liposomes (Fig. 5E1). Both of these cell types are known to express syndecan 1.
Figure 5. Immunohistochemical staining of tissues reveals that treatment of lung tumor xenografts with IL-12 liposomes promotes the expansion and survival of leukocytes within the tumor microenvironment.
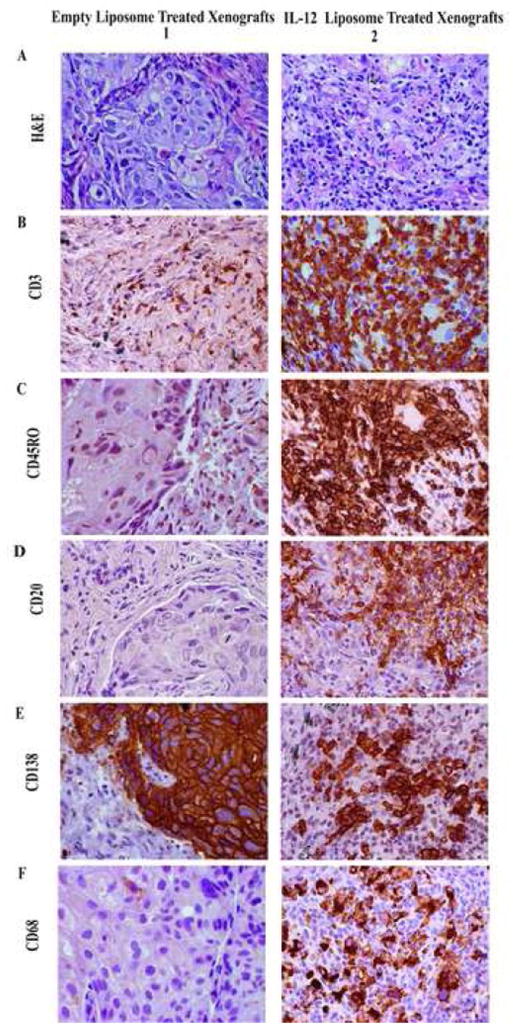
Mice were implanted with a human primary lung tumor, and at 2 weeks post IL-12 or empty liposome treatment mice were sacrificed and the xenografts removed. A. H&E stain of xenografts treated with empty liposomes (1) and IL-12 liposome treated xenografts. B-C, Immunohistochemical staining revealed the presence of CD3+ CD45RO+ cells in the IL-12 liposome treated xenografts (B2-C2) but negligible staining in the control treated xenografts (B1-C1). D-F, In contrast to the diffuse accumulation of T cells throughout the IL-12 treated xenograft, fewer foci of CD20+ B cells (D2), CD138+ plasma cells (E2) and CD68+ macrophages (Fig F2) are present in the cytokine treated xenografts. Very few B cells (D1), plasma cells (E1) or macrophages (F1) are observed in any of the xenografts treated with control liposomes. All images were taken at 400X magnification.
Very similar results, i.e. IL-12 dependent eradication of tumor cells, and florid accumulations of CD3+ CD45RO+ T cells with fewer CD20+ B cells, CD138+ plasma cells and CD68+ macrophages were observed in the IL-12 liposome treated xenografts of human breast carcinomas two weeks after treatment (data not shown).
Most IL-12 Delivered by Liposomes to the Tumor Xenograft Does Not Enter the Circulation
To determine whether the IL-12 delivered to tumor xenografts following a single injection of IL-12 loaded liposomes leaves the xenografts and enters the blood, human lung tumor xenografts were established in 20 SCID mice by the implantation of tumor biopsy tissue from an adenocarcinoma of the lung. One week after tumor tissue engraftment, 15 mice were injected intratumorally with IL-12 liposomes (10 μg IL-12/mouse), and 5 mice were injected intratumorally with control (no IL-12) liposomes. Mice were bled periodically over a period of 5 days and the serum assayed for human IL-12. Very little of the total 10 μg of IL-12 injected into the tumor was detected in the circulation over the 5 day period of observation. Serum levels of IL-12, (4 – 8 h after the IL-12 liposome injection) reached a high of 18–19 ng/ml (Fig. 6). At 12h the average serum IL-12 level dropped to 16 ng/ml, and by day 3 and 5, IL-12 levels were very low or undetectable (Fig. 6). No human IL-12 was detected at any time point in the sera of xenograft bearing mice injected with control (no IL-12) liposomes (Fig. 6). We conclude from these results that the IL-12 multilamellar liposomes deliver the cytokine to the tumor xenografts in mostly a local and sustained fashion. These results suggest that the IL-12 loaded liposomes reported here would provide a safe way to deliver IL-12 into solid tumors and a therapeutically effective way to reactivate resident T cells to kill tumor cells in situ.
Figure 6. The majority of the IL-12 released from the liposomes in situ does not reach the circulation.

Twenty xenografts were established by the implantation of biopsy tissues from a patient with an adenocarcinoma of the lung. Fifteen mice were injected with the IL-12 liposomes (10μg/mouse) and the remaining mice with empty control liposomes. Mice were bled periodically starting at 4 hours post treatment and continued over a period of 5 days. The quantity of IL-12 protein in the serum was determined by ELISA. There were very low or undetectable levels of IL-12 in the sera following the treatment of xenografts with IL-12 liposomes.
DISCUSSION
The ability to load IL-12 efficiently without loss of bioactivity, the prolonged shelf life of the IL-12 loaded liposomes and the demonstrated safety and efficacy of liposomes for the delivery of other biologically active factors in humans provides a rationale for our development and testing of an effective liposome delivery system for the local and sustained release of IL-12 into tumor microenvironments.
We demonstrate here that with mild and reversible denaturation of IL-12 it is possible to load this heterodimeric complex protein into large multilamellar liposomes and have it released in vivo with its biological activity preserved. Our initial objectives were to enhance the amount of protein loaded, and to generate liposomes that would release the loaded protein in a slow and sustained fashion by intercalating a portion of the IL-12 molecule within and between the lipid bilayers of the liposomes. These objectives were achieved and confirmed by our results that showed differences in the fluorescence emission patterns and differential effects of a collisional fluorescence quencher with free IL-12 and IL-12 within liposomes. Our results are consistent with the possibility that the p40 subunit of IL-12 is intercalated into the lipid bilayers via its 10 fluorescent tryptophan residues. The possibility exists that the mild heat denaturation of IL-12 also exposes other hydrophobic residues contributing further to protein lipid hydrophobic associations.
Our data showing that the IL-12 liposomes, but not control empty liposomes, induce tumor resident lymphocytes to secrete IFN-γ, proliferate, and eradicate tumor cells in xenografts confirm our previous studies showing that IL-12 delivered by biodegradable microspheres reverses the hyporesponsiveness of tumor-associated T-cells and initiates an IFN-γ-dependent, T-cell-mediated in situ killing of tumor cells (29, 30). While we have not conducted extensive comparsions of the efficacy of biodegradable microspheres and the multilamellar liposomes for the local delivery of IL-12 in tumors, our preliminary results suggest that liposome delivery is as therapeutically efficacious, if not more, than microspheres, for activating tumor- associated T cells in situ and for arresting or eradicating tumors in xenografts established by the implantation of fresh, non disrupted human tumor tissues into SCID mice. We implanted a serous ovarian carcinoma into SCID mice and the mice were either treated with IL-12 liposomes, IL-12 microspheres or the control mice were left untreated. All of the mice treated with either the IL-12 liposomes or IL-12 microspheres responded by producing human IFN-γ at Day 5 post treatment, whereas the control mice had no detectable levels of IFN-γ (data not shown). When the tumor xenografts were removed 14 days post treatment, there were significant morphological differences in the xenografts (as determined blindly by a pathologist at Roswell Park Cancer Institute). As compared to the microsphere treated mice, the xenografts from the IL-12 liposome mice had a greater number of inflammatory cells (including lymphocytes and plasma cells), less tumor burden and extensive areas of fibrosis and sclerosis. A similar finding was observed with a breast carcinoma xenograft treated with either IL-12 loaded microspheres or IL12 loaded liposomes.
Following the intratumoral injection of the IL-12 liposomes very little of the IL-12 enters the peripheral blood circulation. These results establish that our liposomes release IL-12 primarily in a local and sustained manner which is critical for the safety and therapeutic efficacy of their ultimate use in cancer patients. Our results also confirm the notion that the IL-12 released from the liposomes in vivo has retained its biological activity.
It has become increasingly apparent that the clinical efficacy of cancer immunotherapeutic protocols will be more dependent upon enhancing the tumoricidal activity of immune effector cells within the tumor microenvironment rather than upon the number of circulating tumor-specific lymphocytes (43–46). Therefore, rather than continuing to rely solely upon designing strategies to induce more potent systemic anti-tumor responses or only upon improving adoptive cellular therapies by increasing the number of tumor-specific cells injected into patients, it is important to begin to focus more upon how the functions of immunocompetent cells become arrested when they enter and are retained within the tumor microenvironment and determine ways in which this arrest can be reversed (47).
T cells derived from both malignant and non-malignant chronic inflammatory microenvironments (including chronic viral infections) have been shown to be hyporesponsive to activation (1, 6, 7, 48, 49). While the events that orchestrate this T cell hyporesponsiveness have not been completely defined, and may include multiple contributing factors, it has been established that a membrane bound form of TGF-β1 contributes to a TCR signaling arrest and that IL-12 reverses this arrest (7, 8).
Tumor immunotherapeutic strategies are envisioned with our IL-12 liposomes in which a local intratumoral injection into one or more accessible primary tumor sites would result in (i) the reactivation of quiescent tumor-associated effector memory T cells with the orchestration of an IFN-γ dependent tumor killing in situ, (ii) the release of tumor antigens into the circulation, (iii) the induction of a systemic T cell mediated anti-tumor response, and (iv) the targeting and T cell killing of tumors at inaccessible and untreated sites throughout the body. These expectations are supported by animal tumor studies in which IL-12 is injected intratumorally into a single tumor site resulting in a systemic anti-tumor immunity with the eradication of distant untreated metastatic tumor nodules (50).
Our data have established that the local and sustained release of IL-12 from liposomes reactivates resident lymphocytes to secrete IFN-γ in three different types of human solid tumors, i.e. (a) non-small cell lung carcinomas (including both adenocarcinomas and squamous cell carcinomas), (b) breast carcinomas, and (c) ovarian carcinomas. These results suggest that intratumoral liposome IL-12 delivery is therapeutically effective with several different epithelial tumor types, and may well work in several other tumor types in which T cells are found to be present, but hyporesponsive.
Acknowledgments
Grant Support: This work was supported in part by U.S public Health Service Grants, R-01-CA10897O, R-01-CA131407 (R.B. Bankert), HL-70227 (S.V. Balu-Iyer), the John R. Oishei Foundation (R.B. Bankert) and National Institutes of Health Research Training Grant T32 AI1007614-07 (M.R. Simpson-Abelson)
The authors would like to thank the pharmaceutical instrumentation facility at SUNY Buffalo for the use of the circular dichroism and fluorescence spectrometer. We would also like to thank the Tissue Procurement Facility at Roswell Park Cancer Institute for distribution of the human lung and ovarian tumor tissues and Dr. Ronald L. Bauer at Sisters of Charity Hospital of Buffalo for providing the breast tumor patient tumor sample. We would also like to acknowledge Dr. Thomas F. Conway for performing the IL-12 bioactivity assay.
Footnotes
Disclosure of Potential Conflicts of Interest
Dr. Richard B Bankert is the president of Therapyx, Inc. No potential conflicts of interest were disclosed.
M.R. Simpson-Abelson and V.S. Purohit are co-first authors for this manuscript. R.B Bankert and S.V. Balu-Iyer are co-corresponding authors for this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Agrawal S, Marquet J, Delfau-Larue MH, et al. CD3 hyporesponsiveness and in vitro apoptosis are features of T cells from both malignant and nonmalignant secondary lymphoid organs. J Clin Invest. 1998;102:1715–1723. doi: 10.1172/JCI3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radoja S, Saio M, Frey AB. CD8+ tumor-infiltrating lymphocytes are primed for Fas-mediated activation-induced cell death but are not apoptotic in situ. J Immunol. 2001;166:6074–6083. doi: 10.4049/jimmunol.166.10.6074. [DOI] [PubMed] [Google Scholar]
- 3.Broderick L, Bankert RB. Memory T Cells in Human Tumor and Chronic Inflammatory Microenvironments: Sleeping Beauties Re-awakened by Cytokine Kiss (Review) Immunol Invest. 2006;35:1–18. doi: 10.1080/08820130600755066. [DOI] [PubMed] [Google Scholar]
- 4.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nature Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 5.Chiou SH, Sheu B-C, Chang W-C, Huang S-C, Hong-Nerng H. Current concepts of tumor-infiltrating lymphocytes in human malignancies. J Reprod Immunol. 2005;67:35–50. doi: 10.1016/j.jri.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Uzzo RG, Rayman P, Kolenko V, et al. Renal cell carcinoma-derived gangliosides suppress nuclear factor-kappaB activation in T cells. J Clin Invest. 1999;104:769–776. doi: 10.1172/JCI6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broderick L, Brooks SP, Takita H, Baer AN, Bernstein JM, Bankert RB. IL-12 reverses anergy to T cell receptor triggering in human lung tumor-associated memory T cells. Clin Immunol. 2006;118:159–69. doi: 10.1016/j.clim.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Broderick L, Bankert RB. Membrane-associated TGF-β1 inhibits human memory T cell signaling in malignant and nonmalignant inflammatory microenvironments. J Immunol. 2006;177:3082–88. doi: 10.4049/jimmunol.177.5.3082. [DOI] [PubMed] [Google Scholar]
- 9.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt CS, Mescher MF. Peptide antigen priming of naïve, but not memory, CD8 T cells require a third signal that can be provided by IL-12. J Immunol. 2002;168:5521–29. doi: 10.4049/jimmunol.168.11.5521. [DOI] [PubMed] [Google Scholar]
- 11.Kieper WC, Prlic M, Schmidt CS, Mescher MF, Jameson SC. IL-12 enhances CD8 T cell homeostatic expansion. J Immunol. 2001;166:5515–21. doi: 10.4049/jimmunol.166.9.5515. [DOI] [PubMed] [Google Scholar]
- 12.Curtsinger JM, Schmidt CD, Mondino A, et al. Inflammatory cytokines provide a third signal for activation of naïve CD4+ and CD8+ cells. J Immunol. 1999;162:3256–62. [PubMed] [Google Scholar]
- 13.Leonard JP, Sherman ML, Fisher GL, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–48. [PubMed] [Google Scholar]
- 14.Xu D, Gu P, Pan PY, Li Q, Sato AI, Chen SH. NK and CD8+ T cell-mediated eradication of poorly immunogenic B16-F10 melanoma by the combined action of IL-12 gene therapy and 4-1BB costimulation. Int J Cancer. 2004;109:499–506. doi: 10.1002/ijc.11696. [DOI] [PubMed] [Google Scholar]
- 15.Kawamura T, Takeda K, Mendiratta SK, et al. Critical role of NK1+ T cells in IL-12-induced immune responses in vivo. J Immunol. 1998;160:16–9. [PubMed] [Google Scholar]
- 16.Estaquier J, Idziorek T, Zou W, et al. T helper type 1/T helper type 2 cytokines and T cell death: preventive effect of interleukin 12 on activation-induced and CD95 (FAS/APO-1)-mediated apoptosis of CD4+ T cells from human immunodeficiency virus-infected persons. J Exp Med. 1995;182:1759–67. doi: 10.1084/jem.182.6.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunda MJ. Interleukin-12. J Leukoc Biol. 1994;55:280–8. doi: 10.1002/jlb.55.2.280. [DOI] [PubMed] [Google Scholar]
- 18.Noguchi Y, Richards EC, Chen YT, Old LJ. Influence of interleukin 12 on p53 peptide vaccination against established Meth A sarcoma. Proc Natl Acad Sci USA. 1995;92:2219–23. doi: 10.1073/pnas.92.6.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou JP, Yamamoto N, Fujii T, et al. Systemic administration of rIL-12 induces complete tumor regression and protective immunity: response is correlated with a striking reversal of suppressed IFN-gamma production by anti-tumor T cells. Int Immunol. 1995;7:1135–45. doi: 10.1093/intimm/7.7.1135. [DOI] [PubMed] [Google Scholar]
- 20.Brunda MJ, Gately MK. Interleukin-12: potential role in cancer therapy. Important Adv Oncol. 1995:3–18. [PubMed] [Google Scholar]
- 21.Car BD, Eng VM, Lipman JM, Anderson TD. The toxicology of interleukin-12: a review. Toxicol Pathol. 1999;27:58–63. doi: 10.1177/019262339902700112. [DOI] [PubMed] [Google Scholar]
- 22.Cohen J. IL-12 deaths: explanation and a puzzle. Science. 1995;270:908. doi: 10.1126/science.270.5238.908a. [DOI] [PubMed] [Google Scholar]
- 23.Orange JS, Salazar-Mather TP, Opal SM, et al. Mechanism of interleukin 12-mediated toxicities during experimental viral infections: role of tumor necrosis factor and glucocorticoids. J Exp Med. 1995;181:901–14. doi: 10.1084/jem.181.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atkins MB, Robertson MJ, Gordon M, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. 1997;3:409–17. [PubMed] [Google Scholar]
- 25.Dranoff G. The use of gene transfer in cancer immunotherapy. Forum (Genova) 1998;8:357–64. [PubMed] [Google Scholar]
- 26.Colombo MP, Vagliani M, Spreafico F, et al. Amount of interleukin 12 available at the tumor site is critical for tumor regression. Cancer Res. 1996;56:2531–4. [PubMed] [Google Scholar]
- 27.Salem ML, Gillanders WE, Kadima AN, et al. Novel nonviral delivery approaches for interleukin-12 protein and gene systems: curbing toxicity and enhancing adjuvant activity. J Interferon & Cytokine Res. 2006;26:593–608. doi: 10.1089/jir.2006.26.593. [DOI] [PubMed] [Google Scholar]
- 28.Bankert RB, Egilmez NK, Hess SD. Human-SCID mouse chimeric models for the evaluation of anti-cancer therapies. TRENDS in Immunol. 2001;22:386–93. doi: 10.1016/s1471-4906(01)01943-3. [DOI] [PubMed] [Google Scholar]
- 29.Hess SD, Egilmez NK, Bailey N, et al. Human CD4+ T cells present within the microenvironment of human lung tumors are mobilized by the local and sustained release of IL-12 to kill tumors in situ by indirect effects of IFN-γ. J Immunol. 2003;170:400–12. doi: 10.4049/jimmunol.170.1.400. [DOI] [PubMed] [Google Scholar]
- 30.Broderick L, Yokota SJ, Reineke J, et al. Human CD4+ effector memory T cells persisting in the microenvironment of lung cancer xenografts are activated by local delivery of IL-12 to proliferate, produce IFN-γ, and eradicate tumor cells. J Immunol. 2005;174:898–906. doi: 10.4049/jimmunol.174.2.898. [DOI] [PubMed] [Google Scholar]
- 31.Derrick TS, Kashi RS, Durrani M, Jhingan A, Middaugh CR. Effect of metal cations on the conformation and inactivation of recombinant human factor VIII. J Pharm Sci. 2004;93:2549–57. doi: 10.1002/jps.20167. [DOI] [PubMed] [Google Scholar]
- 32.Stern AS, Podlaski FJ, Hulmes JD, et al. Purification to homogeneity and partial characterization of cytotoxic lymphocyte maturation factor from human B-lymphoblastoid cells. Proc Natl, Acad, Sci. 1990;87:6808–6812. doi: 10.1073/pnas.87.17.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. New York: Kluwer Academic/Plenum Publishers; 1999. [Google Scholar]
- 34.Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ, Jr, Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol. 2007;178:5552–62. doi: 10.4049/jimmunol.178.9.5552. [DOI] [PubMed] [Google Scholar]
- 35.Simpson-Abelson MR, Sonnenberg GF, Takita H, et al. Long-term engraftment and expansion of tumor-derived memory T cells following the implantation of non-disrupted pieces of human lung tumor into NOD-scid IL2Rγnull mice. J Immunol. 2008;180:7009–18. doi: 10.4049/jimmunol.180.10.7009. [DOI] [PubMed] [Google Scholar]
- 36.Oussoren C, Zuidema J, Crommelin DJ, Storm G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid composition and lipid dose. Biochim Biophys Acta. 1997;1328:261–72. doi: 10.1016/s0005-2736(97)00122-3. [DOI] [PubMed] [Google Scholar]
- 37.Van Slooten ML, Boerman O, Romoren K, Kedar E, Crommelin DJ, Storm G. Liposomes as sustained release system for human interferon-gamma: biopharmaceutical aspects. Biochim Biophys Acta. 2001;1530:134–45. doi: 10.1016/s1388-1981(00)00174-8. [DOI] [PubMed] [Google Scholar]
- 38.Allen TM, Hansen C, Rutledge J. Liposomes with prolonged circulation times: factors affecting uptake by reticuloendothelial and other tissues. Biochim Biophys Acta. 1989;981:27–35. doi: 10.1016/0005-2736(89)90078-3. [DOI] [PubMed] [Google Scholar]
- 39.Ahl PL, Chen L, Perkins WR, et al. Interdigitation-fusion: a new method for producing lipid vesicles of high internal volume. Biochim Biophys Acta. 1994;1195:237–44. doi: 10.1016/0005-2736(94)90262-3. [DOI] [PubMed] [Google Scholar]
- 40.Balasubramanian SV, Ramani K, Straubinger RM. Patent WO/2006/002195. Method of complexing a protein by the use of a dispersed system and proteins thereof.
- 41.Balasubramanian SV, Bruenn J, Straubinger RM. Liposomes as formulation excipients for protein pharmaceuticals: a model protein study. Pharm Res. 2000;17:344–50. doi: 10.1023/a:1007561308498. [DOI] [PubMed] [Google Scholar]
- 42.Yoon C, Johnston SC, Tang J, Stahl M, Tobin JF, Somers WS. Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. Embo J. 2000;19:3530–41. doi: 10.1093/emboj/19.14.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 44.Appay V, Jandus C, Voelter V, et al. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J Immunol. 2006;177:1670–8. doi: 10.4049/jimmunol.177.3.1670. [DOI] [PubMed] [Google Scholar]
- 45.Rosenberg SA, Sherry RM, Morton KE, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 46.Offringa R. Cancer. Cancer immunotherapy is more than a numbers game. Science. 2006;314:68–9. doi: 10.1126/science.1133893. [DOI] [PubMed] [Google Scholar]
- 47.Simpson-Abelson M, Bankert RB. Targeting the TCR signaling checkpoint: a therapeutic strategy to reactivate memory T cells in the tumor microenvironment. Expert Opin Ther Targets. 2008;12:477–90. doi: 10.1517/14728222.12.4.477. [DOI] [PubMed] [Google Scholar]
- 48.Zajac AJ, Blattman JN, Murali-Krishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–13. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gallimore A, Glithero A, Godkin A, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–93. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hill HC, Conway TF, Jr, Sabel MS, et al. Cancer immunotherapy with interleukin-12 and granulocyte-macrophage colony-stimulating factor-encapsulated microspheres: Coinduction of innate and adaptive antitumor immunity and cure of disseminated disease. Cancer Res. 2002;62:7254–63. [PubMed] [Google Scholar]