

Abstract
Clearance of cellular debris is a critical feature of the developing nervous system, as evidenced by the severe neurological consequences of lysosomal storage diseases in children. An important developmental process, which generates considerable cellular debris, is synapse elimination, in which many axonal branches are pruned. The fate of these pruned branches is not known. Here, we investigate the role of lysosomal activity in neurons and glia in the removal of axon branches during early postnatal life. Using a probe for lysosomal activity, we observed robust staining associated with retreating motor axons. Lysosomal function was involved in axon removal because retreating axons were cleared more slowly in a mouse model of a lysosomal storage disease. In addition, we found lysosomal activity in the cerebellum at the time of, and at sites where, climbing fibers are eliminated. We propose that lysosomal activity is a central feature of synapse elimination. Moreover, staining for lysosomal activity may serve as a marker for regions of the developing nervous system undergoing axon pruning.
Keywords: synapse elimination, lysosome, autophagy, degradation, axon pruning, retraction bulb
Introduction
Clearance of cellular debris is an important aspect of cellular homeostasis. Through normal cellular metabolism, there is a continual buildup of byproducts that are removed via degradative pathways. Most cells in multicellular organisms undergo continual renewal such that old cells die and are replaced by new ones, meaning that the amount of material accumulated in a cell is typically small. Most neurons, however, are postmitotic and do not divide for years or even decades. They are thus particularly vulnerable to the deleterious effects of debris accumulation, and many lysosomal storage disorders manifest with neurological symptoms, suggesting that nervous tissue is especially susceptible to buildup of storage material (Nixon and Cataldo, 1993). Indeed a number of studies suggest that disruption of axonal degradative pathways is linked to axonopathies and neurodegeneration (Yue et al., 2008). Problems with glial digestion pathways, although less frequently described, have been linked to demyelination (Fortun et al., 2003).
In the developing nervous system, synapse elimination involves a massive loss and eventual disappearance of cellular material. This developmental reorganization causes a large majority of nascent synaptic terminals and their associated axonal branches to be removed. In the peripheral nervous system, synapse elimination has been documented in early postnatal life at the neuromuscular junction (Redfern, 1970) and in autonomic ganglia (Lichtman, 1977; Purves and Lichtman, 1980). At the same developmental stages, analogous reorganizations have been found to occur in the cerebellum (Crepel et al., 1976; Lohof et al., 1996), the retinogeniculate system (Chen and Regehr, 2000), and the somatosensory system (Arsenault and Zhang, 2006), suggesting that there may be a common mechanism of axon pruning throughout the peripheral nervous system and CNS. Because of the accessibility and large size of neuromuscular junctions, synapse elimination has been studied most extensively in muscle. Time-lapse imaging and serial electron microscopy at the neuromuscular junction shows that retreating motor axon branches are pruned by a shedding process in which axonal fragments (“axosomes”) trail along the path of retreat (Bishop et al., 2004). Electron microscopy shows that these axosomes are engulfed by Schwann cells sheathing the retreating motor axon, but the fate of these axonal remnants within the glial cells remains unclear. The large debris load that glia have to accommodate in early postnatal life suggests that this developmental stage would be particularly sensitive to the effects of disorders in debris clearance.
Here we examine the role of the lysosomal pathway in glia and neurons during the developmental phase, when naturally occurring synapse elimination takes place. We show that regions of the nervous system undergoing synapse elimination can be highlighted with a vital marker that labels late endosomes and lysosomes. We also demonstrate that in a molecular disorder of debris clearance (Cao et al., 2006), retreating axons are removed more slowly. We further provide evidence that by highlighting the lysosomal pathway in the developing brain, regions of axon pruning in the CNS can be identified.
Materials and Methods
Transgenic mice.
We used Thy-1 YFP-16 transgenic mice to visualize motor axons as described previously (Feng et al., 2000). Double-transgenic mice were generated for some experiments by crossing Thy-1 CFP-5 mice with S100-GFP transgenic mice to obtain spectrally distinct axonal and Schwann cell labeling (Zuo et al., 2004). GFP-LC3 transgenic (Mizushima et al., 2004) and Cln3Δex7/8 knock-in mouse tissues (Cotman et al., 2002) were received from S.L.C. The Cln3Δex7/8 mice have the most common human CLN3 mutation knocked into the mouse genome. For studying climbing fibers in the cerebellum, we crossed Thy-1 CFP-5 mice with Thy-1 YFP-16 mice to visualize Purkinje cells and climbing fibers, respectively. Of note, a very small subset of climbing fibers express YFP in the Thy-1 YFP-16 line; thus climbing fiber branches innervating adjacent Purkinje cells are likely to originate from the same climbing fiber axon. Thy-1 CFP-5 × S100-YFP double-transgenic mice were crossed to obtain parallel labeling of Purkinje cells and Bergmann glia. Brainbow line I (Livet et al., 2007) crossed with Cre-ER mice were used to obtain membrane-labeled Bergmann glial expression. Tamoxifen (50 μg) was injected intraperitoneally in postnatal day 4 (P4) pups.
Ex vivo imaging in nerve–muscle explants.
We used tissue from Thy-1 YFP-16 or double-transgenic mice (P8–P13) as described previously (Bishop et al., 2004). The triangularis sterni muscle and its innervating nerves were dissected in oxygenated (95% O2/5% CO2) Neurobasal A medium (Invitrogen) at room temperature. The tissue was then pinned into a Sylgard-coated 3.5 cm dish. LysoTracker Red DND-99 (1:5000; Invitrogen) in oxygenated Neurobasal A medium was added to the explant dish and incubated for 2–3 min while being continuously perfused with oxygenated medium. The LysoTracker Red DND-99 containing media was then washed out with new oxygenated Neurobasal A medium to remove excess dye. The tissue was kept heated to 33–35°C and continuously perfused with oxygenated Neurobasal A medium at a perfusion rate of 0.5–1.0 ml/min during imaging. Ex vivo imaging was continued for up to 5–6 h, while the tissue remained healthy and no signs of Wallerian degeneration were noted [for details, see Bishop et al. (2004)].
Time-lapse images were taken with a wide-field Olympus microscope equipped with a 20×/0.5 numerical aperture (NA) water and a 100×/1.0 NA water objective lens, and Retiga EXi CCD camera (Qimaging) controlled by MetaMorph software (Molecular Devices). Time-series images were aligned using Autoquant software (Media Cybernetics) to correct for drifts in the image series. Other time series were acquired on an Olympus FV1000 confocal microscope using the above-mentioned water objectives. Images were edited in Adobe Photoshop. To obtain images free of superimposition, in-focus information was manually extracted from individual images of an image stack. When not illustrating quantitatively accurate results but merely the presence of labeling, we used gamma correction to compress the dynamic range of intensities (for example, see Fig. 1A).
Figure 1.

LysoTracker associated with retreating axons at developing neuromuscular junctions. Three patterns of LysoTracker (LysoT) staining were observed associated with retreating axons in developing muscle. A, A pair of neuromuscular junctions from a P12 mouse and one retraction bulb (arrow), which is surrounded by strong LysoT staining (red). Arrowheads point to weak LysoT-positive staining in the neuromuscular junction. B, In other instances, LysoT staining was observed exclusively inside the axoplasm of the retreating axon (P10). C, In most cases, LysoT staining was observed both in and around retraction bulbs (P13). C′, C″, The same single confocal section through the middle of the bulb shown in C. Arrowheads point to LysoT particles inside the bulb. Note also a LysoT ring-shaped structure, which surrounds a faintly fluorescent region of the retreating bulb. D, An axon undergoing axosome shedding was surrounded by LysoT staining, which was often distributed in multiple ring-like structures (P12). Many of these rings surrounded small axosomes (arrowhead), whereas others (arrow) contained no axonal fluorescence (see Results). Scale bars: A, 10 μm; B–D, 5 μm.
Confocal imaging and immunostaining.
Confocal images of fixed tissue were taken using the Olympus FV1000 microscope equipped with a 20×/0.8 NA, a 40×/1.3 NA, and a 60×/1.45 NA oil objective. Confocal image stacks were acquired sequentially, and images were processed with MetaMorph and Adobe Photoshop.
To obtain colabeling of LysoTracker with anti-LAMP-2 antibody, tissue stained with LysoTracker was immediately fixed in 4% paraformaldehyde, which preserves most large but not all small accumulations of LysoTracker staining. This tissue was subsequently stained with a polyclonal anti-LAMP-2 (1:50–100; Zymed) and a Cy5-labeled goat anti-rabbit secondary antibody (1:1000; Invitrogen).
For visualizing axons and synapses in Cln3Δex7/8 knock-in mice (P5, P10, and P14), the sternomastoid muscle was removed from terminally anesthetized animals and then fixed for 30 min with 4% paraformaldehyde. The tissue was immunostained with mouse monoclonal anti-neurofilament SMI-312 antibody (1:1000; Covance) and mouse monoclonal anti-synapsin antibody (1:1000; Synaptic Systems) and incubated with Alexa568-labeled goat anti-mouse secondary antibodies to obtain axonal staining. Alexa488-conjugated bungarotoxin (5 μm; Invitrogen) was used to obtain postsynaptic acetylcholine receptor labeling. More than 300 total neuromuscular junctions were counted to calculate the incidence of retreating axons in homozygous Cln3Δex7/8 and wild-type littermate pups at each age.
Iontophoretic labeling and EM reconstruction.
After ex vivo imaging, tissues were immersion fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 m PBS at pH 7.4. Previously imaged bulbs with LysoTracker staining were then targeted for serial electron microscopic reconstruction. To create fiducial markers to locate the bulb, sharp electrodes were filled with 1% solutions of DiI (Invitrogen) in 100% methylene chloride and positioned on muscle fibers near the area of interest. Electrical pulses (200 ms, 1–10 V, 1 Hz) were applied to inject a small DiI crystal into the muscle membrane. Photoconversion of the DiI crystals deposited an electron-dense precipitate on muscle fibers flanking the area of interest. Photoconverted tissue was then osmicated in ferrocyanide-reduced osmium for 1 h, dehydrated in ascending ethanol and propylene oxide, and embedded in EMBed812 (Electron Microscopy Sciences). Thin sections (1 μm) were cut and examined until the photoconverted DiI crystals could be located. Serial ultrathin sections (∼70 nm) were cut through the region, collected on Pioloform slot grids, and counterstained with aqueous uranyl acetate and Reynold's lead citrate for 30 min each. Sections were examined at 75 kV using a Hitachi H600 transmission electron microscope. Electron micrographs were captured on film and scanned at a resolution of 1000 ppi. Serial sections were aligned and items of interest were segmented using Reconstruct (http://synapses.clm.utexas.edu/) (Fiala, 2005).
Cerebellar tissue preparation and LysoTracker quantification.
Briefly, pups (P5–P30) were anesthetized with a ketamine and xylazine mixture and decapitated, and transverse cerebellar slices (100–200 μm) were obtained using a Vibratome VT1000S (Leica Microsystems) [for details, see Beierlein and Regehr (2006)]. Slices were cut in ice-cold oxygenated sucrose-containing Ringer's solution (in mm: 81.2 NaCl, 23.4 NaHCO3, 69.9 sucrose, 23.3 glucose, 2.4 KCl, 1.4 NaH2PO4, 6.7 MgCl2, and 0.5 CaCl2). Slices were incubated at 32°C for 30 min in sucrose solution, then transferred for 30 min in 32°C artificial CSF (ACSF) solution (in mm: 125 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 25 glucose, 2 CaCl2, and 1 MgCl2), then incubated in ACSF solution at room temperature for 60 min. LysoTracker Red DND-99 (1:5000; Invitrogen) was added to the slices for 10 min during the incubation at room temperature, and the tissue was subsequently fixed in 4% PFA overnight. All solutions during the slicing and incubating steps were oxygenated (95% O2/5% CO2).
After fixation, the tissue was washed in PBS and mounted for confocal imaging and analysis. Purkinje cells in lobes 3 and 9 were imaged and subsequently analyzed for LysoTracker staining. A single optical section for each Purkinje cell was chosen at random, and the total number of LysoTracker-positive structures exclusively surrounding that Purkinje cell soma but not within the Purkinje cell was counted.
Cerebellum image tracings and reconstructions.
Reconstruct Program (http://synapses.clm.utexas.edu/) (Fiala, 2005) was used to trace confocal data stacks of fixed LysoTracker-stained cerebellar images. Tracings were performed by hand, and three-dimensional reconstructions were created with Reconstruct.
Results
Acidic organelles associated with neuromuscular synapse elimination
We studied the fate of eliminated axonal branches during development using transgenic mice that express fluorescent proteins in the cytoplasm of axons (Feng et al., 2000). Motor axon branches were imaged during postnatal life at the time the final transitions from multiple to single innervation of neuromuscular junctions are occurring. We observed that at some neuromuscular junctions, more than one axon was still present: one that innervated the postsynaptic site and, nearby, a second axon that ended in a bulb. Previous studies showed that bulb-tipped axons are in the midst of retreating after synaptic disconnection (Walsh and Lichtman, 2003) and are often accompanied by a line of small fluorescent fragments (axosomes) marking the axon's path of retreat. The axosomes are filled with mitochondria and synaptic vesicles and are completely surrounded by nearby Schwann cells [see below and Bishop et al. (2004)]. These glial cells contained axon-derived fluorescent proteins in their cytoplasm, raising the possibility that axonal material might become functionally incorporated into glia. Alternatively, axonal material might be digested within the glial cell, as is known to happen during Wallerian degeneration after axonal trauma (Hirata and Kawabuchi, 2002). To explore this latter possibility, we developed methods to visualize digestive compartments at sites of developmental motor axon removal.
In acutely explanted muscles from developing mice undergoing synapse elimination (P8–P13), we applied a BODIPY (boron-dipyrromethene) fluorophore derivative, LysoTracker Red, which partitions into acidic organelles, most notably, the lysosomes and late endosomes of living cells (Zhang et al., 1994). We found that LysoTracker staining was present in these developing muscles and was distributed in several different patterns around retreating axon branches. At neuromuscular junctions with a bulb-tipped axon, we often observed LysoTracker staining present in the immediate vicinity surrounding the bulb (Fig. 1A; supplemental Movie 1, available at www.jneurosci.org as supplemental material) (38.0%; n = 11 of 29 bulbs). Less often, we observed staining exclusively within the axoplasm of the axonal bulb (Fig. 1B; supplemental Movie 2, available at www.jneurosci.org as supplemental material) (10.3%; n = 3 of 29 bulbs). Finally, at approximately one-half of the neuromuscular junctions, we observed staining both in and around the retreating axon (Fig. 1C) (51.7%; n = 15 of 29 bulbs). These latter two results were surprising, because lysosomes in neurons are described to be commonly located only in the soma and at nodes of Ranvier (Holtzman and Novikoff, 1965). However, we observed accumulations of LysoTracker-positive material within the axon terminal at singly innervated neuromuscular junctions (Fig. 1A, arrowheads). We therefore suspect that extant LysoTracker-positive organelles can fuse with debris within the distal axon, increasing the volume of the digestive compartment.
LysoTracker staining around retreating motor axons was often considerable. The total area of LysoTracker-positive material near retreating axons (45.4 μm2; SD = 35.0; n = 15) was significantly greater than the LysoTracker-positive areas associated with nonretreating axons (1.60 μm2; SD = 1.69; n = 15; p < 0.001, t test) at the same developmental age. Furthermore, the LysoTracker-positive material surrounding retraction bulbs often appeared as ring-like structures (Fig. 1D). Within some of these rings, we observed fluorescence derived from the axon that we infer to be axosomes (Bishop et al., 2004); in other cases, the rings surrounded a nonfluorescent core (Fig. 1D, arrowhead vs arrow). It is possible that the nonfluorescent cores were axosomes that were slightly further along a degradative process causing the loss of fluorescence. No rings were seen at any singly innervated developing neuromuscular junctions.
We were interested to learn the temporal sequence of lysosomal activity that gave rise to LysoTracker labeling both within and around retreating axons. In earlier developmental stages (P4–P6), there was a higher incidence of intra-axonal labeling, whereas a few days later (P9–P13), lysosomal activity showed a shift to the extra-axonal compartment surrounding retreating axons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results suggest that the degradative events begin within the axon and then are completed within the surrounding glial cells.
Degradative organelles associated with axon retraction
We next turned to time-lapse imaging of retraction bulbs undergoing axosome shedding to see what became of LysoTracker-ringed axosomes. We observed the fluorescence of ringed axosomes to slowly diminish over 1–2 h (Fig. 2A). The loss in intensity was not explained by photobleaching, because within the same field, different axosomes lost fluorescence intensity at different rates. LysoTracker-ringed axosomes lost fluorescent protein intensity faster than axosomes not associated with LysoTracker (Fig. 2A, arrowhead and arrow vs asterisk). This difference suggests that LysoTracker staining is associated with the disappearance of axosomes. LysoTracker labels material solely based on its acidic properties (Zhang et al., 1994). Therefore, to confirm that lysosomal organelles are indeed present at sites of LysoTracker staining, we labeled motor axons in developing muscles using an antibody to LAMP-2, a lysosomal membrane protein specific for late endosomes and lysosomes (Cuervo and Dice, 1996). LysoTracker-stained compartments, including ring-like structures, were outlined by LAMP-2 staining (Fig. 2B). However, LAMP-2 did not mark all LysoTracker-stained structures, suggesting that LysoTracker also labeled a small subset of acidic structures other than lysosomes. This result is consistent with previous studies showing that LysoTracker labels multivesicular and multilamellar bodies (Watts et al., 2004) and autolysosomes (Wang et al., 2006).
Figure 2.

Degradation of axonal material associated with LysoTracker (LysoT). A, An 80 min time lapse of LysoT labeling (red) in a nerve–muscle explant from a P13 mouse. Brightly (arrowhead) and dimly (arrow) YFP-fluorescent axosomes (green) are visible at the tip of this retreating axon. Bottom, Monochromatic panels show that the dim axosome disappears completely, whereas the bright axosome becomes progressively faint. Interestingly, axosomes not ringed by LysoT (asterisk) do not lose YFP fluorescence over the same time period. B, The presence of lysosomes was confirmed by immunohistochemistry for LAMP-2, a lysosomal membrane protein specific for late endosomes and lysosomes. Axosomes stained by LysoT reveal staining similar but not identical to that of LAMP-2 (arrowhead; see Results). C, D, To confirm the role of autophagy during axon pruning, we used transgenic mice expressing fluorescent protein tethered to LC3, which accumulates on autophagic membranes. We observed LC3-positive autolysosomes in both the associated Schwann cell (C) and in the axoplasm of retreating axons (D; P11). C, Inset, A monochromatic panel showing the pronounced and polarized LC3-accumulation in a Schwann cell surrounding an axonal bulb. D, Insets, Single confocal sections through the middle of the retraction bulb in D, demonstrating LC3-positive, autolysosomes devoid of axoplasmic staining inside the retraction bulb (arrowheads). Scale bars: A, 5 μm; B, D (for C, D), 2 μm.
Because we saw LysoTracker within the retreating axons, we explored the possibility that autophagy plays a role in axon pruning. We examined developing transgenic mice that contain green fluorescent protein fused to microtubule-associated protein light-chain 3 (LC3), which is known to accumulate on autophagic membranes (GFP-LC3 mice) (Mizushima et al., 2004) and to colabel LysoTracker-positive autophagic compartments (Wang et al., 2006). Indeed, we observed evidence of autolysosomes both in retreating axons and the surrounding Schwann cells (Fig. 2C,D). The staining pattern found in GFP-LC3 mice is similar to that obtained with LysoTracker. Together, these results corroborate that LysoTracker highlights heterophagic as well as autophagic processes during synapse elimination at the neuromuscular junction.
To confirm that degradation was occurring in these LysoTracker-positive compartments, we compared light and electron microscopy images of the same axonal bulb showing LysoTracker staining (Fig. 3A, inset). Both a large spherical (arrowhead) and an oblong-shaped structure (arrow) were LysoTracker positive. We relocated these structures in the electron microscope. The retraction bulb was completely sheathed by a Schwann cell (identified by its basal lamina), as were the LysoTracker-positive compartments. We observed a large electron-dense primary lysosome (Fig. 3B) that aligned with the spherical LysoTracker-positive compartment from the fluorescence image (Fig. 3A, inset, arrowhead). In addition, we observed a vesicle-laden structure that coincided with the location of the other LysoTracker-positive structure (Fig. 3C). Consistent with our light microscopic observations of axonal autophagic processes (Fig. 2C), we observed autolysosomes in retraction bulbs at the ultrastructural level (Fig. 3D–I). These results confirm LysoTracker's ability to detect both axonal and glial degradative processes during axon pruning.
Figure 3.

Ultrastructural evidence of degradation of axonal remnants as axons are pruned during development. A, A surface rendering from 74 serial electron micrographs of a retraction bulb from a P13 mouse previously stained with LysoTracker (LysoT; inset). Distal to the tip of the retraction bulb are two LysoT-positive compartments. Electron micrographs were placed within the compartment and magnified in B and C. The arrowhead shows the fluorescence image of the ultrastructure with the yellow dots, and the arrow points to the fluorescence of the images with blue dots in A, B, and C. B, A large electron-dense primary lysosome is surrounded by glial cytoplasm. C, An electron-dense vesicle-laden axosome within a glial cell is in the process of degradation. D, A confocal projection of two neuromuscular junctions and a retraction bulb (arrowhead) within the adjacent nerve fascicle (P9). E, A surface rendering of 91 serial electron micrographs of the retraction bulb (arrowhead in D). F, A single electron micrograph through the center of the bulb. A red dot appears in the top right corner of the electron micrograph for orientation to E. The boxed region in F is magnified as three serial sections in G–I, which reveals an axonal autolysosome (arrowheads in G and H) and also signs of earlier stages of digestion (autophagosome; arrowhead in I). [The data in E and F are derived from further analysis of a serial section electron microscopy stack in Bishop et al. (2004), their Fig. 6.] Scale bars: A (inset), D, 5 μm; B, C, 1 μm; F, 2 μm; (in G) G–I, 0.5 μm.
The sheathing of both bulbs and LysoTracker-positive material within Schwann cells suggests that glia are an important component in the process of axonal removal. Indeed, time-lapse imaging in transgenic mice that express fluorescent proteins in Schwann cells showed directly that axon retreat and axosome shedding occur entirely within the confines of Schwann cells (Fig. 4A; supplemental Movie 3, available at www.jneurosci.org as supplemental material). Importantly, the motor axon's silhouette remained negative during the period of time-lapse imaging, indicating that no fluorescent glial material (e.g., lysosomes) was transferred back to the axon during this process. Moreover, in double-transgenic mice in which both axons and Schwann cells are labeled, we observed colocalization of LysoTracker and axosomes within Schwann cells (Fig. 4B). This LysoTracker-positive material in glia included ring-like structures (Fig. 4C and see above). Interestingly, in some instances axonal bulbs still connected by a thin stalk were nearly completely ringed by LysoTracker within the Schwann cell (Fig. 4D). Retreating axons with this morphology were previously suggested to be at the stage when formation of a large axosome is imminent (Bishop et al., 2004). A phagocytic role of glial cells therefore seems highly likely. In contrast, however, macrophages [as labeled in CX3CR1 knock-in mice (Jung et al., 2000)], which are also potential phagocytic cells and are present in developing muscle, were not observed in association with retreating axons, suggesting that they have no role in axon removal at the developing neuromuscular junction (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 4.

LysoTracker (LysoT) activity is in Schwann cells sheathing retreating axons. A, Frames from a time-lapse movie of fluorescently labeled glial cells (S100-GFP) at a P10 neuromuscular junction, in which one axon is being withdrawn. Both the axon that remains at the junction (arrow) and the axon that is retreating (arrowhead) appear dark. At a slightly higher magnification (red box), frames are shown from the movie (approximately every 25 min). Shed axosomes appear and rapidly disappear in the glial cytoplasm (see supplemental Movie 3, available at www.jneurosci.org as supplemental material). B, LysoT particles (red) are just distal to the tip of a CFP-expressing axon bulb (blue). The LysoT-positive structure furthest from the bulb (arrow) does not show CFP fluorescence, whereas the LysoT-positive structure located closer to the bulb (arrowhead) colocalizes with an axosome that still maintains CFP fluorescence (see monochromatic panels on right). Both of these LysoT-positive structures are embedded in the cytoplasm of a Schwann cell (SC; green). C, A withdrawing axon with a thin stalk undergoing axosome shedding (arrowhead) beneath an axon (arrow) that innervates a P13 neuromuscular junction to the right (data not shown). The retreating axon is surrounded by rings of LysoT within the cytoplasm of the sheathing SC. D, An example of a retreating bulb nearly ringed by LysoT in the surrounding SC. Scale bars: A, 10 μm; B, 2 μm; C, D, 5 μm.
Retreating axons disappear more slowly in a mouse model of a lysosomal storage disease
Evidence of autolysosomes in the retreating axons and nearby glial cells (Fig. 2C,D) prompted us to investigate whether disrupting autophagic pathways would delay axon pruning. We examined axon pruning in a mouse model of juvenile neuronal ceroid lipofuscinosis (also known as Batten's disease), a fatal pediatric neurodegenerative disease in which autophagy is reduced as a result of a mutation in a membrane trafficking protein [CLN3 (Cao et al., 2006)]. In particular, we examined whether partial disruption in autophagy is sufficient to cause a delay in axon removal at developing neuromuscular junctions.
In developing skeletal muscle of homozygous Cln3Δex7/8 mice (Cotman et al., 2002), the incidence of axons in the midst of retreating at P5 and P10 was significantly greater than wild-type littermates (at P5, 41.3%, SD = 14.6, 8 pups vs 17.8%, SD = 4.26, in 5 pups; p < 0.01, t test; and at P10, 9.07%, SD = 2.69, in 8 pups vs 4.50%, SD = 1.46, in 7 pups; p < 0.0005, t test) (Fig. 5A–C; supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Moreover, sometimes two or three retreating axons (n = 6 in 16 muscles) were observed at a single neuromuscular junction (Fig. 5D), a result never obtained in wild-type animals (n = 0 in 10 muscles). The presence of multiple retreating axons per neuromuscular junction is consistent with the idea that there is a delay in the clearance of axonal material in these mice. Because ordinarily only one retreating axon from a neuromuscular junction is observed at any time, it is likely that axons that are removed from a neuromuscular junction do so in a sequential way. In the affected pups, however, the removal process is slowed such that multiple withdrawing axons are present at the same time. Neuromuscular junction structure per se showed no obvious phenotype at P5, P10, and P14. Similarly the degree of multiple innervation (i.e., the number of axons that innervate a synapse) was normal (data not shown). By P14, there was no significant difference in the number of retreating axons, and nearly all neuromuscular junctions were singly innervated as in wild-type littermates (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). These results suggest that although there is no defect in synapse formation or elimination per se, disrupting autophagy leads to a transient delay in axonal branch removal.
Figure 5.

Axon branch removal is delayed in a lysosomal storage disease. A, B, At P5, we observed a higher incidence of retraction bulbs (arrowheads) in a mouse model of juvenile neuronal ceroid lipofuscinosis (JNCL; Cln3Δex7/8 mice; A), a lysosomal storage disease, compared with wild-type (WT) littermates of the same age (B). C, At some neuromuscular junctions, an unusually high number of retraction bulbs (pseudocolored white by manual tracing) were observed. D, In some instances, two retreating axons (arrowheads) were observed withdrawing from the same neuromuscular junction (P5). This is unusual, because axons normally withdraw sequentially from neuromuscular junctions. Scale bars: B (for A, B), D, 10 μm; C, 5 μm. NFT, Neurofilament; Syn, synapsin; BTX, bungarotoxin.
Lysosomal activity associated with synapse elimination in the cerebellum
We next tested whether lysosomal involvement is a feature of developmental synapse elimination in other regions of the nervous system. We examined LysoTracker staining in the Purkinje cell layer of acutely sliced cerebella of neonatal mice at the time climbing fiber inputs to Purkinje cells are lost (Kano et al., 1997). LysoTracker-positive structures were most prevalent during the second postnatal week (Fig. 6A) (2.5 LysoTracker-positive structures per Purkinje cell at P12; n = 42 total Purkinje cells). In contrast, at both P5 and P30, there were fewer LysoTracker-positive structures (0.26 LysoTracker-positive structures per Purkinje cell at P5, n = 53 Purkinje cells; 0.37 per Purkinje cell at P30, n = 71 total Purkinje cells). The increased density of LysoTracker labeling parallels the time course of climbing fiber synapse elimination, which is largely complete by 3–4 weeks of age (Kano et al., 1997). Moreover, most of the LysoTracker staining was localized to the Purkinje cell layer and the nearby deep region of the molecular layer, consistent with the location of climbing fibers that transiently innervate Purkinje cells during development (Scelfo et al., 2003) (Fig. 6B–B″). In several cases, we observed robust LysoTracker staining directly apposed to the site of a climbing fiber that innervated a Purkinje cell (Fig. 6C–C′). In contrast to most of the observed climbing fibers that were smooth in appearance, we identified a subpopulation of climbing fibers during the period of climbing fiber removal that had a fragmented appearance (see supplemental Fig. 4, available at www.jneurosci.org as supplemental material). For example, a typical terminal branch of a “fragmented” climbing fiber was on average made up of six disconnected fluorescent pieces (6.0 pieces; SD = 2.05; n = 12 climbing fibers in 4 pups), whereas normally a terminal branch was continuous, i.e., consisted of less than two pieces (1.41 pieces; SD = 0.65; n = 24 climbing fibers in 6 pups; p < 0.02, t test) (i.e., see examples in Fig. 6D′,E′). In one case, we fully reconstructed two climbing fibers (one fragmented, one not) that innervated adjacent Purkinje cells (Fig. 6D,E). Given the rarity of fluorescent protein expression in climbing fibers in the mouse line we used (see Materials and Methods), these two climbing fiber branches probably originated from the same neuron. The climbing fiber branch that had a fragmented appearance was associated with fourfold more LysoTracker staining (414.4 μm2 total area of LysoTracker staining) than the climbing fiber that was not fragmented (110.9 μm2). A significant difference between the amount of LysoTracker associated with normal appearing and fragmented climbing fibers was a general finding (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). The LysoTracker staining we observed near Purkinje somata and their proximal dendritic trees often appeared in ring-like shapes (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These rings are reminiscent of the labeling pattern that we observed at developing neuromuscular junctions during synapse elimination (Figs. 1D, 4C). In the cerebellum, these rings appear to be within Bergmann glia, which intimately wrap both Purkinje cells and climbing fibers (supplemental Fig. 5B,C, available at www.jneurosci.org as supplemental material).
Figure 6.
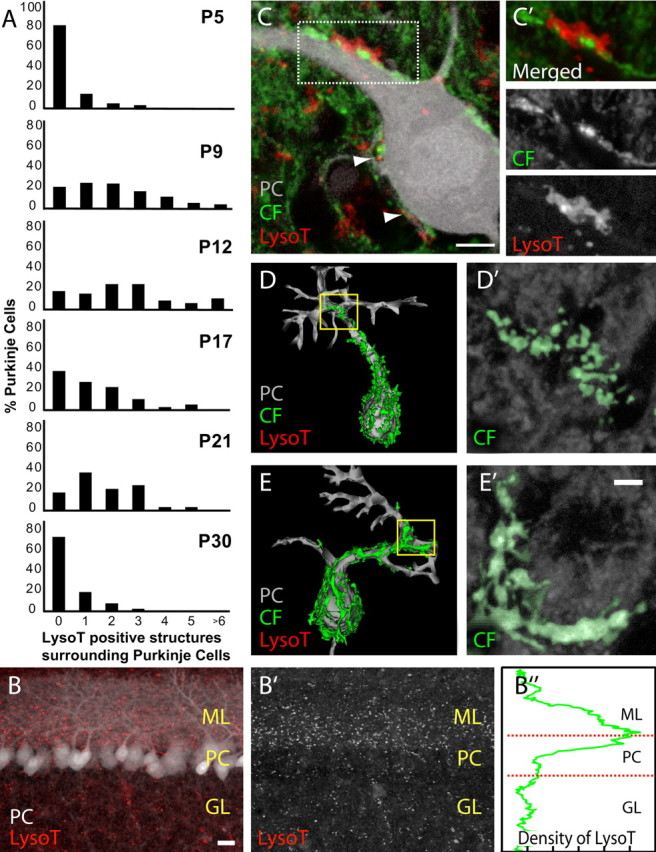
Lysosomal activity associated with cerebellar synapse elimination. A, During the second postnatal week of development, we observed an increase in lysosomal activity around developing Purkinje cells (PCs; gray) that diminished by the fourth week of development. At P5 and P30, ∼80–90% of PCs revealed little or no LysoTracker (LysoT) staining. In comparison, at P9 and P12, an increase in the number of LysoT-positive structures surrounding each PC was observed. B, There is an enrichment of LysoT staining (red) at the interface between the molecular layer (ML) and PC layer of the developing cerebellum but not in the granular cell layer (GL; P9). B′, LysoT staining alone. B″, The relative density of LysoT staining in B. A large increase in density of LysoT staining is observed in the layers in which climbing fibers (CFs) are being eliminated. C–C′, LysoT staining surrounding a YFP-labeled CF closely apposed to the PC's proximal dendritic shaft and soma (arrowheads; P9). C′, A magnified view of the boxed region in C. D, E, CF reconstructions revealed two morphologies. D, A reconstruction of the same PC and CF shown in C. D′, A branch of the CF's terminal arbor (yellow box), which appears fragmented. E, In comparison, a CF on an adjacent PC appears more continuous. E′, A branch of this CF's terminal arbor (yellow box). Both the CFs in D′ and E′ have been manually pseudocolored green on monochromatic confocal panels for better visualization. Scale bars: B, 20 μm; C, 5 μm; E′ (for D′ and E′), 2 μm.
Discussion
We show that the digestive mechanisms known as autophagy and heterophagy are involved in the removal of motor axons during postnatal development of the neuromuscular system. The digestion of axonal remnants induces a local increase of lysosomal activity both within retreating axons and their sheathing glial cells. We found that this activity can be highlighted using LysoTracker, a fluorescent vital dye that is trapped by and fluoresces strongly in acidic compartments such as lysosomes. We also assayed the developing cerebellum at the time that climbing fibers are eliminated. Here too we observed a transient increase in LysoTracker staining that closely paralleled climbing fiber loss. Our findings suggest that probes for increased lysosomal activity may generally mark retreating axons during development.
Our work provides several insights into the process of axon removal during development. First, the presence of LysoTracker surrounding the tips of retreating axons is consonant with previous results of a local piecemeal dismantling of axons by nearby glial cells (Bishop et al., 2004). This idea contrasts with the previous suggestions of wholesale branch degeneration (Rosenthal and Taraskevich, 1977) or absorption of axonal material in a strictly retractive process (Riley, 1981). The upregulation of heterophagic (Figs. 3, 4) and perhaps autophagic (Fig. 2C) mechanisms in glial cells near retreating axons suggests that a significant amount of axonal material is isolated and digested by Schwann cells, rather than integrated into the glial cytoplasm. Our previous work showed that glial cells near retreating fluorescent axons sometimes became fluorescent themselves, suggesting that at least some soluble material from axosomes may end up in the glial cytoplasm (Bishop et al., 2004). Perhaps different axonal components are processed differently in glia.
Second, some axonal material appears to be digested within the axon itself. We observed accumulations of LysoTracker-positive material inside retreating axons (Fig. 1B), and this was consistent with ultrastructural evidence of axonal autolysosomes (Fig. 3D–I). Axonal autophagy may explain the striking atrophy of axons that often precedes synaptic detachment (Bernstein and Lichtman, 1999). These results thus implicate a role for autophagy in developmental axon remodeling. The link between developmental axonal refinements and autophagy is strengthened by our observations of a transient delay in the clearance of axonal material in homozygous Cln3Δex7/8 mice. Studies show that although lysosomal enzyme activity levels in homozygous Cln3Δex7/8 mice are normal, a molecular defect in membrane-trafficking leads to a partially deficient autophagic pathway (Fig. 5) (Cao et al., 2006). This partial deficiency may explain why synapse elimination in homozygous Cln3Δex7/8 mice is resolved by P14. Furthermore, many lines of evidence suggest that compensatory changes occur when particular degradative pathways are disturbed. These include upregulation of lysosomal enzymes (Mitchison et al., 1999) as well as potentially increasing trafficking of degradative organelles through other uninhibited pathways. For example, studies show that when the ubiquitin-proteasome system is disrupted, autophagy compensates by upregulating digestion of intracellular protein aggregates (Pandey et al., 2007).
The intracellularly sequestered degradative mechanisms used for axon clearance seem distinct from the extracellular proteolytic mechanisms that appear to play important roles during an earlier stage of synapse elimination when axons are thought to detach from the postsynaptic site (Akaaboune et al., 1998; Nelson et al., 2003; VanSaun et al., 2003). Once axons have detached, they become entirely contained within glial cells and would seem to be less vulnerable to enzymes with extracellular modes of action.
Based on the results presented here, we formulate a model of how developing motor axon branches might be removed. This sequential process involves a reiterating cycle that progresses in a distal-to-proximal direction (Fig. 7A). Once an axon is disconnected from the postsynaptic site, autophagy is initiated at its tip. The axon then begins to shed axosomes, which are digested in the adjacent Schwann cell cytoplasm. These events result in the loss of the most distal segment of the axon, leaving behind a Schwann cell that no longer sheaths an axon. This Schwann cell now situated just distal to the axon tip continues to digest shed axosomes (Fig. 7B). The proximal stump of the axon is sheathed by a different glial cell. Our data suggest that the whole process is then repeated on the more proximal segment. What remains unknown is the fate of the Schwann cells after an axon has retreated. In adult animals, we observe only one chain of Schwann cells leading to a neuromuscular junction. Thus the glia associated with the lost axons must be removed. However, their ultimate fate is not presently known.
Figure 7.

Model for lysosomal activity associated with axon pruning. A, We propose a recurring cycle of events, in which axonal digestion starts at the distal axon tip with intra-axonal autophagy (red dots in axon), followed by axosome shedding and glial heterophagy (red dots in Schwann cell). As axosomes are shed, intra-axonal lysosomes are reduced in number. At the same time, the next cycle of digestion is initiated in a more proximal position, which might coincide with the next glial cell situated proximally. The fate of the “orphaned” glial cell (dotted) is not known. B, Glial cells increase their lysosomal activity as they digest the remaining remnants of the disappearing axon (P13). This micrograph illustrates step 3 in the model presented in A. Scale bar, 5 μm.
A number of lines of evidence suggest that synapse elimination in the peripheral neuromuscular system is analogous to events occurring in the developing CNS (Tapia and Lichtman, 2008). We investigated the possibility that climbing fiber branch removal in the developing cerebellum might occur by mechanisms that are similar to those that remove axons in the neuromuscular system. At precisely the developmental stage and site of climbing fiber elimination, we observed an increase in lysosomal activity in Bergmann glia that was highly reminiscent of the labeling we described in Schwann cells.
In conclusion, we used a functional marker of lysosomal activity to highlight axons undergoing branch withdrawal during development. Using this method, we characterized the roles of heterophagic and autophagic mechanisms in glia and axons leading to axon removal. Although at present only several areas of the brain are known with certainty to undergo developmental reorganization, many neurobiologists suspect that axonal reorganization may be a general feature of mammalian development (Huttenlocher and Dabholkar, 1997; Lichtman and Colman, 2000). Having a means of marking where developmental reorganization is occurring could provide a wealth of information about where and when experience-based alterations in brain connectivity are manifest.
Footnotes
This work was supported by the National Institutes of Health Grants R01NS020364-25 (J.W.L.) and 1R15NS048055 (D.L.B.). J.W.S. received support from the Alpha Omega Alpha Carolyn L. Kuckein Medical Student Fellowship. T.M. acknowledges support from the Alexander von Humboldt Foundation, the Institute of Advanced Study–Technical University Munich, the Deutsche Forschungsgemeinschaft, and the Center of Integrated Protein Science Munich. Y.C. received support from Batten Disease Support and Research Association. We thank N. Mizushima and RIKEN for GFP-LC3 mice, members of the W. Regehr lab for cerebellar acute section techniques, and Sara Haddad, Kate Mahoney, and Edith Robbins for mouse facility care. We acknowledge constructive suggestions and help from our laboratory colleagues.
References
- Akaaboune M, Hantaï D, Smirnova I, Lachkar S, Kapsimali M, Verdière-Sahuqué M, Festoff BW. Developmental regulation of the serpin, protease nexin I, localization during activity-dependent polyneuronal synapse elimination in mouse skeletal muscle. J Comp Neurol. 1998;397:572–579. doi: 10.1002/(sici)1096-9861(19980810)397:4<572::aid-cne9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Arsenault D, Zhang ZW. Developmental remodeling of the lemniscal synapse in the ventral basal thalamus of the mouse. J Physiol. 2006;573:121–132. doi: 10.1113/jphysiol.2006.106542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Local interneurons regulate synaptic strength by retrograde release of endocannabinoids. J Neurosci. 2006;26:9935–9943. doi: 10.1523/JNEUROSCI.0958-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein M, Lichtman JW. Axonal atrophy: the retraction reaction. Curr Opin Neurobiol. 1999;9:364–370. doi: 10.1016/s0959-4388(99)80053-1. [DOI] [PubMed] [Google Scholar]
- Bishop DL, Misgeld T, Walsh MK, Gan WB, Lichtman JW. Axon branch removal at developing synapses by axosome shedding. Neuron. 2004;44:651–661. doi: 10.1016/j.neuron.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, Cotman SL. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. 2006;281:20483–20493. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Developmental remodeling of the retinogeniculate synapse. Neuron. 2000;28:955–966. doi: 10.1016/s0896-6273(00)00166-5. [DOI] [PubMed] [Google Scholar]
- Cotman SL, Vrbanac V, Lebel LA, Lee RL, Johnson KA, Donahue LR, Teed AM, Antonellis K, Bronson RT, Lerner TJ, MacDonald ME. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet. 2002;11:2709–2721. doi: 10.1093/hmg/11.22.2709. [DOI] [PubMed] [Google Scholar]
- Crepel F, Mariani J, Delhaye-Bouchaud N. Evidence for a multiple innervation of Purkinje cells by climbing fibers in the immature rat cerebellum. J Neurobiol. 1976;7:567–578. doi: 10.1002/neu.480070609. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke S, Reichenbach A. Quantitative-morphometric aspects of Bergmann glial (Golgi epithelial) cell developments in rats. Anat Embryol. 1987;177:183–188. doi: 10.1007/BF00572543. [DOI] [PubMed] [Google Scholar]
- Hirata K, Kawabuchi M. Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc Res Tech. 2002;57:541–547. doi: 10.1002/jemt.10108. [DOI] [PubMed] [Google Scholar]
- Holtzman E, Novikoff AB. Lysomes in the rat sciatic nerve following crush. J Cell Biol. 1965;27:651–669. doi: 10.1083/jcb.27.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron. 1997;18:71–79. doi: 10.1016/s0896-6273(01)80047-7. [DOI] [PubMed] [Google Scholar]
- Lichtman JW. The reorganization of synaptic connexions in the rat submandibular ganglion during post-natal development. J Physiol. 1977;273:155–177. doi: 10.1113/jphysiol.1977.sp012087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron. 2000;25:269–278. doi: 10.1016/s0896-6273(00)80893-4. [DOI] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, Lichtman JW. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Lohof AM, Delhaye-Bouchaud N, Mariani J. Synapse elimination in the central nervous system: functional significance and cellular mechanisms. Rev Neurosci. 1996;7:85–101. doi: 10.1515/revneuro.1996.7.2.85. [DOI] [PubMed] [Google Scholar]
- Mitchison HM, Bernard DJ, Greene NDE, Cooper JD, Junaid MA, Pullarkat RK, de Vos N, Breuning MH, Owens JW, Mobley WC, Gardiner RM, Lake BD, Taschner PE, Nussbaum RL. Targeted disruption of the Cln3 gene provides a mouse model for Batten Disease. Neurobiol Dis. 1999;6:321–334. doi: 10.1006/nbdi.1999.0267. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PG, Jia M, Li MX. Protein kinases and hebbian function. Neuroscientist. 2003;9:110–116. doi: 10.1177/1073858403252226. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM. The lysosomal system in neuronal cell death: a review. Ann N Y Acad Sci. 1993;679:87–109. doi: 10.1111/j.1749-6632.1993.tb18291.x. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Purves D, Lichtman JW. Elimination of synapses in the developing nervous system. Science. 1980;210:153–157. doi: 10.1126/science.7414326. [DOI] [PubMed] [Google Scholar]
- Redfern PA. Neuromuscular transmission in new-born rats. J Physiol. 1970;209:701–709. doi: 10.1113/jphysiol.1970.sp009187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley DA. Ultrastructural evidence for axon retraction during the spontaneous elimination of polyneuronal innervation of the rat soleus muscle. J Neurocytol. 1981;10:425–440. doi: 10.1007/BF01262414. [DOI] [PubMed] [Google Scholar]
- Rosenthal JL, Taraskevich PS. Reduction of multiaxonal innervation at the neuromuscular junction of the rat during development. J Physiol. 1977;270:299–310. doi: 10.1113/jphysiol.1977.sp011953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scelfo B, Strata P, Knöpfel T. Sodium imaging of climbing fiber innervation fields in developing mouse Purkinje cells. J Neurophysiol. 2003;89:2555–2563. doi: 10.1152/jn.00884.2002. [DOI] [PubMed] [Google Scholar]
- Tapia JC, Lichtman JW. Synapse elimination. In: Squire LR, Berg D, Bloom FE, du Lac S, Ghosh A, Spitzer NC, editors. Fundamental neuroscience. Burlington, MA: Elsevier; 2008. p. 469. [Google Scholar]
- VanSaun M, Herrera AA, Werle MJ. Structural alterations at the neuromuscular junctions of matrix metalloproteinase 3 null mutant mice. J Neurocytol. 2003;32:1129–1142. doi: 10.1023/B:NEUR.0000021907.68461.9c. [DOI] [PubMed] [Google Scholar]
- Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts RJ, Schuldiner O, Perrino J, Larsen C, Luo L. Glia engulf degenerating axons during developmental axon pruning. Curr Biol. 2004;14:678–684. doi: 10.1016/j.cub.2004.03.035. [DOI] [PubMed] [Google Scholar]
- Yue Z, Wang QJ, Komatsu M. Neuronal autophagy: going the distance to the axon. Autophagy. 2008;4:94–96. doi: 10.4161/auto.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Diwu Z, Mao F, Leung W, Haugland RP. Novel fluorescent acidic organelle-selective dyes and mitochondrion-selective dyes that are well retained during cell fixation and permeabilization. Mol Biol Cell. 1994;5:113a. [Google Scholar]
- Zuo Y, Lubischer JL, Kang H, Tian L, Mikesh M, Marks A, Scofield VL, Maika S, Newman C, Krieg P, Thompson WJ. Fluorescent proteins expressed in mouse transgenic lines mark subsets of glia, neurons, macrophages, and dendritic cells for vital examination. J Neurosci. 2004;24:10999–11009. doi: 10.1523/JNEUROSCI.3934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]