

Abstract
Purpose
Kidney stone formation is associated with the deposition of hydroxyapatite (HA) either as sub-epithelial plaques or tubular deposits in the renal papillae. We investigated the effect of renal epithelial exposure to HA crystals in vitro to develop an insight into the pathogenesis of kidney stones.
Materials and methods
NRK52E cells were exposed to 67 μg/cm2 or 133 μg/cm2 of HA or calcium oxalate monohydrate crystals. In some studies cells were also exposed to crystals from the basal side. After exposure of 3 or 6 hours media were analyzed for, lactate dehydrogenase (LDH), 8-isoprostane (8-IP) and hydrogen peroxide (H2O2). Media collected after cells’ exposure on the apical side was also analyzed for the production of monocyte chemoattractant-1 (MCP-1) and prostaglandin E2 (PGE2). Cells were stained with DAPI to determine apoptotic activity and examined by SEM to observe crystal-cell interaction.
Results
Cell exposure to HA resulted in the production of H2O2 and 8-IP as well as release of LDH. Apical exposure appeared more provocative and injurious than the basal exposure. An exposure to HA for 6 hours, resulted in increased apoptotic activity. Apical exposure also resulted in increased production of MCP-1 and PGE2.
Conclusion
Cell exposure to HA crystals induces oxidative stress, lipid peroxidation, and caused upregulation of mediators of inflammation that may be responsible for the inflammation in kidneys of stone patients associated with tubular deposition of HA. They may also play a role in eruption of subepithelial Randall’s plaques to the papillary surface.
Keywords: Apatite, Calcium Phosphate, Reactive Oxygen Species, Kidney Stones, Inflammation, Monocyte Chemoattractant Protein-1, Nephrolithiasis
Introduction
Morphological studies have provided evidence that idiopathic calcium oxalate (CaOx) stones develop on subepithelial plaques of calcium phosphate (1, 2). Calcium phosphate precipitates as carbonate apatite (HA) in basement membrane of the loops of Henle and from there extends outwards through the renal interstitium to just under the papillary surface urothelium (2). In other types of kidney stones including cystine, brushite, distal tubular acidosis, and CaOx secondary to intestinal bypass surgery, inner medullary collecting ducts are shown to be plugged with HA. Thus in the course of stone formation, renal epithelial cells become exposed to HA either on the luminal or the basal side.
We have shown earlier that renal epithelial cells react to the presence of brushite and CaOx crystals in time and concentration dependent manner (3, 4). Longer exposure to high concentrations of brushite or CaOx crystals is injurious to the renal epithelial cells. Crystal induced cell injury is suggested to play a significant role in the development of kidney stones by providing the sites for crystal attachment and retention within the kidneys (5, 6) and promoting subsequent eruption of retained crystals to papillary surfaces for the development of stone nidus (7). However similar studies have not been performed to investigate renal epithelial response to apatite crystals.
To determine the renal epithelial reaction to an exposure to HA we investigated the interaction between renal epithelial cells in culture and HA crystals. We exposed a renal epithelial cell line NRK52E, which is derived from rats, to two different amounts of HA crystals and investigated the release of LDH into the medium as a marker of cell injury. Studies of oxalate and CaOx crystal induced injury of renal epithelial cells have shown them to be under oxidative stress, which eventually leads to lipid peroxidation (8). Therefore we also examined the production of hydrogen peroxide (H2O2), one of the reactive oxygen species (ROS), and 8-isoprostane (8-IP), a marker for oxidative stress. Since renal epithelial cells exposed to CaOx crystals produce significant amounts of inflammatory mediators, monocyte chemoattractant protein-1 (MCP-1) (4) and prostaglandin-E2 (PGE2) (5, 12) by the exposed cells we also investigated their production. As mentioned above, Randall’s plaques start in the basement membrane and renal interstitium resulting in the tubular epithelial exposure from the basal side. Therefore we also investigated renal epithelial response to the crystals present on the basal side. Our results indicate that HA crystals, when present on either side of the renal epithelial cells, are injurious and reactive oxygen species are most likely involved in the HA crystal induced injury and inflammation.
Materials and Methods
NRK52E (ATCC, Gaithersburg, MD, Cat # CRL-1571) were maintained in growth media (500 ml DMEM/F-12, 50 ml of newborn calf serum, 10 ml of antibiotic-antimytotic) in 75 cm2 Corning T-Flasks. The cells were sub-cultured by disassociation with 0.05% trypsin and 0.25% ethylene-di-amine-tetra-acetic acid (EDTA) and seeded onto cell culture inserts (BD Falcon, Fisher Scientific, Cat # 08–771) in a 6 well Nunc tissue culture plates. After reaching confluence, the cells were weaned from growth media to acclimatization media (500 ml DMEM/F-12, 10 ml of antibiotic-antimytotic solution, 5 ml of Insulin/Transferrin/Selenium mix, 1 ml of Hydrocortisone, 3.4 ml of Triiodo-L-Thyronine, 1mL of Prostaglandin E1) for 8–12 hours.
We have shown earlier that renal epithelial cells react to the presence of brushite and CaOx crystals in time and concentration dependent manner (3, 4). Cells responded in consistent manner to the crystals at concentrations of 67 μg/cm2 and 133 μg/cm2 for 3 or 6 hours. Based on the results of these studies, we exposed the cells to 67 μg/cm2 and 133 μg/cm2 HA (Sigma-Aldrich, St. Louis, MO, Cat # 21223) or CaOx monohydrate (BDH Limited, Poole, England, Cat # 27609) crystals for 3 or 6 hours. Before exposure the acclimatization media was removed and fresh acclimatization media was added with either COM or HA crystals from the top or below the insert for apical or basal exposure. Media was collected for lactate dehydrogenase (LDH), hydrogen peroxide (H2O2) and 8-Isoprostane (8-IP). Media from the cells exposed apically to HA was also collected for prostaglandin E2 (PGE2) and monocytes-chemoattractant protein-1 (MCP-1) analysis.
Microscopic Analysis
Calcium oxalate crystals have been shown to induce apoptosis in the exposed renal epithelial cells. Therefore we evaluated the morphology of crystal exposed cells in culture by fluorescence microscopy following staining with 4′–6-diamidino-2-phenylinedole (DAPI). DAPI permeates the plasma membrane and stains the chromatin. Viable cells display normal nuclear size and blue fluorescence, while apoptotic cells show condensed chromatin and fragmented nuclei.
Scanning electron microscopy (SEM) was also utilized to observe the crystal cell interaction. Cells were grown in inserts, exposed to 133μg/cm2 of HA for 3 or 6 hours and processed and examined as described in our earlier publications (9).
Lactate Dehydrogenase (LDH)
Media was aliquoted to designated wells of a 96 well plate (Fisher Scientific, Norcross, GA Cat # 21–377–205). The CytoTox 96 Non-Radioactive Cytotoxicity assay kit (Fisher Scientific, Norcross, GA, Promega Cat # PR-G1780) was used to determine LDH percent release. Substrate (supplied with kit) was added to all samples, positive control (MDCK cells lysed with lysis solution-supplied with kit), and blanks (acclimazation media). The plate was incubated at room temperature for 30 minutes in the dark. Stop solution (supplied with kit) was added to all samples, positive control, and blanks. Optical density absorbency was read at 490 nm on a Bio-Rad 3550 microplate reader (Bio-Rad, Hercules, CA).
Reactive Oxygen Species, Hydrogen Peroxide(H2O2)
After incubation period, media was removed and placed into microcentifuge tubes. Hydrogen peroxide quantification was carried out using a colormetric assay obtained from Pierce (PeroXoquant Quantitative Peroxide Assay Kit: Cat # 23280). An aliquot was taken (amount specific to manufacture’s protocol) and a 1:100 dilution and 1:1 dilution of sample was first measured according to assay protocol to determine optimum sample volume for parameters of standard curved set by kit protocol. Once determination was made for the necessary dilution of sample, the following kit protocol was followed. In a 96-well plate, 10 μl of working reagent (WR: diluted to necessary concentration from stock supplied from kit) was added to 100 μl of sample, standard and reagent blank. The plate was mixed using a plate mixer and incubated at room temperature for up to 20 minutes (or until a sufficient color of blue was seen in highest concentration of standard). Absorbance was measured at 570 nm (Assay wavelength range 560–600 nm) using a Bio-Rad microplate reader (Model 3550). The concentration of hydrogen peroxide in the sample was calculated by reference to its assay absorbance compared to the standard curve.
Lipid Peroxidation - 8-IP
Determination of lipid peroxidation was carried out using a kit from Oxford Biomedical Research (Urinary Isoprostane: Cat # EA85). Briefly, after incubation with COM or HA, media was collected. Media samples were spun to pellet solid materials and a 100 μl of standard (supplied by kit), samples, and reagent blank (Enhanced dilution buffer supplied by kit) was added to designated wells in a 96-well plate (supplied by kit). 100 μl of diluted 15-isoprostane F2t HRP conjugate (supplied by kit) was added to all well except reagent blank. Plate was incubate at room temperature for 2 hours. After incubation, plate was washed 3 times with wash buffer (supplied by kit). 200 μl of substrate (supplied by kit) was added to each well and incubated at room temperature for 20–40 minutes (Or until appreciable blue hue was observed in standard blank). Addition of 3 M sulfuric acid was added to each well to stop reaction. Plate was then read using a Bio-Rad 3550 microplate reader at 450 nm. The concentration of isoprostane in the sample was calculated by reference to its assay absorbance compared to the standard curve.
Inflammatory Response - PGE2 assay
PGE2 quantification was carried out using an assay kit obtained from GE Healthcare (Cat # RPN222). First pipette 100 μl of diluted assay buffer into the non-specific binding wells (NSB). Then pipette 50 μl of diluted assay buffer in to the zero standard wells. Next 50 μl of serial diluted standards (diluted with culture media) and samples were pipetted into designated wells. 50 μl of of diluted antibody (supplied with kit) was added to all wells except blank and NSB wells. Next 50 μl of diluted conjugate (supplied with kit) was added to all wells except blank. Plate was incubated at room temperature for 1 hour on a microplate shaker. The wells were then aspirated and washed four times with wash buffer (supplied with kit). Immediately 150 μl of enzyme substrate (supplied with kit) was added to all wells. Plate was covered and incubated at room temperature for 30 minutes on a microplate shaker. After incubation, 100 μl of 1 M sulfuric acid was added to all wells and the plate was read on a Bio-Rad 1550 microplate reader at 450 nm within 30 minutes of addition of sulfuric acid. Data was calculated by the percent bound for each standard and sample using the %B/B0.
Inflammatory Response, Monocyte Chemoattractant Protein-1 (MCP-1)
To quantify the amount of MCP-1 protein present at the end of incubation with COM or HA, we used the Pierce MCP-1 Rat ELISA (Cat # ERMCP1). Briefly, 50 μl of standards and samples were added to designated wells of a 96 well plate (supplied with kit) and then incubated at room temperature for 1 hour. The plate was washed three times with wash buffer (supplied with kit) and 50 μl of biotinylated antibody reagent (supplied with kit) was added to all wells and then incubated at room temperature for 1 hour. Plates were then washed three times with wash buffer and 100 μl of prepared streptavidin-HRP solution (supplied with kit) to each well. Then incubate plate at room temperature for 30 minutes. After incubation stop solution (supplied with kit) was added to each well and the plate was read at 450 nm using Bio-Rad 1550 microplate reader.
Statistical Analysis
All analysis for statistical significance was carried out using GraphPad Prism software. A student t-test and descriptive analysis (Two-way ANOVA) between control vs COM or HA exposed cells and COM or HA vs duration of exposure was carried out to determine significance between all variables.
Results
Microscopic Observations
We examined the nuclear morphologies of exposed and non-exposed cells by staining with DAPI, a fluorescent DNA-binding agent. Nuclei of many cells exposed to either concentrations of HA for 3 or 6 hours showed typical morphological features of apoptosis (Figs. 1A, B, C). The nuclei had highly condensed chromatin and many of them had broken into apoptotic bodies. Control cells did not show any signs of apoptosis.
Figure 1.
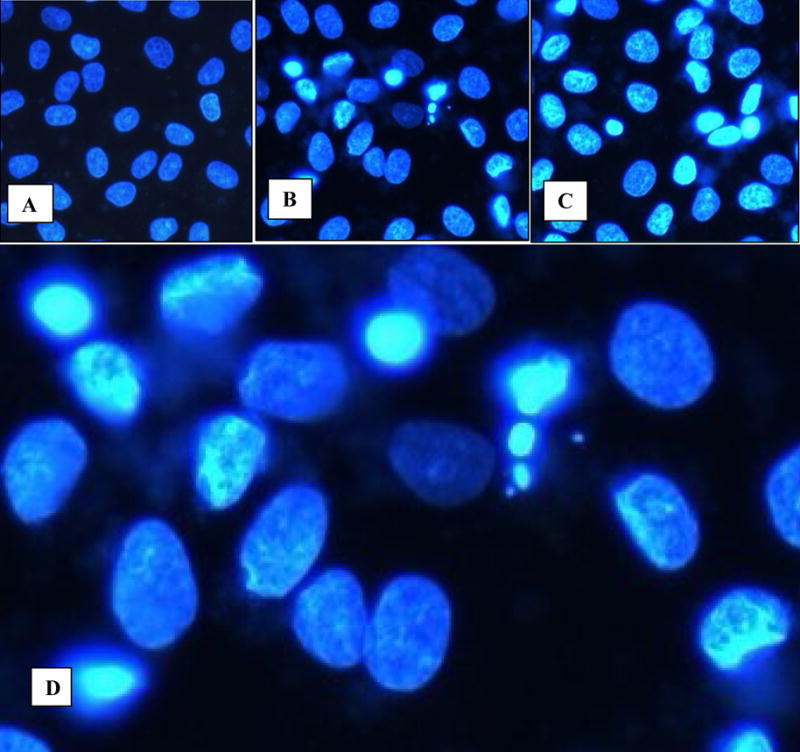
Cells in culture examined by fluorescence microscopy following staining with 4′–6-diamidino-2-phenylinedole. A. Normal NRK52E cells showing normal nuclei. Reduced from X20. B, C. Cells exposed to HA for 3 and 6 hours show condensed chromatin. Reduced from X20. D. High mag of Fig 1B showing both normal appearing nuclei as well as nuclei with condensed chromatin, and formation of apoptotic bodies. Reduced from X40.
Scanning electron microscopy of the cells in culture showed normal squamous epithelial morphology with short stubby microvilli that were sparsely distributed on cell surfaces (Fig. 2A, B). Exposed cells showed clumps of crystals (Fig 2C) being probed by slender finger like cellular extensions (Fig. 2D) that looked similar to the microvilli but were much longer. Some cells appeared to endocytose the crystal mass (Fig. 2E), with the cell body growing over them. Still other cells appeared to have already taken the crystals in (Fig. 2F).
Figure 2.

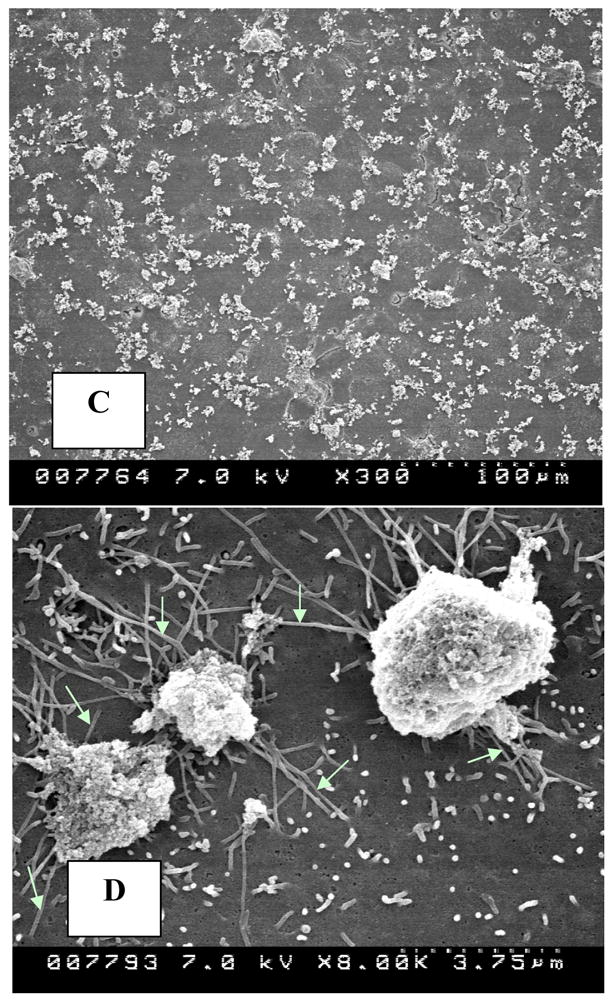

Cells in culture examined by scanning electron microscopy. A, B. Normal epithelial cells showing squamous epithelial surface morphology with sparse stubby microvilli. C. Cells exposed to 133μg/cm2 HA show crystal clumps distributed on cell surface. D. Slender extensions of cells appear to probe crystal clumps (white arrows). E. A cell is endocytosing a crystal clump with cell margin growing over the crystals. F. Crystal is endocytosed. Edge of the endocytosing cell (arrows) can still be distinguished from the underlying cell. (Magnifications-Figure 2A X1500, 2B X8000, 2C X300, 2D X8000, 2E X4500K, 2F X7000)
Lactate Dehydrogenase (LDH) Release
Figures 3A and B show LDH release by cells as percent increase against each control on exposure at apical or basal side to 67μg/cm2 or 133μg/cm2 of HA or CaOx monohydrate (COM) crystals respectively, for 3 or 6 hours. Exposure to both HA and COM from both the apical and basal side resulted in significant LDH release into the medium. LDH release was however, generally higher when cells were supplied the crystals from the apical side than when cells were exposed from the basal side. Percentage increase in LDH release was time and concentration dependent in case of apical exposure only.
Figure 3.

LDH release by cells exposed to HA (A) or COM (B) crystals. Cells were exposed to 67 or 133μg/cm2 of crystals for 3 or 6 hours from the apical or basal side. Both apical and basal treatments resulted in significant increase in LDH release. Apical exposure appears to be more stimulating.
Production of the Reactive Oxygen Species, H2O2
Exposure to both HA and COM crystals resulted in the production of significantly more H2O2 than the controls except for a 3 hour exposure to 67μg/cm2 of HA from the basal side. Exposure to HA produced more H2O2 when crystals were supplied from the apical side except for an exposure to 133μg/cm2 when basal exposure produced significantly more H2O2 than the apical exposure (Fig. 4A). In the case of COM crystals exposure of cells from the basal side produced significantly more H2O2 than when cells were exposed from the apical side except an a 3 hours exposure to 67μg/cm2 of COM from the basal side (Fig. 4B).
Figure 4.

Production of hydrogen per oxide by cells exposed to HA (A) or COM (B) crystals. Cells were exposed to 67 or 133μg/cm2 of crystals for 3 or 6 hours from the apical or basal side. Both apical and basal treatments resulted in significant production of H2O2.
Production of 8-Isoprostane (8-IP)
As shown in Figures 5A and B exposure to both HA and COM crystals from either the apical or basal side resulted in the production of 8-IP. A 3 hour exposure to both the 67μg/cm2 or 133μg/cm2 of HA resulted in significant increase in 8-IP production. But there was no significant difference in 8-IP production whether cells were exposed to HA crystals from the bottom or the top. The 6 hour exposure to HA at 67μg/cm2 from the basal side resulted in significant increase in 8-IP production. On the other hand at 133μg/cm2 of HA for 6 hours, significant increase was seen when cells were exposed from the apical side.
Figure 5.

Production of 8-isoprostane by cells exposed to HA (A) or COM (B) crystals. Cells were exposed to 67 or 133μg/cm2 of crystals for 3 or 6 hours from the apical or basal side. Both apical and basal treatments resulted in significant production of 8-IP.
In case of COM crystals, exposure from the basal side was more reactive. There was significantly more 8-IP produced when cells were exposed from the basal side to 67μg/cm2 or 133μg/cm2 COM crystals. Only an exposure to 133μg/cm2 from the apical side produced significantly higher amount of 8-IP.
Inflammatory Response
Earlier studies have shown that an exposure to CaOx crystals provokes an inflammatory response from the renal epithelial cells. Cells exposed to the crystals produce MCP-1 as well as PGE2 (Figs. 6A, B). Results of the current study also show increased production of MCP-1 and PGE2 by NRK52E cells exposed apically to 67μg/cm2 or 133μg/cm2 COM crystals for 6 hours. In case of MCP-1, production was dose dependent. Exposure to 133μg/cm2 COM or HA crystals produced significantly more MCP-1 than an exposure to 67μg/cm2. HA crystals appear more reactive provoking significantly more MCP-1 than COM crystals at the same concentrations.
Figure 6.

A. Production of monocyte chemoattractant protein-1 (MCP-1) by cells exposed to hydroxyapatite HA or calcium oxalate monohydrate COMcrystals. Cells were exposed to 67 or 133μg/cm2 of crystals for 6 hours from the apical side. The treatments resulted in significant production of MCP-1. B. Production of prostaglandin E2 (PGE-2) by cells exposed to hydroxyapatite HAor calcium oxalate monohydrate (COM) crystals. Cells were exposed to 67 or 133μg/cm2 of crystals for 6 hours from the apical side. The treatments resulted in significant production of PGE-2.
Discussion
Even though CaOx is the majority crystal in most kidney stones, almost all stones contain some CaP, mostly in the form of hydroxyapatite (HA). These observations are not surprising since urine is often supersaturated with HA (10). HA crystals are frequently encountered in the urine and are a good substrate for the nucleation of CaOx. Stone formation however, does not only require crystal formation but also their retention within the kidneys (11). There are two major theories about crystal retention. According to one theory, stone formation begins with deposition of apatite crystals in the basement membrane of the loops of Henle (1, 2). The apatite crystal deposits then grow through the renal interstitium to subepithelial location in the renal papillae. These papillary subepithelial deposits called Type 1 Randall’s plaques become the nidi for the development of CaOx stones. Because the initial event is in the interstitium as opposed to moving urine in the renal tubules, attachment is not a factor for crystal retention. An alternate theory of stone formation considers stone formation as an intratubular event requiring crystal attachment to the cell surfaces and some form of cell membrane alteration or injury for the retention of crystals (5, 6).
A variety of renal epithelial cells including LLC-PK1, MDCK, HK-2 (human kidney), NRK (normal rat kidney) and RPTEC (human renal proximal tubular epithelial cells), when exposed to high levels of oxalate, or brushite or CaOx crystals, show signs of membrane damage, and release LDH and enzymes such as γ-glutamyl trasnspeptidase, and N-acetyl-β-glucoseaminidase (4, 6, 12, 13).
It has also been proposed that exposure of renal epithelial cells to higher than normal levels of calcium and oxalate can perturb the plasma membrane, causing lateral and trans-membrane migration of phospholipids, sequestering them in specific domains. Migration of the acidic phospholipids such as phosphatidylserine from the inner leaflet of the plasma membrane to the outside (14) promotes adhesion of CaOx crystals to the epithelial cells. Crystal attachment to the inner medullary collecting duct cells has also been correlated with membrane fluidity (14).
Recent studies have provided evidence that both Ox and CaOx crystals selectively activate p38 MAPK signal transduction pathways (15). In addition p38 MAPK is essential for re-initiation of the induced DNA synthesis. Ox exposure also causes modest activation of JNK as determined by c-Jun phosphorylation. Apparently the renal epithelial response to oxalate involves signal transduction via MAP kinases, similar to the cellular responses to many other challenges. Cytosolic phospholipase A2 (cPLA2) is released upon the activation of MAP kinases and translocated to the cell membrane. cPLA2 preferentially hydrolyses arachidonoyl phospholipids generating a number of byproducts including arachidonic acid and lysophospolipids. Exposure of MDCK cells to oxalate produces a time and concentration dependent increase in cPLA2 activity (16). Inhibition of cPLA2 activity blocks the oxalate-induced upregulation of Egr-1, c-jun and c-myc genes.
A limited analysis of renal epithelial cell response to HA exposure has also been carried out. The HA has been shown to adhere to clumps of cells in primary cultures of rat renal papillary collecting tubule (RPTC) rather than the cells in the monolayer (14). It was concluded that a sub-lethal injury caused a loss of tight junction integrity and separation of cells exposing binding sites on cell surfaces. Results also indicate that CaOx and apatite crystals share the same binding sites. Monkey kidney epithelial cells of non-transformed BSC-1 cell line endocytose the CaOx, apatite or brushite crystals (17). The reaction of non-renal cell types to HA has also been investigated because the deposition of calcium pyrophosphate dihydrate (CPPD) and a variety of other calcium phosphates including hydroxyapatite, carbonate apatite, octacalcium phosphate and tricalcium phosphate collectively termed basic calcium phosphate (BCP), causes many diseases of the joints (18). BCP and to some extent CPPD crystals induce mitogenesis, stimulate production of prostaglandin E2 (PGE2), activate phospholipase C, promote the synthesis of metalloproteinases (MMP’s) and induce proto-oncogenes c-fos and c-myc.
The results of the current study show that exposure to apatite crystals, the most common crystals found in the human urine, stones and the Randall’s plaques injures the renal epithelial cells in culture as demonstrated by the release of LDH into the medium from the NRK52E cells. There were signs of apoptotic cell death. On apical exposure cells endocytosed the crystals present on the surface. Thus the cellular response to HA crystals was similar to that of the COM crystals (6, 9, 17), which have been shown to be endocytosed, induce apoptosis and cause cell injury. Exposure to HA on both the apical and basal surfaces resulted in significantly increased release of LDH. However the release was markedly higher when cells were exposed to the HA and COM crystals from the apical side than when exposed on the basal side. More LDH was released when exposed to a crystal concentration of 133μg/cm2 for longer duration of 6 hours.
Results also show the generation of free radicals during the interaction between HA crystals and epithelial cells as evidenced by the production of H2O2. The amount of H2O2 produced was significantly higher when cells were exposed to higher amounts of HA or COM crystals for a longer duration. H2O2 has been shown to cause lipid peroxidation, DNA damage and in the end, cell death. 8-Isoprostane is produced by the random oxidation of tissue phospholipids by reactive oxygen species. Our data show that 8-IP was elevated in cells exposed to 67μg/cm2 or 133μg/cm2 HA or COM. With respect to the production of 8-IP, there were similar cell responses to both HA and COM crystals. However basal exposure appeared to be more effective than the apical exposure except in the case of 133μg/cm2 HA crystals when significantly less 8-1P was produced after basal exposure than after the apical exposure. Exposure to CaP crystals also resulted in the production of MCP-1 and PGE2, two important mediators of inflammation. In this respect too exposure to HA crystals was similar to the brushite and CaOx crystals. Reactive oxygen species are also shown to be involved in the CaOx and brushite crystal induced production of MCP-1. MCP-1 is a major mediator of monocyte/macrophage movement to the site of inflammation. CaOx crystal deposits in both the human and rat kidneys have been shown to be surrounded by the macrophages and giant cells (5, 6).
It is apparent that both apatite and CaOx crystals are injurious to renal epithelial cells, whether supplied from the apical or basal side. In vivo animal model studies as well as human studies have also shown CaOx to be injurious to the renal epithelium. Urinary excretion of many enzymes regarded as markers of renal tubular injury is increased by rats with CaOx nephrolithiasis (8) as well as human CaOx stone formers (19). In addition CaOx nephrolithiais in both rats and humans is associated with increased urinary excretion of lipid peroxides (8, 19). Administration of antioxidants to hyperoxaluric rats leads to a decrease in renal injury, the production of lipid peroxides and CaOx crystal deposition in the kidneys indicating the involvement of reactive oxygen species in the crystal induced renal injury.
Recent morphological studies of the papillae obtained from human kidney stone patients showed that human kidneys respond differently to different type of stones (2). No morphological renal injury was detected in kidneys of idiopathic stone formers even though apatite crystals were present in their interstitium and stones were seen attached to the apatitic subepithelial Randall’s plaques. On the other hand, kidneys of brushite stone formers showed interstitial apatite deposits without any signs of cell injury while crystal filled medullary collecting ducts had extensive cell injury and interstitial fibrosis. The kidneys of intestinal bypass patients with calcium oxalate stones, on the other hand, showed only apatite crystals in their inner medullary collecting ducts and the ducts of Bellini. Tubules filled with apatite crystals showed extensive cell injury and death. Thus human renal data from stone formers indicate that Randall’s plaques in the form of interstitial apatite crystals with basal cell exposure are not noticeably injurious and inflammatory however injury and inflammation occurs when there are tubular deposits with apical cell exposure. What are the reasons for reported cellular injury in the presence of intratubular apatite crystals and the absence of injury and inflammation when apatite crystals are present as Randall’s plaque in the renal interstitium? We can only speculate at this time. Individual apatite crystals are extremely small and like all the other crystals produced in the biological systems are always coated with organic molecules. The presence of this organic coat around the crystals may interfere with the crystal cell interaction. Tubular crystals formed in the flowing urine may have a thinner coat which may allow a better cell crystal contact. In addition the intratubular crystals completely filled the inner medullary ducts and blocked the urinary movement, which may be partially responsible for the observed injury and inflammation. The crystals produced in the stationary milieu of the interstitium are likely to be slow growing and thus contain a thicker organic coating and a better barrier between the crystals and the cell membrane. We have recently shown that biological crystals obtained from the kidney stones are less injurious to the renal epithelial cells than the inorganic crystals (20). Alternatively, the interstitial crystals may provoke the production of pro-inflammatory molecules by the renal epithelium without leading to migration of inflammatory cells, the histological marker of inflammation. Thus inflammation and injury associated with sub-epithelial apatite of Randall’s plaque may not be morphologically visible. Our results presented here and elsewhere clearly show that an exposure of renal epithelial cells to CaP as well as CaOx crystals leads to the production of inflammatory molecules. This apatite induced injury and inflammation may be involved in the eruption of subepithelial plaques to the papillary surface, establishing a substrate for the deposition of CaOx and formation of kidney stones.
Acknowledgments
Supported by NIH grant # RO1 DK 065658 and UF Center for the Study of Lithiasis.
KEY OF DEFINITIONS OF ABBREVIATIONS
- HA
hydroxyapatite
- MCP-1
monocyte chemoattractant protein-1
- PGE-2
prostaglandin E-2
- CaOx
calcium oxalate
- COM
calcium oxalate monohydrate
- EDTA
ethylene-di-amine-tetra-acetic acid
- LDH
lactate dehydrogenase
- H2O2
hydrogen peroxide
- DAPI
4–6-diamidino--2-phenylenedole
- SEM
scanning electron microscopy
- 8-IP
8-isoprostane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Randall A. Papillary pathology as precursor of primary renal calculus. J Urol 1940. 1940;44:580. [Google Scholar]
- 2.Miller NL, Evan AP. Lingeman JE, Pathogenesis of renal calculi. Urologic Clinics of North America. 34:295–313. doi: 10.1016/j.ucl.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Aihara K, Byer KJ, Khan SR. Calcium phosphate induced renal epithelial injury and stone formation: involvement of reactive oxygen species. Kidney Intl. 2003;64:1283–1291. doi: 10.1046/j.1523-1755.2003.00226.x. [DOI] [PubMed] [Google Scholar]
- 4.Umekawa T, Chegini N, Khan SR. Increased expression of monocyte chemoattractant protein-1 (MCP-1) by renal epithelial cells in culture on exposure to calcium oxalate, phosphate and uric acid crystals. Nephrol Dial Transplant. 2003;18:664–669. doi: 10.1093/ndt/gfg140. [DOI] [PubMed] [Google Scholar]
- 5.Verkoelen CF, Verhulst A. Proposed mechanisms in renal tubular crystal retention. Kidney Intl. 2007;72:13–18. doi: 10.1038/sj.ki.5002272. [DOI] [PubMed] [Google Scholar]
- 6.Khan SR. CaOx crystal interaction with renal epithelium, mechanism of crystal adhesion and its impact on stone development. Urol Res. 1995;23:71–79. doi: 10.1007/BF00307936. [DOI] [PubMed] [Google Scholar]
- 7.Khan SR, Glenton PA, Byer KJ. Modeling of hyperoxaluric calcium oxalate nephrolithiasis: experimental induction of hyperoxaluria by hydroxyl-L-proline. Kidney Intl. 2006;70:914. doi: 10.1038/sj.ki.5001699. [DOI] [PubMed] [Google Scholar]
- 8.Thamilselvan S, Hackett RL, Khan SR. Lipid peroxidation in ethylene glycol induced hyperoxaluria and calcium oxalate nephrolithiasis. J Urol. 1997;157:1059–1063. [PubMed] [Google Scholar]
- 9.Khan SR, Byer KJ, Thamilselvan S, Hackett RL, McCormack WT, Benson NA, et al. Crystal-cell interaction and apoptosis in oxalate-associated injury of renal epithelial cells. J Am Soc Nephrol. 1999;10:S457–S463. [PubMed] [Google Scholar]
- 10.Tiselius HG. Is precipitation of calcium phosphate an important factor for the evelopment of calcium oxalate stones in the urinary tract. Front Biosci. 2003;8:S326–332. doi: 10.2741/1053. [DOI] [PubMed] [Google Scholar]
- 11.Kok DJ, Khan SR. Calcium oxalate nephrolithiasis, a free or fixed particle disease. Kidney Int. 1994;46:847–854. doi: 10.1038/ki.1994.341. [DOI] [PubMed] [Google Scholar]
- 12.Schepers MSJ, van Ballegooijen ES, Bangma CH, Verkoelen CF. Crystals cause acute necrotic cell death in renal proximal tubule cells. But not in collecting tubules. Kidney Intl. 2005;68:1543–1553. doi: 10.1111/j.1523-1755.2005.00566.x. [DOI] [PubMed] [Google Scholar]
- 13.Habibzadegah-Tari P, Byer K, Khan SR. Reactive oxygen species mediated calcium oxalate crystal-induced expression of MCP-1 in HK-2 cells. Urol Res. 2006;34:26–36. doi: 10.1007/s00240-005-0007-3. [DOI] [PubMed] [Google Scholar]
- 14.Mandel NS. Crystal-membrane interaction in kidney stone disease. J Am Soc Nephrol. 1994;5:S37–S45. doi: 10.1681/ASN.V55s37. [DOI] [PubMed] [Google Scholar]
- 15.Chaturvedi LS, Koul S, Sekhon A, Bhandari A, Menon M, Koul HK. Oxalate selectively activates the p38 mitoge-activated protein kinase and c-Jun N-terminal kinase signal transduction pathway in renal epithelial cells. J Biol Chemistry. 2002;277:13321–13330. doi: 10.1074/jbc.M108203200. [DOI] [PubMed] [Google Scholar]
- 16.Jonassen JA, Cao LC, Honeyman T, Scheid CR. Mechanisms mediating oxalate-induced alterations in renal cell functions. Crt Rev in Eukar Gene Expr. 2003;13:55–72. doi: 10.1615/critreveukaryotgeneexpr.v13.i1.50. [DOI] [PubMed] [Google Scholar]
- 17.Lieske JC, Walsh-Reitz MM, Toback FG. Calcium oxalate monohydrate crystals are endocytosed by renal epithelial cells and induce proliferation. Am J Physiol. 1992;262:F622–F630. doi: 10.1152/ajprenal.1992.262.4.F622. (renal Fluid Electrolyte Physiol 31) [DOI] [PubMed] [Google Scholar]
- 18.Cheung HS. Biologic effects of calcium-containing crystals. Curr Opin Rheumatol. 2005;17:336–40. doi: 10.1097/01.bor.0000155362.83990.80. [DOI] [PubMed] [Google Scholar]
- 19.Boonla C, Wunsuwan R, Tungasanga K, Tosukhowong P. Urinary 8-hydroxydeoxyguanosine is elevated in patients with nephrolithiasis. Urol Res. 2007;35:185–91. doi: 10.1007/s00240-007-0098-0. [DOI] [PubMed] [Google Scholar]
- 20.Escobar C, Byer KJ, Khan SR. Naturally produced crystals obtained from kidney stones are less injurious to renal tubular epithelial cells than the synthetic crystals. BJU International. 2007;100:891–897. doi: 10.1111/j.1464-410X.2007.07002.x. [DOI] [PubMed] [Google Scholar]