

Abstract
Regulation of B cell receptor signaling is essential for the development of specific immunity while retaining tolerance to self. Systemic lupus erythematosus (SLE) is characterized by a loss of B cell tolerance and the production of anti-self antibodies. Accompanying this break down in tolerance are alterations in B cell receptor signal transduction including elevated induced calcium responses and increased protein phosphorylation. Specific pathways that negatively regulate B cell signaling have been shown to be impaired in some SLE patients. These patients have reduced levels of the kinase Lyn in lipid raft microdomains and this reduction is inversely correlated with increased CD45 in lipid rafts. Function and expression of the inhibitory immunoglobulin receptor FcγRIIB is also reduced in Lupus IgM- CD27+ memory cells. Because the relative contribution of different memory and transitional B cell subsets can be abnormal in SLE patients, we believe studies targeted to well defined B cell subsets will be necessary to further our understanding of signaling abnormalities in SLE. Intracellular flow cytometric analysis of signaling is a useful approach to accomplish this goal.
Keywords: B cells, Systemic Lupus Erythematosus, Signal Transduction, Human
Take home message.
B cells in SLE patients have elevated induced calcium responses and increased tyrosine phosphorylation.
The lipid raft localization of signaling molecules is altered in some SLE patients. Lyn is decreased in lipid rafts and CD45 increased.
Expression and function of the inhibitory immunoglobulin receptor FcγRIIB is reduced in memory B cells in SLE patients.
The composition of memory B cell subsets is altered in SLE patients and they have a higher prevalence of activated cells.
B cell signaling in well defined memory subsets can be studied using intracellular flow cytometric analysis.
1. Introduction
Signal transduction links membrane B cell receptor (BCR) and T cell receptor antigen binding to intracellular changes in protein function and gene expression. Fine modulation of antigen-mediated signal transduction is necessary for B and T cells to develop appropriate effector function in response to pathogens while remaining tolerant to self antigens [1, 2]. Systemic lupus erythematosus (SLE) in which both T cell and B cell autoimmune responses develop against intracellular and nuclear self antigens provides a clear example of the consequences of tolerance break down. Over the past decade considerable progress has been made in defining signaling aberrations in SLE T cells and how they may be contributing to disease. (reviewed in [3–5]). In contrast, despite many studies showing multiple abnormalities in B cell homeostasis, tolerance, and function (both as the source of auto-antibodies and as immunomodulators) [6], very little is known about B cell receptor signaling in human SLE (reviewed in [7]). Yet, the importance of understanding the intrinsic properties and regulation of BCR signaling in human SLE is underscored by multiple demonstrations in animal models that abnormalities in these pathways may result in systemic autoimmunity resembling SLE [8–11].
2. Studies of B cell receptor signaling in human SLE
The first observation of abnormal BCR signaling in lupus patients was made by Liossis and colleagues using unfractionated B cells [12]. This study revealed elevated calcium flux and, using conventional biochemical measurements, increased tyrosine phosphorylation after BCR stimulation through anti-IgM or IgD antibodies, irrespective of disease activity or treatment. In addition, limited data have been published only in review form using intracellular flow cytometry suggesting that unfractionated SLE B cell immediately ex vivo may contain a higher percentage of B cells with increased basal phosphorylated mitogen activated protein kinases [13].
3. Lyn, CD45, and lipid rafts in SLE
Transgenic mice deficient in the src-family protein tyrosine kinase Lyn develop an SLE-like picture with auto-antibodies and severe nephritis [14, 15]. Consistent with this observation, a subset of SLE patients have reduced levels of Lyn [16] as a result of both reduced mRNA [17] and ubiquitin mediated degradation [18]. However, the cellular consequences of these changes are not immediately obvious as in addition to mediating BCR signaling, Lyn may also attenuate BCR signaling by phosphorylating both immunoreceptor tyrosine based inhibition motif (ITIMs) on negative regulators of B cell signaling such as FcγRIIB and the regulatory tyrosine on Syk [19].
The potential importance of Lyn functional abnormalities in SLE has recently been highlighted by the observation that in Lupus B cells there is a concomitant decrease of Lyn and a significant increase of CD45 in lipid raft microdomains [20]. CD45 can dephosphorylate both the activating pY-396 and negative regulatory pY-507 tyrosines in Lyn [21]. B cells from SLE patients displayed modestly higher phosphorylation at both tyrosines after stimulation with anti-BCR coated beads. In these experiments, Lyn was recruited more slowly in Lupus B cells to the bead interface but then was retained there for indefinitely as compared to control cells which quickly localized to the interface but were then excluded after 10 minutes. Although the regulation of Lyn by CD45 is unclear, one potential model of these findings is that in SLE B cells CD45 dephosphorylates the fraction of Lyn that is localized in lipid rafts. Thus the Lyn that is in proximity to relevant substrates would be inactive, static, and unable to down regulate BCR signaling.
4. Impaired FcγRIIB expressions and activity in SLE
One of the targets of Lyn, the immunoglobulin binding receptor FcγRIIB is a strong candidate for genetic differences that might account for intrinsic changes in SLE B cell signaling [22]. FcγRIIB contains an ITIM that upon phosphorylation recruits SH2 containing inositol phosphate phosphatase which destabilizes and down regulates the BCR signaling complex [23]. The importance of FcγRIIB in providing negative feedback is demonstrated by the generation of enhanced IgG autoantibody responses and spontaneous autoimmunity in susceptible strains of FcγRIIB deficient mice [24]. Interestingly, two genetic polymorphisms that may directly impact FcγRIIB function have been found to be more prevalent in SLE patients. A polymorphism in the transmembrane domain that is associated with SLE Asian and African populations [25–27] renders the FcγRIIB unable to localize to lipid rafts and inhibit BCR signaling in transfected cells [28, 29]. A different single nucleotide polymorphism in the FcγRIIB promoter is associated with SLE in European-Americans. B cells from individuals homozygous for this polymorphism expressed less FcγRIIB after stimulation and transcription was reduced in reporter construct transfected cells [30]. It should be noted that while both these polymorphisms are found in only a subset of SLE patients more generalized functional differences can also be demonstrated. Thus, B cells from SLE patients show less inhibition of calcium responses after stimulation with whole anti-IgM as compared to F(ab′)2 anti-IgM, suggesting that they are less susceptible to FcγRIIB-mediated feedback inhibition of BCR stimulation [31]. Although this study found no difference in FcγRIIB expression subsequent studies have shown decreased FcγRIIB expression in CD27+ B cells from SLE patients [32]. However, both these studies need to be interpreted in light of the substantial alterations in the distribution of different B cell subsets commonly seen in SLE [33]. Indeed, CD27+ IgM+ unswitched memory cells express the brightest level of FcγRIIB whether this is measured by global anti-CD32 staining [34] or by our studies with the FcγRIIB (CD32b)-specific antibody 4F-5 (data not shown). As a result, at least part of the low expression in CD27+ B cells seen in SLE patients may be due to the low number of unswitched memory cells seen in these patients.
Even given this caveat, this decrease is clearly functionally relevant in switched memory cells since B cells from high FcγRIIB expressing patients showed inhibited whole anti-IgG induced calcium flux while B cells from low expressing patients showed no difference between whole IgG and F(ab′)2 stimulatory antibodies [32]. This is also supported by the lower anti-CD32 staining found in SLE CD27+ IgM- memory cells [34].
5. Challenges in the study of BCR signaling in SLE
Several mature and transitional populations of B cells can be recognized in human peripheral blood and these population differ in their BCR responses. SLE patients can have expanded populations of both switched memory B cells and CD27- memory cells [35] as well as increased frequencies of transitional cells and dramatically reduced numbers of unswitched IgD+ CD27+ memory cells [33]. These populations vary in their magnitude and duration of anti-BCR induced signaling responses. Tyrosine phosphorylation after anti-IgG stimulation of memory cells is sustained longer than anti-IgM induced signaling [36] and we have found unswitched memory cells demonstrate increased anti-IgM induced signaling and decreased anti-IgD responses as compared to naïve cells (data not shown).
The ex vivo activation status of SLE B cells is also altered. SLE patients have a higher proportion of IgD+, IgM low, CD23+ B cells that based on increased size and high levels of costimulatory molecules resemble an activated naïve population [37]. There is also evidence of increased activation later during B cell differentiation. Some patients have a higher frequency of CD19 high B cells enriched for autoreactive specificity that have a pre-plasma cell like transcriptional profile. These cells are clearly activated in vivo since they have elevated basal phosphorylation of Syk and ERK [38] and this finding is consistent with the earlier report of a population of SLE B cells with high basal phosphoryaltion of ERK [13]. It is obvious therefore that the relative contribution of specific subsets to the total B cell compartment is frequently abnormal in SLE patients and may significantly vary from patient to patient due to disease heterogeneity (possibly genetically determined and impacting the quality of B cell signaling), disease activity and pharmacological treatment. Consequently, we believe that a better understanding of B cell signaling abnormalities in SLE as compared to normal subjects or other diseases will require studies targeted to well characterized B cell subsets as well as the selection of patients with homogenous disease phenotype and shared genetic abnormalities.
6. Flow cytometric analysis of phosphorylation can effectively measure signaling in SLE B cell subsets
An important part of the challenges discussed above can be overcome by studying cell signaling using multi-color flow cytometry [36]. This approach offers the major advantage that cells can be stained for cell surface markers after fixation and thus B cells populations can be differentiated without altering cell function. We have examined proximal BCR signaling of cells stimulated directly ex vivo and fractionated on the basis of CD27 and IgD or IgM expression. These studies confirm that considerable heterogeneity exists in patient responses. In many patients phosphoryaltion after stimulation is similar or slightly decreased compared to healthy subjects but some patients have substantially higher or lower induced phosphorylation. Two examples are shown in Figure 1. Panel A shows phosphorylation of ERK after anti-IgD stimulation and demonstrates that phospho-ERK (pERK) staining increased more in SLE 1 (3.5 fold) than in the healthy control (3.1 fold). It’s apparent that while all IgM+ CD27- cells have greater pERK after stimulation there is a subpopulation of pERK very bright cells that is more prevalent in the SLE patient (48%) than the healthy control (20%). What distinguishes this population is unknown but they express more IgM and may be transitional cells. In Panel B the healthy control B cells showed strong phospo-BLNK staining after stimulation (8.1 fold) while B cells from SLE patient 2 were only slightly above the unstimualted cells (1.5 fold). Interestingly, this SLE patient also had high levels of 9G4+ anti-lymphocyte antibodies that bind B220 on naïve B cells [39]. These antibodies may decrease signaling by directly modulating phosphatase activity or by compromising cell viability [40]. The presence of SLE-specific autoantibodies that bind to modulatory molecules further illustrates the complexity of the interpretation of signaling and functional studies in SLE lymphocytes.
Figure 1.
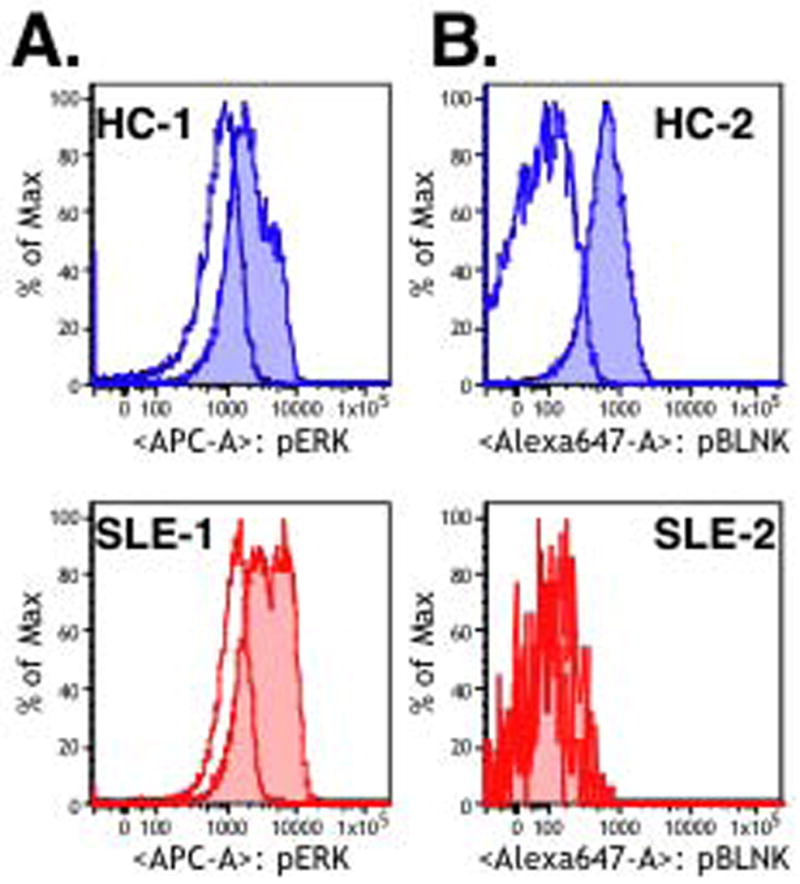
PBL from SLE patients (red) or from healthy control subjects (blue) were stimulated for five minutes with 25 μg/ml anti-IgD (A) or 10 μg/ml anti-IgM (B) (shaded histogram) or control antibody (open histogram). After stimulation cells were fixed immediately with paraformahldehyde and permeabilized with methanol. Cells were then stained with antibodies against CD20, IgM (A) or IgD (B), CD27, and antibodies specific for phosphorylated forms of ERK (pY204/pT202) (A) or BLNK (pY84) (B). Phosporylation for the IgM/IgD+ CD27- naïve populations is shown.
7. Conclusions
It is clear that B cells in SLE patients have altered BCR signaling. Defects in protein expression and function in specific pathways including Lyn and FcγRIIB have begun to provide mechanistic explanations of the observed changes. There remain several open questions. To what extent are changes in SLE B cell signaling intrinsic or are they the consequence of the disease process? Can signaling alterations provide a useful biomarker to measure disease activity or guide patient treatment? Given the complexity of SLE, it is likely that there is also heterogeneity in signaling and that a better comprehension of this heterogeneity should help us understand disease initiation and progression in different groups of patients. To answer these questions the ability to differentiate B cell populations will be essential. As the examples in figure 1 demonstrate, the flow cytometric analysis of phosphorylation can simultaneously study cell signaling states and phenotypic markers in unmanipulated B cells. This and other single cell approaches will be instrumental in furthering our understanding of how BCR signaling dysregulation contributes to SLE. Moreover, these studies should identify specific pathways and molecules that could be specifically targeted by new therapeutic strategies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–7. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 2.Ferraccioli G, Tolusso B. Infections, B cell receptor activation and autoimmunity: different checkpoint impairments lead to autoimmunity, clonal B cell expansion and fibrosis in different immunological settings. Autoimmun Rev. 2007;7:109–13. doi: 10.1016/j.autrev.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Crispin JC, Kyttaris VC, Juang YT, Tsokos GC. How signaling and gene transcription aberrations dictate the systemic lupus erythematosus T cell phenotype. Trends Immunol. 2008;29:110–5. doi: 10.1016/j.it.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Tenbrock K, Juang YT, Kyttaris VC, Tsokos GC. Altered signal transduction in SLE T cells. Rheumatology (Oxford) 2007;46:1525–30. doi: 10.1093/rheumatology/kem154. [DOI] [PubMed] [Google Scholar]
- 5.Katsiari CG, Tsokos GC. Transcriptional repression of interleukin-2 in human systemic lupus erythematosus. Autoimmun Rev. 2006;5:118–21. doi: 10.1016/j.autrev.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Renaudineau Y, Pers JO, Bendaoud B, Jamin C, Youinou P. Dysfunctional B cells in systemic lupus erythematosus. Autoimmun Rev. 2004;3:516–23. doi: 10.1016/j.autrev.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 7.Pugh-Bernard AE, Cambier JC. B cell receptor signaling in human systemic lupus erythematosus. Curr Opin Rheumatol. 2006;18:451–5. doi: 10.1097/01.bor.0000240353.99808.5f. [DOI] [PubMed] [Google Scholar]
- 8.Wu T, Qin X, Kurepa Z, Kumar KR, Liu K, Kanta H, et al. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. J Clin Invest. 2007;117:2186–96. doi: 10.1172/JCI30398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anolik J, Sanz I. B cells in human and murine systemic lupus erythematosus. Curr Opin Rheumatol. 2004;16:505–12. doi: 10.1097/01.bor.0000133660.52599.f6. [DOI] [PubMed] [Google Scholar]
- 10.Hasler P, Zouali M. B cell receptor signaling and autoimmunity. FASEB J. 2001;15:2085–98. doi: 10.1096/fj.00-0860rev. [DOI] [PubMed] [Google Scholar]
- 11.Cornall RJ, Goodnow CC, Cyster JG. The regulation of self-reactive B cells. Curr Opin Immunol. 1995;7:804–11. doi: 10.1016/0952-7915(95)80052-2. [DOI] [PubMed] [Google Scholar]
- 12.Liossis SN, Kovacs B, Dennis G, Kammer GM, Tsokos GC. B cells from patients with systemic lupus erythematosus display abnormal antigen receptor-mediated early signal transduction events. J Clin Invest. 1996;98:2549–57. doi: 10.1172/JCI119073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grammer AC, Fischer R, Lee O, Zhang X, Lipsky PE. Flow cytometric assessment of the signaling status of human B lymphocytes from normal and autoimmune individuals. Arthritis Res Ther. 2004;6:28–38. doi: 10.1186/ar1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, et al. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity. 1995;3:549–60. doi: 10.1016/1074-7613(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 15.Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 1995;83:301–11. doi: 10.1016/0092-8674(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 16.Huck S, Le Corre R, Youinou P, Zouali M. Expression of B cell receptor-associated signaling molecules in human lupus. Autoimmunity. 2001;33:213–24. doi: 10.3109/08916930109008048. [DOI] [PubMed] [Google Scholar]
- 17.Liossis SN, Solomou EE, Dimopoulos MA, Panayiotidis P, Mavrikakis MM, Sfikakis PP. B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Investig Med. 2001;49:157–65. doi: 10.2310/6650.2001.34042. [DOI] [PubMed] [Google Scholar]
- 18.Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA. Decreased Lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:3955–65. doi: 10.1002/art.21416. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005;22:9–18. doi: 10.1016/j.immuni.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA. Altered lipid raft-associated proximal signaling and translocation of CD45 tyrosine phosphatase in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2007;56:291–302. doi: 10.1002/art.22309. [DOI] [PubMed] [Google Scholar]
- 21.Shrivastava P, Katagiri T, Ogimoto M, Mizuno K, Yakura H. Dynamic regulation of Src-family kinases by CD45 in B cells. Blood. 2004;103:1425–32. doi: 10.1182/blood-2003-03-0716. [DOI] [PubMed] [Google Scholar]
- 22.Gergely P, Jr, Isaak A, Szekeres Z, Prechl J, Erdei A, Nagy ZB, et al. Altered expression of Fcgamma and complement receptors on B cells in systemic lupus erythematosus. Ann N Y Acad Sci. 2007;1108:183–92. doi: 10.1196/annals.1422.020. [DOI] [PubMed] [Google Scholar]
- 23.Sohn HW, Pierce SK, Tzeng SJ. Live cell imaging reveals that the inhibitory FcgammaRIIB destabilizes B cell receptor membrane-lipid interactions and blocks immune synapse formation. J Immunol. 2008;180:793–9. doi: 10.4049/jimmunol.180.2.793. [DOI] [PubMed] [Google Scholar]
- 24.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–85. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Wu J, Carter RH, Edberg JC, Su K, Cooper GS, et al. A novel polymorphism in the Fcgamma receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum. 2003;48:3242–52. doi: 10.1002/art.11313. [DOI] [PubMed] [Google Scholar]
- 26.Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, et al. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. 2002;46:1242–54. doi: 10.1002/art.10257. [DOI] [PubMed] [Google Scholar]
- 27.Clatworthy MR, Willcocks L, Urban B, Langhorne J, Williams TN, Peshu N, et al. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proc Natl Acad Sci U S A. 2007;104:7169–74. doi: 10.1073/pnas.0608889104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, et al. FcgammaRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet. 2005;14:2881–92. doi: 10.1093/hmg/ddi320. [DOI] [PubMed] [Google Scholar]
- 29.Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. 2005;11:1056–8. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 30.Blank MC, Stefanescu RN, Masuda E, Marti F, King PD, Redecha PB, et al. Decreased transcription of the human FCGR2B gene mediated by the -343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum Genet. 2005;117:220–7. doi: 10.1007/s00439-005-1302-3. [DOI] [PubMed] [Google Scholar]
- 31.Enyedy EJ, Mitchell JP, Nambiar MP, Tsokos GC. Defective FcgammaRIIb1 signaling contributes to enhanced calcium response in B cells from patients with systemic lupus erythematosus. Clin Immunol. 2001;101:130–5. doi: 10.1006/clim.2001.5104. [DOI] [PubMed] [Google Scholar]
- 32.Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, et al. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med. 2006;203:2157–64. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanz I, Wei C, Lee FE, Anolik J. Phenotypic and functional heterogeneity of human memory B cells. Semin Immunol. 2008;20:67–82. doi: 10.1016/j.smim.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isaak A, Gergely P, Jr, Szekeres Z, Prechl J, Poor G, Erdei A, et al. Physiological up-regulation of inhibitory receptors Fc gamma RII and CR1 on memory B cells is lacking in SLE patients. Int Immunol. 2008;20:185–92. doi: 10.1093/intimm/dxm132. [DOI] [PubMed] [Google Scholar]
- 35.Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. 2007;178:6624–33. doi: 10.4049/jimmunol.178.10.6624. [DOI] [PubMed] [Google Scholar]
- 36.Irish JM, Czerwinski DK, Nolan GP, Levy R. Kinetics of B cell receptor signaling in human B cell subsets mapped by phosphospecific flow cytometry. J Immunol. 2006;177:1581–9. doi: 10.4049/jimmunol.177.3.1581. [DOI] [PubMed] [Google Scholar]
- 37.Chang NH, McKenzie T, Bonventi G, Landolt-Marticorena C, Fortin PR, Gladman D, et al. Expanded population of activated antigen-engaged cells within the naive B cell compartment of patients with systemic lupus erythematosus. J Immunol. 2008;180:1276–84. doi: 10.4049/jimmunol.180.2.1276. [DOI] [PubMed] [Google Scholar]
- 38.Nicholas MW, Dooley MA, Hogan SL, Anolik J, Looney J, Sanz I, et al. A novel subset of memory B cells is enriched in autoreactivity and correlates with adverse outcomes in SLE. Clin Immunol. 2008;126:189–201. doi: 10.1016/j.clim.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cappione AJ, Pugh-Bernard AE, Anolik JH, Sanz I. Lupus IgG VH4.34 antibodies bind to a 220-kDa glycoform of CD45/B220 on the surface of human B lymphocytes. J Immunol. 2004;172:4298–307. doi: 10.4049/jimmunol.172.7.4298. [DOI] [PubMed] [Google Scholar]
- 40.Bhat NM, Bieber MM, Hsu FJ, Chapman CJ, Spellerberg M, Stevenson FK, et al. Rapid cytotoxicity of human B lymphocytes induced by VH4–34 (VH4.21) gene-encoded monoclonal antibodies, II. Clin Exp Immunol. 1997;108:151–9. doi: 10.1046/j.1365-2249.1997.d01-976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]