

Abstract
Immobilized single horseradish peroxidase enzymes were observed by confocal fluorescence spectroscopy during catalysis of the oxidation reaction of the nonfluorescent dihydrorhodamine 6G substrate into the highly fluorescent product rhodamine 6G. By extracting only the non-Markovian behavior of the spectroscopic two-state process of enzyme-product complex formation and release, memory landscapes were generated for single-enzyme molecules. The memory landscapes can be used to discriminate between different origins of stretched exponential kinetics that are found in the first-order correlation analysis. Memory landscapes of single-enzyme data shows oscillations that are expected in a single-enzyme system that possesses a set of transient states. Alternative origins of the oscillations may not, however, be ruled out. The data and analysis indicate that substrate interaction with the enzyme selects a set of conformational substates for which the enzyme is active.
Understanding the dynamics of complex biological molecules depends on continuously improved experiments, especially those performed on the single molecule level. Data from dynamic processes of individual biological molecules such as proteins or DNA are becoming assessable. Predictions (1–3) about possible origins and behavior of single molecules are now judged for processes like enzyme catalysis (4, 5), folding-unfolding (ref. 6 and references therein), and conformational or spectral fluctuations (7–10). Many more advances in the field of experimental single-molecule analysis in condensed matter environments can be found in a recent review article by W. E. Mörner and M. Orrit (11).
Non-ergodic properties of a process of a molecule (12, 13) as well as non-exponential state transition probabilities for a single process (5) are both indicators of a complex behavior. Enlarged dynamic models on the single molecule level are then required as compared with models derived from standard chemical kinetics of an ensemble of molecules.
Catalysis of the oxidation of the dihydrorhodamine 6G into rhodamine 6G by the enzyme horseradish peroxidase on the single enzyme level has recently been observed at room temperature (5). Horseradish peroxidase is a 44-kDa heme protein (14, 15) and is an effective catalyst of the decomposition of hydrogen peroxide (H2O2) in the presence of hydrogen donors (14, 15). The reaction is monitored by existing experimental methods (8) based on confocal fluorescence spectroscopy (16, 17). We used the nonfluorescent substrate dihydrorhodamine 6G, which after oxidation yields the highly fluorescing rhodamine 6G fluorophore. Hence, direct observation of successive single substrate turnovers into product is made possible by means of fluorescence microscopy if a single enzyme molecule is observed. The enzyme, the substrate, and the enzyme-substrate complex are nonfluorescent. However, the enzyme-product complex (EP) (18) is fluorescent and is formed as the result of the substrate being oxidized while still bound to the enzyme; the enzyme-substrate complex transforms into a fluorescent EP. For each catalytic cycle, a new substrate is bound to the enzyme and is turned over into a product (EP), after which the product dissociates from the enzyme. Then, another substrate attaches to the enzyme, is turned over into a product (EP), and so on. The average binding time of the product (lifetime of EP) was determined in ref. 5 to be approximately 50 ms. The observable state from a spectroscopic viewpoint is the EP. All other states of the enzyme are nonfluorescent. It is assumed that the spectroscopic properties of the EP are unaffected by the oxidation state of the enzyme (e.g., 4+ or 5+ oxidized state) because we directly monitor the product and not the enzyme (5). Hence, from a spectroscopic viewpoint, the observable is a reversible two-state process with one fluorescent state (EP) and one nonfluorescent state (representing all other states of the enzyme).
The notion of a memory in a dynamic system refers to the temporal dependency of a future prediction on the history. The Markov assumption means that a process forgets its past and that the best possible future prediction is made only from the most recent information, regardless of information stemming from earlier times. Deviations from Markovian behavior for a given process can have many origins. Here, non-Markovian emission fluctuations of a single enzyme performing catalysis are studied with aim to get more kinetic detail of the enzyme. In ref. 5, it was concluded that the emission fluctuations emerging from product formation and release on a single enzyme molecule was widely distributed. The enzyme is fluctuating in its activity over time scales ranging from milliseconds to seconds. Other experiments on single protein dynamics report similar kinetic characteristics with largely distributed kinetics. For example, the conformational fluctuations of the GCN4 peptide (19) and of the staphylococcal nuclease (20) show a broad range of transition rates. Also, experiments done in the bulk on myoglobin association and dissociation to carbon monoxide (CO) show conformational reconfigurations of myoglobin that obey a stretched exponential law with β = 0.1 at room temperature (21).
The observed distributed kinetics may have different origins (Fig. 1) that are not possible to resolve by the use of the kind of one-time correlation analysis performed in (5). In this paper, we therefore introduce higher order correlation analysis that gives information about the non-Markovian properties of the emission fluctuations as a tool to differentiate between situations like those in Fig. 1.
Figure 1.

Descriptions of two possible origins of the stretched exponential kinetics in the one-time autocorrelation of the fluorescence intensity as observed in ref. 5. The schemes in A and B are two special cases of the Grand scheme in ref. 5. In A, there are a number of intermediate states (Ek, k = 1,2,3,… ,n) that the enzyme may traverse before a new product is formed (EP). In B, transitions to the EP state are exponential for each substrate turnover; however, each turnover may occur via any of the n channels, each with a different state transition probability.
Materials and Methods
Measurement of a Single Enzyme.
For experimental detail, the reader is referred to ref. 5; however, for clarity, a brief overview is given. The biotinylated enzyme is bound to a streptavidinized glass coverslip surface. The substrate solution is applied as a “hanging droplet.” To find a single-enzyme molecule, a scanning procedure is conducted in which the open volume element from where the fluorescence is detected is moved in a direction parallel to the coverslip surface until a single enzyme is detected (Fig. 2A). The signature of a single enzyme molecule is that of fluctuations in the fluorescence intensity traces combined with a clear signal in the autocorrelation function of the intensity fluctuations (Fig. 2 B and C). When no enzyme is present the fluorescence intensity trace show only background signal, and the fluorescence intensity autocorrelation function is flat (Fig. 2 D and E). Another control experiment shows a blank in the absence of H2O2, but with all other ingredients present (not shown). In ref. 5, we hence concluded: (i) The control experiments make it possible to conclude that the fluctuations in the presence of enzyme must originate from the enzyme interaction with the substrate; (ii) the finding that the average fluorescence intensity is continuously increasing inside the sample solution when enzyme is bound to the glass surface, but not otherwise (when no enzyme is present), indicates that the surface bound enzymes are active; (iii) additional control assays done in the bulk indicate that the average substrate turnover rate is 34 s−1, which is roughly in line with the average of the observed substrate turnover rates, and product dissociation rates from single enzyme molecules.
Figure 2.

(A) A surface scan provides a fluorescence image of single enzyme molecules. (B and C) The signature of a single enzyme performing catalysis is that of fluctuations in the intensity trace (B) combined with a clear signal in the autocorrelation function (C). (D and E) A control experiment in which no enzyme is present (but with everything else held constant) shows only background signal in the intensity trace (D) and no autocorrelation signal (E).
The above facts combined make us conclude that we observe single enzymes catalyzing the substrate formation into product (5). In the present paper we take the analysis a step further, investigating the origin of the distributed kinetics. The experiments were carried out at substrate (dihydrorhodamine 6G) concentration of 130 nM, H2O2 concentration of 120 mM, in 100 mM potassium phosphate buffer at pH 7.0.
Memory Landscapes.
With aim to expand our knowledge about the kinetic detail of the enzyme, we present a data evaluation approach based on the calculation of non-Markovian properties of the spectroscopic two-state process. An investigation regarding non-Markovian behavior has already been done by Lu et al. for the case of the cholesterol oxidase enzyme for consecutive enzyme state transitions between its oxidized and reduced states as a reflection of product formation (4). In the case of a single cholesterol enzyme molecule, the enzyme itself emits fluorescence with different magnitudes, depending on whether the enzyme is in its oxidized or reduced state. In the present paper, we monitor the catalytic cycle by direct observation of the enzyme-product complex. The observed signal is different between the present case and the case of a single cholesterol enzyme molecule (4); however, they are both similar in that they report on the kinetics of the enzymatic cycle.
We assess the reaction dynamics by analyzing the time series of the recorded fluorescence from a single-enzyme molecule. Higher order statistics (22) such as higher order correlation functions (23) may be used to unveil the origin of non-Markovian behavior. Single-molecule higher order statistical analysis is introduced as a simple and useful expression sensitive to memory patterns in the experimental data. It is based on divergence from the Markov assumption (24). The Markov assumption states that optimal prediction of the probability for a value of a process at a certain time is achieved by considering only the latest information available. This means that information from any time earlier but the latest information time is superfluous. Define {Xt} as a stochastic process. {Xt} is binary in the sense that its event room W contains only two elements: W = {0,1}. {Xt} is stationary in the sense that its expectation value E{Xt} = m, where 0 < m < 1 is a constant (not time dependent). If dt is considered a very small time interval, the two possible events “Xt = 0” and “Xt = 1” represent the event that zero or one photon was registered in the time interval [t,t +dt], respectively. The Markov assumption can then formally be written
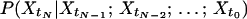 |
1 |
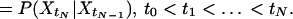 |
If Eq. 1 is valid, we also have the following weaker but still valid statement:
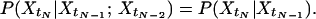 |
2 |
However, if Eq. 2 is not valid for all values of tN−2 < tN−1 < tN, neither is Eq. 1. Hence, if Eq. 2 is not true, the Markov assumption Eq. 1 must also be violated.
The non-Markovian function (NMF) for the observed process {Xt} is given by
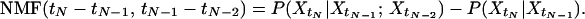 |
3 |
Because {Xt} is a stationary process, NMF has only two arguments (instead of three in the more general case if {Xt} is not stationary) that equal the times differences between the three observation times.
Consider the normalized first and second order autocorrelation of {Xt}. Let E(.) denote the expectation value of a random variable. Set tN − tN−1 = τ1 and tN−2 − tN−1 = τ2. The time τ2 is, hence, the time in addition to the time τ1 from the reference time tN, which we set arbitrarily to zero because the process is stationary. We get, by definition,
 |
4 |
 |
and
 |
 |
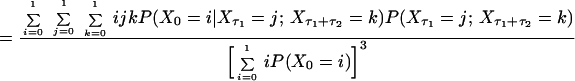 |
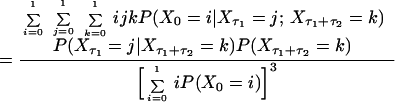 |
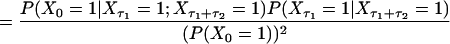 |
5 |
By comparison of Eqs. 3, 4, and 5, we then obtain
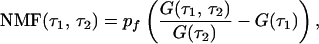 |
6 |
if we set pf = P(X0 = 1). Hence, the NMF can be related to the normalized first and second order correlation functions that are used in fluorescence correlation spectroscopy (23, 25).
The assumption that the bin size is small enough so that only zero or one photon is registered per bin is not met in the present experiments. This restriction means that no two-state emission dynamics can be monitored on faster time ranges than the inverse of the bin size (50 s−1). However, for two-state dynamics that have larger characteristic times than the inverse of the bin-size, the NMF correctly displays deviations from Markovian dynamics.
The NMF measures a degree of divergence from the Markov-assumption, and we are therefore motivated to call the two-dimensional plot of NMF for some experimental data the “memory landscape” (ML) of that particular process.
Results and Discussion
In Fig. 3 A–C, the ML are shown for three horseradish peroxidase molecules observed for 110 s. Many molecules have been observed; Fig. 3 shows examples. Indeed, the ML show non-Markovian behavior on the 2.5-s time scale. Apart from a clear memory at shorter times (<100 ms), there are structures in the memory landscape for all molecules in the range of seconds. It is also evident that, even though the 110-s ML are not identical, they all have a characteristic pattern with elongated valleys and peaks diagonally in the ML. A peak or a valley in which NMF ≠ 0 indicates that the knowledge of the spectroscopic state at the additional historical time τ2 influences the state probability at time 0.
Figure 3.

Memory landscapes (ML) are shown for three molecules observed for 110 s in A, B, and C. The standard deviation of the mean for each point in the ML was calculated.† The relative errors are less than ±3%, ±4.5%, and ±3% for all points in the ML in A, B, and C, respectively. (D) A ML generated from measurement data for the case when no enzyme is present (110-s measurement time). (E) ML as generated from simulated data of a two-state Markovian process (110-s simulation time).
In contrast to the ML generated from the data from the single enzymes performing catalysis, ML from data taken in the absence of enzyme (but everything else held constant) show a flat unstructured landscape with values close to zero (Fig. 3D). Also, data taken from a simulation of a Markovian two-state process show a noisy landscape with all values close to zero (Fig. 3E). These controls certify that the structures in the ML as obtained from the single enzyme data are caused by the properties of the enzyme and not by artificial effects or background effects.
Among the two origins in Fig. 1, we conclude in favor of that of Fig. 1A for time scales larger than 20 ms (the bin size of the experimental data's time series). On a more detailed level than the general one of Fig. 1A, an explanation to the oscillations in the NMF of a single enzyme (Fig. 3) that is consistent with theoretical predictions of the kinetics of single-enzyme systems (26–28) may be formulated. It is based on the idea that the dynamic process of a single enzyme performing catalysis is not an equilibrium process. This is so, because there is a continuous flow through the system. (Observe that the system is defined as the single-enzyme molecule and all substrate as well as product molecules interacting with the single enzyme.) The flow consists of substrate molecules that enter the system and irreversibly leave the system as products. If a kinetic model of such a non-equilibrium system is made with at least one intermediate state and one EP state, the eigenvalues to the corresponding rate matrix may be complex, leading to sine and cosine solutions (26–28). Such oscillations are clearly non-Markovian and hence will be visible via application of the NMF.
To assess the above hypothesis about the origin of the oscillations, we make a simulation. The simulated data are generated by the use of a Markovian state transition model with a predefined number of states between which state transition probabilities (pij) of a transition from state i to state j (i ≠ j) are specified. Each state is set as either fluorescent (corresponding to the EP state) or nonfluorescent (corresponding to all states but the EP state). Observe that, even though each state transition is Markovian, the spectroscopic transitions between the fluorescent state and a nonfluorescent state may be non-Markovian if there is more than one state that belongs to the same spectroscopic state.
The simulation considers a constant level of background fluorescence of 4 kHz. The fluorescent intensity of the fluorescent state is set to 8 kHz (4 kHz of fluorescent signal + 4 kHz of background fluorescence). By the use of random numbers, a stochastic kinetic pathway is generated in which the kinetic system makes transitions between the defined states with probabilities (pij) according to the rate constants from state i to state j (kij). Each state transition describes the probability to make transitions within the time period of the bin size, b. The time period consumed by an iteration of the simulation is accordingly defined as b, then; we relate the rate constants in Scheme 1 with the state transition probability via the expression
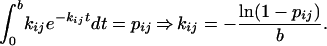 |
7 |
The simulation model in Scheme 1 takes into account the finding of activity fluctuations of the enzyme (5) as well as the above discussion of the origin to the NMF result. Scheme 1 considers activity fluctuations (5) by defining one active state (E8) of the enzyme that correspond to those conformational substates (CS) (3) in which the enzyme can perform catalysis. We then define a set of intermediate non-active states of the enzyme (E1, E2, E3, E4, E5, E6, E7) that interconnect in a serial fashion as demanded by the theoretical framework of transient states in single-enzyme systems (26–28). In the model, we assume that the substrate is involved in both the enzymatic cycle as well as a driver for the CS transitions through the intermediate states.
In Fig. 4 A and B, ML are shown as observed by NMF analysis of simulated data using Scheme 1 for two different values of the rate k. The ML show clear oscillations, similar to the ML observed for single enzymes. 
Figure 4.

ML are shown for two cases of simulations using Scheme 1 in A and B (500,000 data points simulated equaling 11,000-s measuring time). The rate constant k is set to 10 s−1 in A and 22 s−1 in B. The rate constants k−1 and k1 are always set to 20 and 15−1, respectively. The bin size is set to 20 ms in accordance with the experimental conditions.
Obviously, there are a large number of possible schemes that may be written and simulated by varying the number of transient states, and also by changing the rate parameter values or by introducing differences in the rate parameters, etc. We will not do such a detailed investigation here; however, we point out that further analysis using fluctuating rate constants (for example) may lead to a better description. Presently, we feel that priority should be given to the basic principles of the enzyme dynamics before models that use, for example, fluctuating rate constants are considered. Scheme 1 may not give the full kinetic picture of the enzyme but may provide a first approximation.
Studies of the energetic connectivity in proteins reveal unexpected dependencies between distant parts (amino acid groups) of the molecule (29). It is likely that the fluctuations of many distant parts of the molecule affect its enzymatic activity; the binding site of the molecule is not an autonomous part with regard to the catalytic properties of the enzyme. The prediction that time correlation between variables specifying different sets of conformations is revealing with regards to catalysis was postulated already in 1974 (18). Hence, the introduction of many CS in which the present enzyme may reside leads to a discussion about the connection between different CS and the activity of the enzyme.
In refs. 30 and 31, the kinetics of peptide binding to class II MHC proteins are investigated. It is shown that the peptide binding makes the protein stabilized in its active state: directly after peptide dissociation, the protein's conformation is such that it is active and may accept binding of a new peptide. The active form of the protein results from dissociation of previously bound enzyme and is quickly inactivated in the absence of new peptide. With the result of the present experiment and analysis in hand, an analogy to the results obtained in refs. 30 and 31 is straightforward: When active, the enzyme is situated in a region of CS in which it may continuously bind its substrates (dihydrorhodamine 6G and H2O2) and transform them into products. By uninterrupted catalytic cycles, the enzyme is stabilized in its active state. The enzyme may, however, make a transition out of the enzymatically active region of CS. The slow oscillations in the ML would then originate from a set of transient CS that the enzyme traverses toward the active region of CS. The prediction would be that, with increasing substrate concentration, the fraction of time the enzyme resides in its active state increases. Evaluation of single enzyme data using first-order correlation analysis shows less distributed kinetics with increasing concentration (5), being in line with this prediction. Hence, substrate interactions with the enzyme select the set of CS in which the enzyme is active. Although our data are based on a single type of protein molecule, we suggest that a similar behavior may be found in other types of proteins, such as ion-channels and ligand-specific receptors.
Acknowledgments
We thank Clas Blomberg, Hermann Haken, Peter Schuster, and Peter Wolynes for discussions. We acknowledge grants from the Swedish Natural Science Research council (NFR) and the Swedish Technical Science Research Council (TFR).
Abbreviations
- EP
enzyme-product complex
- NMF
non-Markovian function
- ML
memory landscape
- CS
conformational substates
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.130589397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.130589397
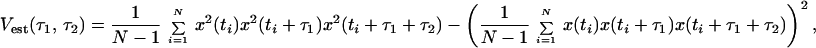 |
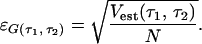 |
 |
 |
References
- 1.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 2.Wang J, Wolynes P G. Phys Rev Lett. 1995;74:4317–4320. doi: 10.1103/PhysRevLett.74.4317. [DOI] [PubMed] [Google Scholar]
- 3.Frauenfelder H, Sligar S G, Wolynes P G. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 4.Lu H P, Xun L, Xie X S. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 5.Edman L, Földes-Papp Z, Wennmalm S, Rigler R. Chem Phys. 1999;247:11–22. [Google Scholar]
- 6.Mehta A D, Rief M, Spudich J A, Smith D A, Simmons R M. Science. 1999;283:1689–1694. doi: 10.1126/science.283.5408.1689. [DOI] [PubMed] [Google Scholar]
- 7.Edman L, Mets Ü, Rigler R. Proc Natl Acad Sci USA. 1996;93:6710–6715. doi: 10.1073/pnas.93.13.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wennmalm S, Edman L, Rigler R. Proc Natl Acad Sci USA. 1997;94:10641–10646. doi: 10.1073/pnas.94.20.10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu H P, Xie X S. Nature (London) 1998;385:143–145. [Google Scholar]
- 10.Smith D E, Babcock H P, Chu S. Science. 1999;283:1724–1727. doi: 10.1126/science.283.5408.1724. [DOI] [PubMed] [Google Scholar]
- 11.Mörner W E, Orrit M. Science. 1999;283:1670–1676. doi: 10.1126/science.283.5408.1670. [DOI] [PubMed] [Google Scholar]
- 12.Edman L, Wennmalm S, Tamsen F, Rigler R. Chem Phys Lett. 1998;292:15–21. [Google Scholar]
- 13.Wennmalm S, Edman L, Rigler R. Chem Phys. 1999;247:61–67. [Google Scholar]
- 14.Willsätter R, Pollinger A. A Liebigs Ann. 1923;430:269–319. [Google Scholar]
- 15.Theorell H, Åkesson A. Ark Kemi Mineral Geol. 1943;16A:1–11. [Google Scholar]
- 16.Rigler R, Mets Ü. SPIE Laser Spectrosc Biomol. 1992;1921:239–248. [Google Scholar]
- 17.Mets Ü, Rigler R. J Fluoresc. 1994;4:259–264. doi: 10.1007/BF01878461. [DOI] [PubMed] [Google Scholar]
- 18.Careri C. In: Quantum Statistical Mechanics in the Natural Sciences. Korsunoglu B, Mintz S L, Windmayer S M, editors. New York: Plenum; 1974. pp. 15–33. [Google Scholar]
- 19.Jia Y, Talaga D S, Lau W L, Lu H S M, DeGrado W F, Hochstrasser R M. Chem Phys. 1999;247:69–83. [Google Scholar]
- 20.Ha T, Ting A Y, Liang J, Deniz A A, Chemla D S, Shultz P G, Weiss S. Chem Phys. 1999;247:107–118. [Google Scholar]
- 21.Frauenfelder H. In: Structure and Dynamics of Glass and Glass Formers. Angell C A, Ngai K L, Kieffer J, Egami T, Nienhaus G U, editors. Pittsburgh: Mater. Res. Soc.; 1997. pp. 343–347. [Google Scholar]
- 22.Zeldovich Y B, Ruzmaikin A A, Sokoloff D D. The Almighty Chance. Singapore: World Scientific; 1990. [Google Scholar]
- 23.Qian H. Biophys Chem. 1990;38:49–57. doi: 10.1016/0301-4622(90)80039-a. [DOI] [PubMed] [Google Scholar]
- 24.Kallenberg O. Foundations of Modern Probability. New York: Springer; 1997. [Google Scholar]
- 25.Elson E, Magde D. Biopolymers. 1974;13:1–27. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 26.Ryde-Pettersson U. Eur J Biochem. 1989;186:145–148. doi: 10.1111/j.1432-1033.1989.tb15188.x. [DOI] [PubMed] [Google Scholar]
- 27.Hirsch M W, Smale S. Differential Equations, Dynamics Systems, and Linear Algebra. New York: Academic; 1974. [Google Scholar]
- 28.Jackson E A. Perspectives of Nonlinear Dynamics. Vol. 1. Cambridge, U.K.: Cambridge Univ. Press; 1989. [Google Scholar]
- 29.Lockless S W, Ranganathan R. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 30.Rabinowitz J D, Vrljic M, Kasson P M, Liang M N, Busch R, Boniface J J, Davis M M, McConnel H M. Immunity. 1998;9:699–709. doi: 10.1016/s1074-7613(00)80667-6. [DOI] [PubMed] [Google Scholar]
- 31.Kasson P M, Rabinowitz J D, Schmitt L, Davis M M, McConnel H M. Biochemistry. 2000;39:1048–1058. doi: 10.1021/bi9921337. [DOI] [PubMed] [Google Scholar]