

Abstract
Microtubule-based transport of mitochondria into dendrites and axons is vital for sustaining neuronal function. Transport along microtubule tracks proceeds in a series of plus and minus end-directed movements that are facilitated by kinesin and dynein motors. How the opposing movements are controlled to achieve effective transport over large distances remains unclear. Previous studies showed that the conserved mitochondrial GTPase Miro is required for mitochondrial transport into axons and dendrites and serves as a Ca2+ sensor that controls mitochondrial mobility. To directly examine Miro's significance for kinesin- and/or dynein-mediated mitochondrial motility, we live-imaged movements of GFP-tagged mitochondria in larval Drosophila motor axons upon genetic manipulations of Miro. Loss of Drosophila Miro (dMiro) reduced the effectiveness of both anterograde and retrograde mitochondrial transport by selectively impairing kinesin- or dynein-mediated movements, depending on the direction of net transport. Net anterogradely transported mitochondria exhibited reduced kinesin- but normal dynein-mediated movements. Net retrogradely transported mitochondria exhibited much shorter dynein-mediated movements, whereas kinesin-mediated movements were minimally affected. In both cases, the duration of short stationary phases increased proportionally. Overexpression (OE) of dMiro also impaired the effectiveness of mitochondrial transport. Finally, loss and OE of dMiro altered the length of mitochondria in axons through a mechanistically separate pathway. We suggest that dMiro promotes effective antero- and retrograde mitochondrial transport by extending the processivity of kinesin and dynein motors according to a mitochondrion's programmed direction of transport.
Introduction
Supplying dendrites and axons with mitochondria is vital for sustaining synaptic function (Li et al., 2004; Guo et al., 2005; Verstreken et al., 2005; Kann and Kovács, 2007; Mattson, 2007; Kang et al., 2008). Mitochondrial transport to synapses depends on microtubules (MTs) in axons and dendrites. MT-based mitochondrial transport displays saltatory bidirectional movement, where moving mitochondria frequently stop, start, and change direction. This bidirectional motility is facilitated by MT plus end-directed kinesin and minus end-directed dynein motors, but how the opposing motor movements are controlled remains unclear. Since both motors are apparently attached to mitochondria at all times, achieving effective net transport must require control mechanisms that favor motor movements in the programmed direction of transport, either antero- or retrograde. Accordingly, movement in one direction can only occur if one motor overpowers the other through a “tug-of-war” scenario. Alternatively, the activities of both motors may be coordinated such that only one motor is active and the processivity (e.g., how long an attached motor can travel along a microtubules track) of the active motor is high (Hollenbeck, 1996; Gross, 2003; Vale, 2003; Mallik and Gross, 2004; Welte, 2004; Hollenbeck and Saxton, 2005; Gross et al., 2007).
The evolutionary conserved mitochondrial GTPase Miro is characterized by the presence of two GTPase domains, two Ca2+ binding domains, and a C-terminal transmembrane domain that tail-anchors Miro in the outer mitochondrial membrane (Fransson et al., 2003; Frederick et al., 2004; Guo et al., 2005; Frederick and Shaw, 2007). Loss of Miro in yeast disrupts the tubular mitochondrial network and reduces mitochondrial inheritance (Frederick et al., 2004, 2008). Mutations in mammalian and Drosophila Miro cause abnormal mitochondrial distributions in all examined cells and impair mitochondrial transport into axons and dendrites of neurons (Fransson et al., 2003, 2006; Guo et al., 2005). Miro binds the adaptor protein Milton/GRIF1/OIP106 to form a complex with the kinesin subunit KIF5 (Stowers et al., 2002; Fransson et al., 2006; Glater et al., 2006; MacAskill et al., 2009a). Miro also binds directly to KIF5 in a Ca2+-dependent manner (MacAskill et al., 2009b). Both binding mechanisms facilitate mitochondrial transport (Glater et al., 2006; Saotome et al., 2008; MacAskill et al., 2009a,b; Wang and Schwarz, 2009). Ca2+ binding by Miro's EF-hand domains arrests bidirectional mitochondrial movements, suggesting that it serves as a Ca2+ sensor controlling mitochondrial mobility (Saotome et al., 2008; MacAskill et al., 2009b; Wang and Schwarz, 2009). Whereas these findings underline a critical and pleiotrophic role of Miro in mitochondrial transport, it remained unclear how Miro affects kinesin-mediated movements and whether it is required for dynein-mediated movements.
To directly address how Miro facilitates effective mitochondrial transport, we analyzed the kinetics of mitochondrial movements in Drosophila motor axons during genetic manipulations of dMiro. Our findings significantly extend the current model of dMiro function, suggesting that is not simply a membrane anchor for kinesin motors but required for selectively extending the duration of kinesin-mediated movements during net anterograde mitochondrial transport and dynein-mediated movements during net retrograde transport.
Materials and Methods
Fly stocks.
Flies were raised on standard medium with dry yeast at 25°C unless otherwise stated. The strain w1118, P[w+; UAS::mito-GFP] (mitoGFP) expressing green fluorescent protein (GFP) tagged by an N-terminal mitochondrial localization signal was obtained from W. Saxton (Indiana University, Bloomington, IN). To express mitoGFP and dMiro, we used the enhancer trap strain w1118; P[w+, OK6::Gal4], which drives expression in motor neurons and some interneurons of the ventral ganglia, salivary glands, wing discs, and a subset of tracheal branches (Aberle et al., 2002). The dmiro null alleles B682 and SD32 are null alleles truncating dMiro in the first GTPase domain at position 105 and 89, respectively (Guo et al., 2005). The transgenic line OE-10 (P[w+, UAS::dMiro-RM]) expresses an untagged dMiro cDNA as described by Guo et al. (2005). The following genotypes were examined: control: w1118; P[w+, OK6-Gal4]/+; P[w+, UAS::mGFP]/+. dmiro null: w1118; P[w+, OK6-Gal4]/+; dmiroSD32, P[w+, UAS::mGFP]/dmiroB682. Rescue: w1118; P[w+, OK6-Gal4]/P[w+, UAS::dMiro-RE]; dmiroSD32, P[w+, UAS::mGFP]/dmiroB682. Overexpression (strains OE-10 and OE-5): w1118; P[w+, OK6-Gal4]/P[w+, UAS::dMiro]; +/+. OE-5 expresses N-terminally myc-tagged but otherwise normal dMiro protein.
Generation of myc-tagged dMiro transgenes.
dMiro cDNA (RE) was sequentially PCR-amplified to replace the original ATG start codon with a consensus Kozak sequence followed by a N-terminal myc tag (primer1: 5′-ATCTCTGAAGAAGATCTGGGACAGTACACGGCGTCG-3′; primer 2: 5′-ATGGAGCAGAAACTCATCTCTGAAGAAGATCTG-3′; primer 3: 5′-AATTAATGCGGCCGCACCATGGAGCAGAAACTCATC-3′). The PCR fragment was then sub-cloned into a pCR2.1-TOPO vector. After confirming the DNA sequence by DNA sequencing, the myc-tagged cDNA was subcloned into a pUAST P element vector and transgenic strains containing P were generated as described (Song et al., 2002).
Immunostainings.
Dissected third instar larvae were fixed with 3.5% paraformaldehyde for 1 h at room temperature (RT), washed twice with PBT (PBS, pH 7.4 containing 0.2% Triton X-100) for 10 min, blocked with PBT containing 1% BSA and 5% normal goat serum (PBTB) for 30 min, incubated with primary antibody in PBTB overnight at 4°C, washed with PBT, incubated with secondary antibody in PBTB for 1–2 h at RT, and washed three times with PBT for 10 min. Antibodies and their dilutions: mouse-anti-Myc 1:100 (Invitrogen); goat anti-HRP Cy3-conjugated 1:500 (Jackson Laboratories); rabbit anti-GFP Alexa488-conjugated 1:500 (Invitrogen).
Time lapse imaging of mitochondria.
Mitochondria in motor neurons of dissected climbing third larvae were imaged as described previously (Louie et al., 2008). The dissected preparation was submersed in HL-6 solution (containing 7 mm l-glutamate and 0.6 mm CaCl2) and viewed with an upright Olympus microscope BX50WI equipped with a confocal laser scanner (FluoView300) and a 60× water-immersion objective (LUMPLANFL; numerical aperture, 0.9). Larvae were orientated such that the ventral ganglion (VG) appeared on the right of the acquired image and segmental nerves were aligned horizontally across the image. The 488 nm excitation line of the multiargon laser (Mellet Griot, 150 mW) was attenuated to 2–4% of its maximum power, with all other lasers at 0%. Fluorescence emission was monitored through a low-pass optical filter with a cutoff at 510 nm (BA510IF, Olympus). The pinhole was fully opened to allow maximum “focal depth.” Images of 1024 × 280 pixels (58.868 × 16.065 μm) were acquired from individual segmental nerves just next to the ventral ganglion. A centered 872 × 278 pixel (50 × 16 μm) region of interest (ROI) was photobleached for 180 s, such that the bleached area was always flanked by nonbleached stationary mitoGFP-positive signals. Immediately after bleaching, 200 images were acquired at a rate of one frame per 1.006 s (zoom factor 2×), and saved as 8-bit multitiff files. Time-lapse images were acquired only from one nerve per animal within 30 min of dissection.
Visualization and analysis of stationary mitochondria.
Motile and stationary mitochondria in larval motor axons were visualized by merging two differently pseudo-colored images taken at time 0 (green) and time (red). This labeled stationary mitochondria in yellow and motile mitochondria in red or green. Green signals indicated mitochondria that were present at this position at time 0 but moved away during the indicated time interval. Threshold of movement was a displacement of approximately the size of an individual mitochondrion (2 μm). The density of motile mitochondria (green) and stationary mitochondrial clusters (yellow) was determined by counting the total number of yellow signals and normalizing it to the examined area of the nerve. In dmiro null mutants, individual immobile mitochondria were distinguished from stationary mitochondrial clusters by the intensity of their normalized mitoGFP fluorescence, using a cutoff of 65 AFU (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Tracking of mitochondrial movements.
Movements of mitochondria into or through the photobleached ROI were tracked by using NIH ImageJ imaging software (Abramoff et al., 2004; Louie et al., 2008) and the plug-in MTrackJ (Meijering, E., University Medical Center of Rotterdam, Netherlands; http://www.imagescience.org/meijering/software/mtrackj/). The displacement of a mitochondrion from one frame to the next was converted from pixels to real distances by calibrating the x–y axes of the analyzed images in MTtrackJ. Up to 12 clearly labeled, motile mitochondria were tracked per animal as long as each remained visible in consecutive frames for no <60 frames. In addition, at least one stationary mitochondrial cluster flanking the bleached ROI was tracked for normalization.
The obtained x–y–t tracking coordinates of mitochondrial movements were analyzed with Excel-based software as described earlier (Louie et al., 2008). Briefly, x–y tracking coordinates of motile mitochondria were normalized to reduce the influence of movement artifacts. Mitochondrial movement was modeled as a three-state system consisting of plusend-directed runs, minus end-directed runs, and stops. The start of a run was defined by a minimal displacement of 0.151 μm/s and the end of a run by a minimal displacement of 0.12 μm/s. After identifying runs and stops, mitochondrial tracks were sorted into two classes: mitochondria showing net anterograde movement (AM) or net retrograde movement (RM). The following motility parameters were calculated: duration, distance, and velocity of plus end-directed and minus end-directed trips and runs, duration and frequency of stops, frequency of reversals in direction, velocity of net transport (total net distance during entire tracking time), and the percentage of time allocated to trips and runs (duty cycles).
Statistical analysis.
Track data of individual mitochondria from one segmental nerve were averaged for each animal and then averaged for all animals of the same genotype. The obtained averages were analyzed for statistical significance by one- or two-way ANOVA testing using Prism software (Graphpad Software, 2007). N and n indicate number of animals and mitochondria, respectively. p values of <0.05, <0.01, and <0.001 were indicated with one, two, and three asterisks (*), respectively.
Results
Lack and OE of dMiro reduce (but do not abolish) the amount of motile mitochondria and stationary mitochondrial clusters in Drosophila motor axons
To better understand the role of dMiro for mitochondrial transport, we live-imaged the movements of GFP-tagged mitochondria (mitoGFP) in motor axons of larval Drosophila nerves during loss and overexpression (OE) of dMiro protein. Since axonal microtubules of these motor neurons exhibit a uniform plus end-out orientation (Stone et al., 2008), and because mitochondria were labeled only in motor neurons but not in glial cells and sensory neurons of the segmental nerve, plus end-directed kinesin-driven movements can be unambiguously distinguished from minus end-directed dynein-driven movements (Pilling et al., 2006; Louie et al., 2008).
To selectively express mitoGFP and dMiro (for OE and rescue) in motor neurons, we used the Gal4/UAS system (Brand and Perrimon, 1993) driving transgenic expression with the Gal4-driver Ok6 (Aberle et al., 2002). MitoGFP fluorescence strongly labeled mitochondria in motor neurons, axons, and neuromuscular junctions (NMJs) of control (Fig. 1A,B). Consistent with earlier findings (Guo et al., 2005), mitochondria were largely absent from distal motor axons (data not shown) and NMJs of dmiro null mutants (dmiroSD32/B682) (Fig. 1A,B), although 1 out of 5 animals showed 1–3 mitochondria at a NMJ. However, dmiro null mutant motor axons did contain mitochondria in close proximity to the ventral ganglion, although their numbers were much reduced (Fig. 2A,C,D).
Figure 1.
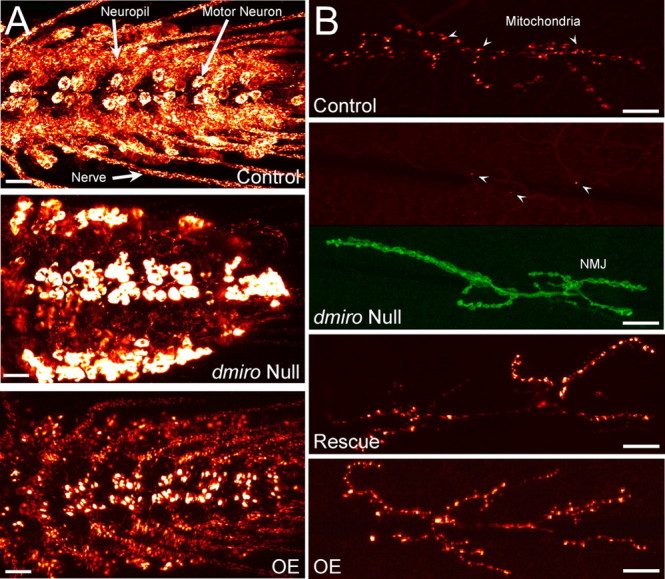
Distribution of GFP-tagged mitochondria in motor neurons during loss and overexpression of dMiro. Mitochondria in larval motor neurons and a few inter neurons were visualized by OK6-driven expression of mitoGFP. A, B, Shown is the mitoGFP fluorescence in ventral ganglia (A) and at larval NMJs of muscle 6/7 (B) of control, dmiro null mutants, and during OE of normal dMiro protein in an otherwise wild-type genetic background. Null mutant NMJs were counterstained with HRP-antibodies (green) to visualize neuronal membranes of the NMJ. Note that mitochondria (arrowheads) were present only rarely at dmiro null mutant NMJs (1 of 5 animals). Scale bars, 20 μm.
Figure 2.
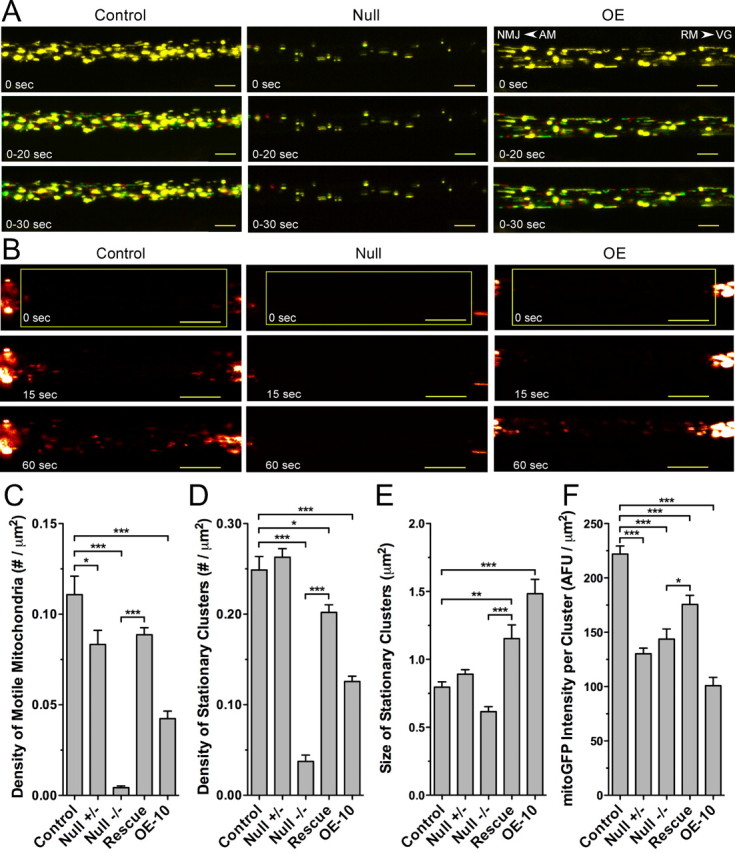
Mutations in dMiro disrupt mitochondrial transport and distribution in larval motor axons. A–F, Mitochondria of larval motor axons were labeled by Ok6-driven expression of mitoGFP. Confocal time-lapse images of a segmental larval nerve exiting the ventral ganglion were acquired at a rate of 1 s−1 without (A) or after photobleaching (B). Time intervals and direction of net AM and net RM are indicated (arrowheads in A, right top). Scale bars: A, 5 μm; B, 10 μm. Error bars represent SEM. A, Distribution of motile (red or green) and stationary mitochondrial clusters (yellow) in proximal larval motor axons of control, dmiro null mutants (Null) and during overexpression of normal dMiro (OE). B, Time lapse images showing mitochondria in larval motor axons entering a photobleached area. Genotypes as in A. Note the reduced number of mitochondria entering the photobleached region in dmiro null mutants and during dMIro OE. C, D, Average density of motile mitochondria (C) and stationary mitochondrial clusters (D) in proximal motor axons of control, heterozygous dmiro null mutants (Null −/+), homozygous dmiro null mutants (Null −/−), homozygous dmiro null mutants during expression of normal dMiro protein (Rescue), and during dMiro overexpression (OE-10). Significant differences between control and mutant genotypes as well as between Null and rescue are indicated by asterisks (p < 0.05; N > 10, one-way ANOVA). E, F, Average size (E) and normalized mitoGFP fluorescence intensity (F) of stationary mitochondrial clusters in proximal motor axons. Genotypes and significant differences are indicated as in C, D.
Typically, less than ∼40% of mitochondria are motile in control fly motor axons; the majority of mitochondria tend to cluster at functionally undefined sites for >180 s (Louie et al., 2008). Heterozygous dmiro null mutant axons showed a significant reduction in the amount of motile (displaced by >2 μm during 60 s) mitochondria (p < 0.5) (Fig. 2C) but a normal density of stationary mitochondrial clusters (Fig. 2D), suggesting that mitochondrial motility is highly sensitive to dMiro protein levels. In homozygous dmiro null mutants, the amount of motile mitochondria was significantly reduced to <4% of control (p < 0.001) (Fig. 2A,C) In contrast to control, dmiro null mutant axons exhibited many mitochondria that were stationary but not clustered, as judged by their size and mitoGFP fluorescence (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This made it necessary to distinguish between individual stationary mitochondria that likely could not move (∼40%) and stationary mitochondria that were clustered. After removing individual stationary mitochondria (see Materials and Methods), the number of stationary mitochondrial clusters was significantly reduced in dmiro nulls to 15% of control (p < 0.001) (Fig. 2D).
Expression of normal dMiro protein in homozygous dmiro null mutants restored the amount of motile mitochondria to a level (80 ± 3.5%) that was statistically indistinguishable from control (p > 0.05) (Fig. 2C). However, the number of stationary mitochondrial clusters was only partially restored to a level that was significantly smaller than control (p < 0.05) (Fig. 2D). OE of dMiro in otherwise wild-type animals significantly reduced the amount of motile mitochondria and the number of stationary mitochondrial clusters in motor axons (p < 0.001) (Fig. 2A,C,D), tentatively indicating that OE of dMiro may have dominant-negative effects.
The size of stationary mitochondrial clusters was not significantly altered in heterozygous and homozygous dmiro null mutants but was increased during expression of normal dMiro protein in dmiro mutants and wild type (p < 0.01) (Fig. 2E). After normalizing for cluster size, mitoGFP fluorescence was significantly reduced in all examined dmiro genotypes (p < 0.001) (Fig. 2F), suggesting that even slight alterations in dMiro protein levels affect the amount of mitochondria clustering at functionally undefined sites in motor axons.
Lack and OE of dMiro reduce the rate of net antero- and retrograde mitochondrial transport
Despite the few motile mitochondria in dmiro null mutant motor axons (only 42 out of 72 imaged nerves/animals showed motile mitochondria; supplemental Movie 1, available at www.jneurosci.org as supplemental material), it was nevertheless feasible to analyze the dynamics of mitochondrial transport with one caveat: since the high density of mitochondria in control axons significantly obscured a precise tracking over large distances (supplemental Movie 2, available at www.jneurosci.org as supplemental material), it was necessary to photobleach a 50-μm-long region of interest before acquiring time-lapse images (Fig. 2B, supplemental Movie 3, available at www.jneurosci.org as supplemental material). However, photobleaching was not a feasible strategy in null mutants (Fig. 2B, supplemental Movie 4, available at www.jneurosci.org as supplemental material) since the rate (flux) of antero- and retrograde mitochondrial transport measured just proximal to the ventral ganglion was reduced by >98% (p < 0.001) (Fig. 3E,F). Even in the best cases, peak values of anterograde flux reached only 0.6 mitochondria/min in dmiro nulls, whereas control showed a mean value of 8.9 ± 0.3 min−1. Both antero- and retrograde fluxes were partially restored by the expression of normal dMiro protein in dmiro null mutant axons (Fig. 3E,F). Like loss of dMiro, OE of dMiro also significantly decreased antero- and retrograde mitochondrial flux to 51 ± 4% and 32 ± 3% of control, respectively (p < 0.01) (Fig. 3E,F).
Figure 3.
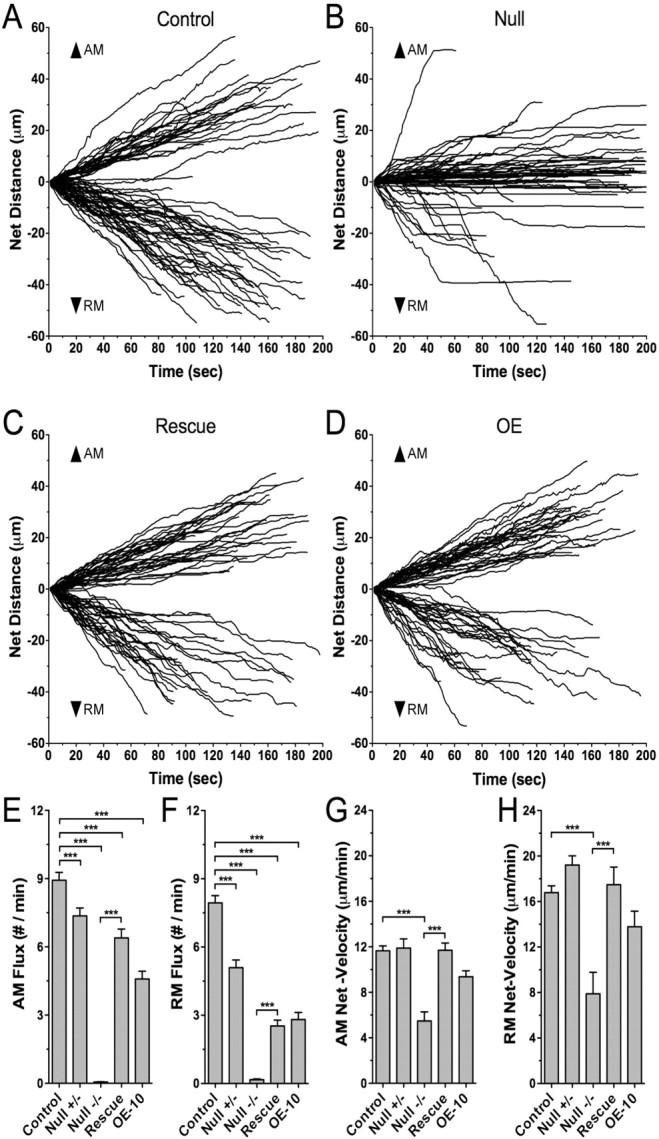
Loss of dMiro reduces the net distance and net velocity of antero- and retrograde mitochondrial transport. A–D, Each plot shows typical tracks of individual mitochondrial movements that were obtained from the analysis of time-lapse images (rate 1.006 s−1, duration 200 s) of proximal larval motor axons in control (A), dmiro null mutants (B), dmiro null mutants expressing normal dMiro protein (C), and during OE of dMiro (D). For comparison, the start of individual tracks was set to zero. Plus end-directed (upward) and minus end-directed movements (runs, downward) are defined by gaining positive or negative distance values, respectively. Note that net antero- (upward) and retrogradely (downward) transported mitochondria exhibited a mix of plus and minus end-directed runs that were separated by pauses or reversals in direction. With a few notable exceptions, dmiro null mutant mitochondria gained little net distance in either direction. E, F, Average anterograde (E) and retrograde mitochondrial flux (F) in proximal motor axons of control, heterozygous dmiro null mutants (Null −/+), homozygous dmiro null mutants (Null −/−), homozygous dmiro null mutants during expression of normal dMiro protein (Rescue), and during dMiro overexpression (OE-10). Significant differences between control and mutant genotypes as well as between Null and rescue are indicated by asterisks (p < 0.001; N > 22, one-way ANOVA). G, H, Average net velocity of net AM (G) and net RM mitochondria (H). Genotypes and significant differences are indicated as in E, F (p < 0.001; N > 19; one-way ANOVA). Error bars represent SEM.
Since both the lack and OE of dMiro protein impaired mitochondrial flux as well as the amount of mitochondria in motor axons, it was unclear whether the partial rescue of mitochondrial flux in dmiro null mutants was attributable to either abnormally high or abnormally low levels of dMiro protein. To distinguish between these possibilities, we examined whether lowering of transgenic dMiro protein expression in null mutant motor neurons may improve the rescue of phenotypes. Lower temperature is known to reduce expression levels of the used Gal4/UAS system since the yeast transcription factor Gal4 is temperature-sensitive (Duffy, 2002). Therefore, we compared the degree of rescuing mitochondrial flux in dmiro null mutants at 25°C (standard) (Fig. 3E,F) and 20°C. The difference in temperature did not significantly affect the rescue of anterograde mitochondrial flux. However, the rescue of retrograde mitochondrial flux was significantly better at 20°C (p < 0.05; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This suggests that the incomplete rescue of dmiro null phenotypes at 25°C is likely attributable to an artificially high expression level of dMiro induced by the OK6 promoter driving Gal4.
Mitochondrial flux was sensitive to the gene doses of dMiro. In heterozygous dmiro null mutants, anterograde and retrograde mitochondrial flux were significantly reduced to 82.5 ± 2.1% and 64.1 ± 4.3% of control, respectively (p < 0.001) (Fig. 3E,F). Together with the distribution of mitochondria to stationary clusters (Fig. 2F), these were the only examined mutant phenotypes that were sensitive to gene doses and only partially rescued after reintroduction of normal dMiro protein (see below). This suggests that mitochondrial flux and distribution in axons are not only determined by transport kinetics but also by other factors. Accordingly, the mechanisms underlying these phenotypes may be to some degree independent of the below-examined role of dMiro controlling plus and minus end-directed mitochondrial movements.
Loss of dMiro reduces the net velocity of antero- and retrograde mitochondrial transport
Bidirectional movements of mitochondria are regulated by a largely unknown programming mechanism that partitions axonal mitochondria into two distinct classes: anterograde-moving (AM) and retrograde-moving (RM) mitochondria (Morris and Hollenbeck, 1993; Hollenbeck, 1996). Earlier studies suggested that directional programming of AM and RM mitochondria is strong in larval motor axons of Drosophila (Pilling et al., 2006; Louie et al., 2008). Consistently, we never observed a wild-type mitochondrion change its direction of net transport (n = 320) (Fig. 3A; supplemental Movies 2–3, available at www.jneurosci.org as supplemental material).
AM and RM mitochondria of control typically gained a net distance of 30–40 μm within 3 min (Fig. 3A). However, the majority of dmiro null mutant mitochondria gained <20 μm (Fig. 3B). Accordingly, net velocities of null mutant AM and RM mitochondria were significantly reduced to 47 ± 7% and 47 ± 11% of control, respectively (p < 0.001) (Fig. 3G,H). Notably, the net velocity of RM mitochondria was still higher than that of AM mitochondria, such that the ratio of antero- and retrograde net velocities in dmiro nulls was normal (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Expression of normal Miro protein in dmiro nulls fully restored normal antero- and retrograde net velocities (p > 0.05) (Fig. 3G,H), confirming that the observed defects are entirely caused by the loss of dMiro protein. Heterozygous null mutants showed no effect on AM and RM net velocities (Fig. 3G,H). These findings provided the first evidence that dMiro may control aspects of both anterograde and retrograde mitochondrial transport.
Loss of dMiro reduces the time spent moving in the programmed direction of transport
Mitochondrial transport consists of alternating plus and minus end-directed movements that are often separated by brief stops. For our analysis, we followed commonly used definitions (Hollenbeck, 1996; Welte et al., 1998; Smith and Simmons, 2001; Maly, 2002; Welte, 2004). We defined a “run” as a period of uninterrupted plus or minus end-directed motor movement. In contrast, a “trip” is defined as a sequence of several runs in the same direction that are briefly interrupted by stops. The “duty cycle of runs” describes the percentage of time that AM and RM mitochondria allocate to stops, plus, and minus end-directed runs (Morris and Hollenbeck, 1993), whereas the “duty cycle of trips” describes the time allocation to plus and minus end-directed trips.
Duty cycles provide an estimate of directional programming by illustrating the bias of mitochondria toward motor movements for one direction. In control, AM mitochondria allocated 91 ± 1% of their time to plus end-directed trips but only 9 ± 1% to minus end-directed trips. This time allocation was essentially reversed for RM mitochondria (Fig. 4A,B; supplemental Table 1, available at www.jneurosci.org as supplemental material). This bias of control AM and RM mitochondria was less pronounced in dmiro null mutants (Fig. 4A,B). Specifically, the time mutant AM mitochondria spent on plus end-directed trips was significantly reduced by 18%, whereas the time spent on minus end-directed trips increased (p < 0.001) (Fig. 4A). Similarly, the time mutant RM mitochondria spent on minus end-directed trips was significantly reduced by 10%, whereas the time spent on plus end-directed trips increased (p < 0.001) (Fig. 4B). All defects in the trip duty cycle were restored by expression of normal dMiro protein (p > 0.05) (Fig. 4A,B). Although dmiro null mutations significantly altered the balance between plus and minus end-directed trips, the observed changes appeared too small to explain the reduced net velocity for both AM and RM mitochondria.
Figure 4.

Effects of dmiro mutations on the time mitochondria allocate to plus and minus end- directed mitochondrial movements. A, B, Average percentage of time AM (A) and RM mitochondria (B) allocated to plus end-directed [(+)end] or minus end-directed [(−)end] trips. Genotypes: control, homozygous dmiro null mutants (Null −/−), homozygous dmiro null mutants during expression of normal dMiro protein (Rescue), and during dMiro overexpression (OE-10). Significant differences among all genotypes are indicated by asterisks (p < 0.05, N > 18, two-way ANOVA). Error bars represent SEM. C, D, Average percentage of time AM (C) and RM mitochondria (D) allocated to plus end-directed [(+)end] runs, minus end-directed [(−)end] runs, and short stationary phases (stops). Genotypes: as in A with the addition of heterozygous dmiro null mutants (Null −/+). Significant differences are indicated (p < 0.05, N > 18, two-way ANOVA). Error bars represent SEM. E, F, Average frequency of stops for AM (E) and RM mitochondria (F). Significant differences indicated (p < 0.05, N > 18, one-way ANOVA). Genotypes are as in A and B. Error bars represent SEM. G, H, Average frequency of reversals in direction for AM (G) and RM mitochondria (H). Significant differences are indicated (p < 0.05, N > 18, one-way ANOVA). Genotypes are as in A and B. Error bars represent SEM.
The nature of the transport defect in dmiro nulls became more apparent after analyzing the duty cycle of runs, which illustrates the time that mitochondria allocate to runs in each direction and to brief stops. In control, AM mitochondria spent ∼63% of their time on plus end-directed runs, 5% on minus end-directed runs, and 32% stationary (Fig. 4C). For RM mitochondria, this time allocation was essentially reversed for plus and minus end-directed runs, whereas the time spent stationary remained the same (Fig. 4D). Loss of dMiro severely disrupted this status quo.
In comparison to control, the time dmiro mutant AM mitochondria allocated to plus end-directed runs was significantly reduced by 53 ± 4%, whereas the time allocated to stops doubled (p < 0.001) (Fig. 4C). However, the time mutant AM mitochondria allocated to minus end-directed runs was normal (p > 0.05). In contrast, the time RM mitochondria allocated to minus end-directed runs was reduced by 54 ± 8% (p < 0.001), whereas the time allocated to plus end-directed runs was not significantly altered (p > 0.05) and the time spent stationary doubled (p < 0.001) (Fig. 4D). All defects were restored by expression of normal dMiro protein in null mutant motor neurons (p > 0.05) (Fig. 4A–D). Heterozygous dmiro null mutants exhibited a normal run duty cycle (Fig. 4C,D).
The defects in the run duty cycle suggest that dMiro selectively regulates both plus and minus end-directed motor activities in the context of the programmed direction of transport. Since this action primarily increases the time spent on a particular motor activity by reducing the time spent stationary, these findings indicate that dMiro may control how long an attached motor can move along a MT track. In contrast, these findings do not support the possibility that dMiro coordinates motor activities by switching one motor “on” and the opposing motor “off”, especially because the frequency of reversals in the direction of runs was not significantly affected (p > 0.05) (Fig. 4G–H).
Loss of dMiro shortens plus end-directed movements during anterograde transport
To understand how dMiro might establish the bias of mitochondria toward motor movements in the programmed direction of transport, we analyzed the effects of dmiro null mutations on mitochondrial trip and run kinetics. AM mitochondria of control typically exhibit long plus end-directed trips driving movement in the programmed net direction, and short minus end-directed trips that are opposite to the net direction (Fig. 5A–F; supplemental Table 2, available at www.jneurosci.org as supplemental material). Loss of dMiro significantly shortened the distance of plus end-directed trips to 52 ± 11% of control (p < 0.001) (Fig. 5A), whereas their duration was not significantly altered (p > 0.05) (Fig. 5B). Hence, this indicates that motor movements for plus end-directed trips are likely shorter and/or stationary phases between movements are longer. Minus end-directed trips opposite to the programmed direction were affected differently, such that they exhibited a 2–3-fold increase in their duration (p < 0.01) (Fig. 5E) but no significant change in their distance (p > 0.05) (Fig. 5D), suggesting that the underlying motor movements are either longer or separated by longer stop periods.
Figure 5.
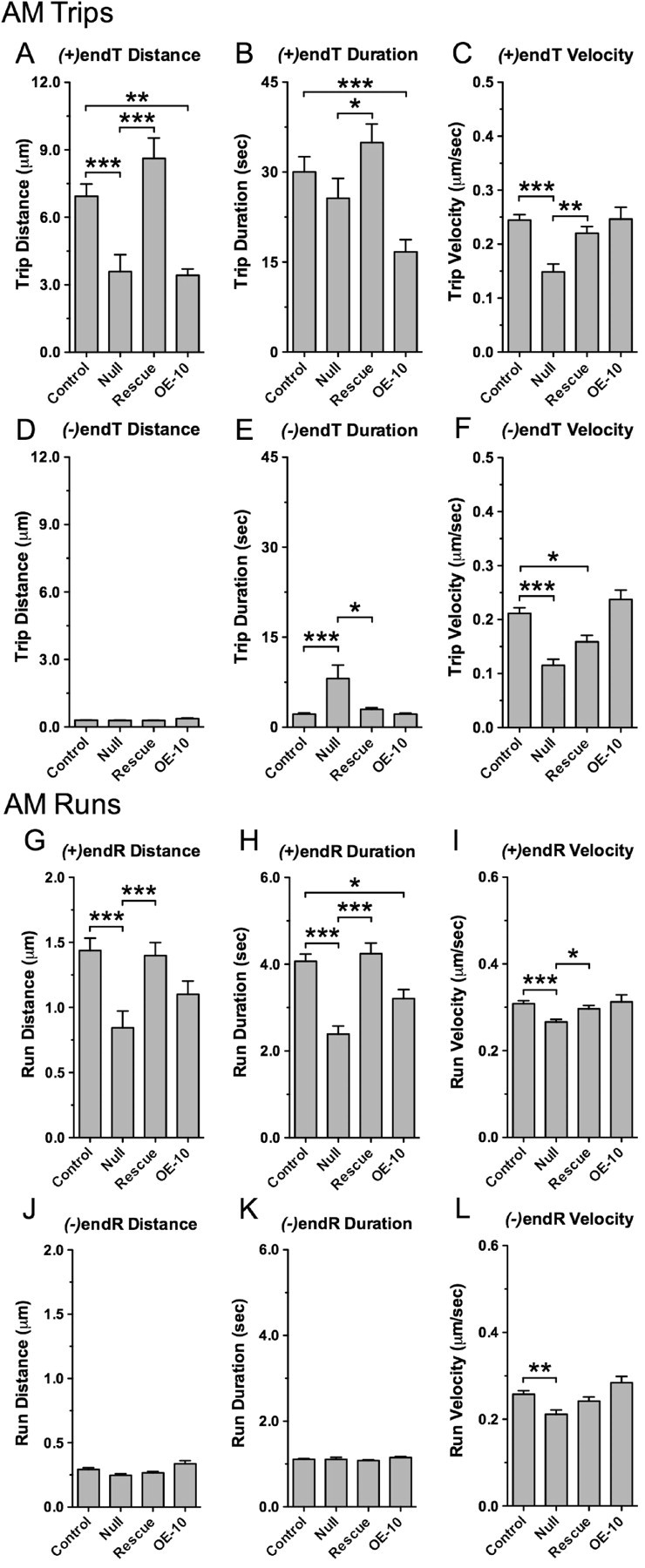
Loss of dMiro shortens plus end-directed trips and runs during net anterograde mitochondrial transport. A–F, Average distances, durations and velocities of plus [(+)endTs] and minus end-directed trips [(−)endTs] for AM mitochondria of control, dmiro null mutants (Null), dmiro null mutants expressing normal dMiro protein (Rescue), and during overexpression of dMiro (OE-10). Asterisks show significant differences among all indicated genotypes (p < 0.05, N > 18, one-way ANOVA). Error bars represent SEM. G–L, Average distances, durations and velocities of plus [(+)endRs] and minus end-directed runs [(−)endRs] for AM mitochondria. Genotypes are as in A. Asterisks show significant differences between indicated genotypes (p < 0.05, N > 18, one-way ANOVA). Error bars represent SEM.
Analysis of individual runs and stop periods substantiated that dMiro promotes plus end-directed motor movements of AM mitochondria. Control exhibited long plus end-directed runs and short minus end-directed runs by AM mitochondria (Fig. 5G–L; supplemental Table 3, available at www.jneurosci.org as supplemental material). Lack of dMiro significantly reduced the distance and duration of the dominating plus end-directed runs by 41% (p < 0.001) (Fig. 5G–H), whereas minus end-directed runs exhibited a normal distance and duration (p > 0.05) (Fig. 5J–K). The reduced duration of plus end-directed runs was inversely correlated to a threefold increase in the duration of stops by AM mitochondria (p < 0.001) (Fig. 7A,C). Heterozygous dmiro null mutants exhibited normal run kinetics of AM mitochondria (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Since all defects in trip and run kinetics of AM mitochondria of homozygous dmiro null mutants were rescued by expression of normal dMiro protein (p > 0.05) (Figs. 5, 7A–C), these findings suggest dMiro selectively increases the duration of plus end-directed kinesin motor movements during anterograde mitochondrial transport by reducing the time spent in short stationary phases.
Figure 7.
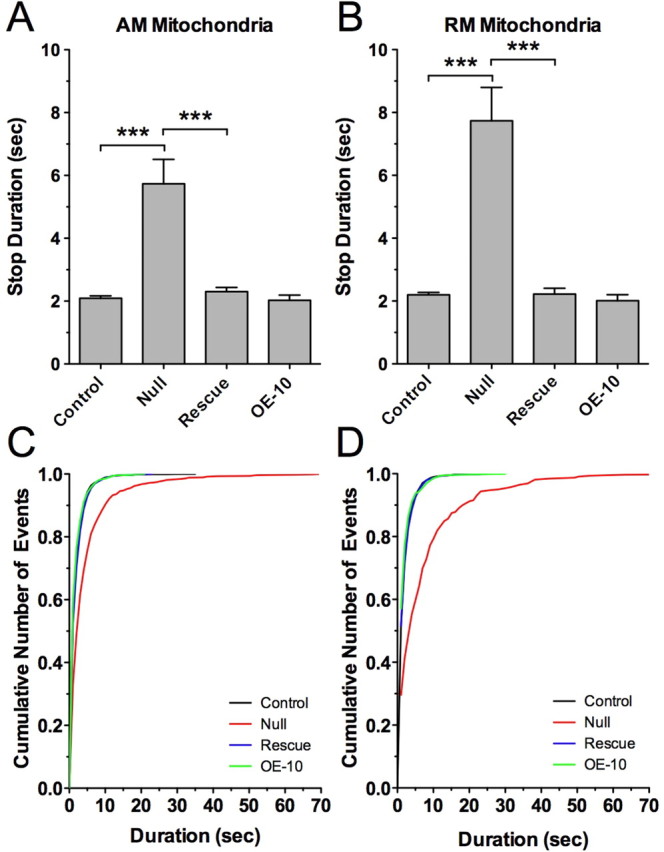
Loss of dMiro increases the duration of stop periods. A, B, Average duration of stops by AM (A) and RM mitochondria (B) in control, dmiro null mutant motor axons (Null), dmiro null mutants expressing normal dMiro protein (Rescue), and during overexpression (OE-10) of normal dMiro protein. Significant differences are indicated by asterisks (p < 0.001; N <18; one-way ANOVA). Error bars represent SEM. C, D, Cumulative frequency distribution for the duration of stops by AM (C) and RM mitochondria (D) of the indicated genotypes.
Loss of dMiro shortens minus end-directed movements during retrograde transport
Normal RM mitochondria exhibit long minus end-directed trips and runs, whereas plus end-directed trips and runs are several-fold shorter (Fig. 6; supplemental Tables 2–3, available at www.jneurosci.org as supplemental material). Lack of dMiro significantly reduced the distance but not duration of the typically dominating minus end-directed trips to 63 ± 21% of control (p < 0.001) (Fig. 6A,B). The duration of plus end-directed trips significantly increased (p > 0.05) (Fig. 6E). The velocity of both plus and minus end-directed trips was slightly but significantly reduced (p < 0.01) (Fig. 6C,F).
Figure 6.
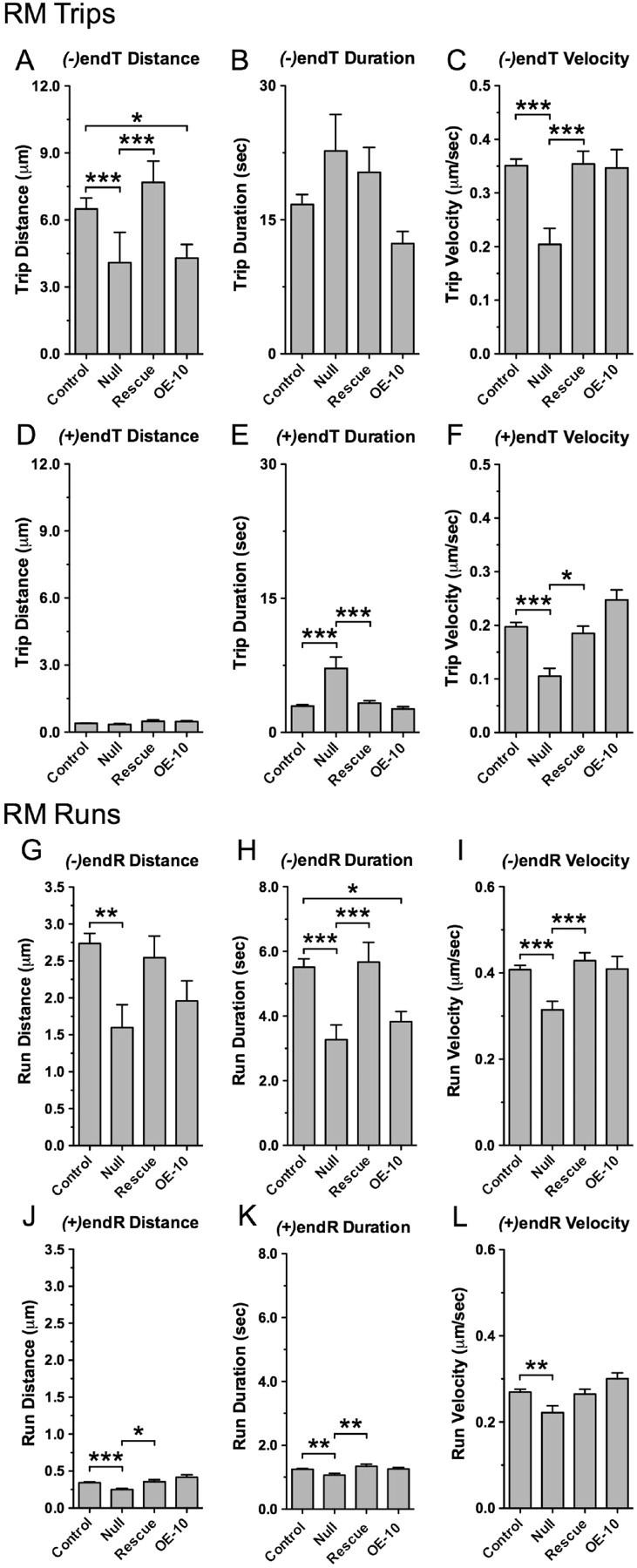
Loss of dMiro shortens minus end-directed trips and runs during net retrograde mitochondrial transport. A–F, Average distances, durations and velocities of minus [(−)endTs] and plus end-directed trips [(+)endTs] for RM mitochondria of control, dmiro null mutant motor axons (Null), dmiro null mutants expressing normal dMiro protein (Rescue), and during overexpression of dMiro (OE-10). Asterisks show significant differences among all genotypes (p < 0.05, N > 18, one-way ANOVA). Error bars represent SEM. G–L, Average distances, durations and velocities of minus [(−)endRs] and plus end-directed runs [(+)endRs] for RM mitochondria. Genotypes as in A. Asterisks show significant differences between indicated genotypes (p < 0.05, N > 18, one-way ANOVA). Error bars represent SEM.
Consistent with effects on trip kinetics, lack of dMiro significantly reduced the distance and duration of minus end-directed runs of RM mitochondria by ∼41% (p < 0.001) (Fig. 6G–H). Plus end-directed runs were also impaired but to a much lesser degree since their distance and duration were only reduced by 27 ± 5% and 15 ± 5%, respectively (p < 0.01) (Fig. 6J–K). Technically, the mutant effect on minus end-directed runs had a much larger impact on the effectiveness of retrograde transport because minus end-directed runs were ∼2.1 μm shorter, whereas plus end-directed runs were only ∼0.1 μm shorter (supplemental Table 3, available at www.jneurosci.org as supplemental material). The velocity of both plus and minus end-directed runs by RM mitochondria was slightly but significantly reduced (p < 0.01) (Fig. 6I,L). The reduced duration of minus end-directed runs inversely correlated with a 3.5-fold increase in the duration of stop periods (p < 0.001) (Fig. 7B,D). In heterozygous dmiro null mutants, run kinetics of RM mitochondria were normal (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). All deficits in trips and runs of dmiro null mutants were fully rescued (p > 0.05) (Figs. 5–6). Together, these findings suggest that dMiro selectively increases the duration of minus end-directed motor movements during retrograde mitochondrial transport by reducing the time spent in short stationary phases.
In conclusion, our findings consistently suggest that dMiro primarily promotes the duration and distance of motor movements by plus end-directed kinesin motors during anterograde mitochondrial transport and minus end-directed dynein motors during retrograde transport (Figs. 5–6). However, this conclusion depends on a proper classification of AM and RM mitochondria, which has been a concern for RM mitochondria of dmiro null mutants. AM and RM mitochondria are primarily classified by the net direction of their movements but are also distinguished by their different net velocities, motor time allocations, and motor kinetics (Pilling et al., 2006; Louie et al., 2008). Based on these criteria, several lines of evidence confirmed a reliable classification: First, even the worst performing AM and RM dmiro null mutant mitochondria still showed a recognizable bias in their time allocation to one motor (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), although most of these mitochondria showed a net displacement of <2–3 μm (Fig. 3B). Second, RM mitochondria typically exhibit a higher net velocity than AM mitochondria. Despite a severe reduction in dmiro nulls, this feature was still preserved since the ratio of AM and RM net velocities was indistinguishable from control (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Third, plus end-directed runs by control AM mitochondria are typically much longer than plus end-directed runs by RM mitochondria (Figs. 5–6). The same is true for minus end-directed runs, which are much longer for RM mitochondria than for AM mitochondria (Figs. 5–6). However, even the worst performing dmiro null mutant mitochondria retained this feature typical of AM or RM mitochondria (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Together, these findings suggest that loss of dMiro impairs but does not abolish directional programming of mitochondrial transport in axons.
Effects of dMiro overexpression on mitochondrial transport
OE of dMiro protein in an otherwise wild-type control background significantly reduced the flux of antero- and retrograde mitochondrial transport and the distribution of mitochondria in axons (Fig. 3E,F). The nature of this negative effect is not clear but may be attributable to a titration of factors that are associated with dMiro, including the Milton-KIF5 complex (Fransson et al., 2006; Glater et al., 2006; Louie et al., 2008). This and subsequent effects of dMiro OE were confirmed by at least one independently derived transgenic strain (data not shown).
Although dMiro OE did not significantly reduce net velocities of antero- and retrograde transport (p > 0.05, N > 18) (Fig. 3G–H), it significantly reduced the time AM mitochondria spent on plus end-directed trips by increasing the time spent on minus end-directed trips to 165 ± 14% of control (p < 0.05) (Fig. 4A). However, dMiro OE had no effect on the time allocation of trips by RM mitochondria (p > 0.05) (Fig. 4B). In addition, OE had no significant effects on the time allocation of runs and the frequency and duration of stops (p > 0.05) (Figs. 4C–F, 7A–D).
dMiro OE significantly reduced the distance and duration, but not velocities, of plus end-directed trips of AM mitochondria to 49 ± 4% and 56 ± 7%, respectively (p < 0.01) (Fig. 5A–C). Minus end-directed trips of AM mitochondria were normal (Fig. 5D–F). Consistently, dMiro OE significantly reduced the duration of plus end-directed runs (p < 0.05) (Fig. 5H), whereas the reduction in run distance was statistically not significant (p > 0.05) (Fig. 5G). The velocity of plus end-directed run was normal (Fig. 5I). The distance, duration, and velocity of minus end-directed runs opposite to direction of net transport were normal (Fig. 5J–L).
For RM mitochondria, dMiro OE significantly reduced the distance of minus end-directed trips to 66 ± 9% of control (p < 0.05) (Fig. 6A). The reduced duration of minus end-directed trips was statistically not significant (p < 0.05) (Fig. 6B) and the velocity was normal (Fig. 6C). Minus end-directed trips of RM mitochondria were normal (Fig. 6D–F). Consistent with the effects on trips, dMiro OE significantly reduced the duration of minus end-directed runs by RM mitochondria (p < 0.05) (Fig. 6H) but not run distance and velocity (p > 0.05) (Fig. 6G,I). Kinetics of plus end-directed runs by RM mitochondria runs were normal during dMiro OE (Fig. 6J–L).
Although the effects of null mutations were more pronounced, loss and OE of dMiro had similar negative effects on antero- and retrograde transport, strengthening the notion that dMiro is required for both modes of transport. There was, however, one critical difference: in contrast to dmiro null mutations, dMiro OE significantly increased the frequency of reversals in the direction of movement for AM and RM mitochondria to 167 ± 21% and 141 ± 18% of control, respectively (p < 0.001) (Fig. 4G–H), whereas the duration of short stationary phases remained normal (Fig. 7). Importantly, this suggests that the effects of OE on trip and run kinetics were entirely caused by the increased rate of reversals, which was not the case for dmiro null mutations where the effects on trips inversely correlated with an increase in the duration of stops. Consequently, the dominant-negative effect of dMiro OE on transport is likely reflecting a titration of unknown factors but not a loss or gain of dMiro activity. The effect of dMiro OE on mitochondrial transport in Drosophila motor neurons is different from that of overexpressing mammalian Miro1 in neuronal cultures, which increases the amount mitochondria being transported into neuronal processes (MacAskill et al., 2009a,b; Wang and Schwarz, 2009). The cause of this difference is not known but could be attributable to expression levels or species-specific differences.
Mutations in dMiro affect mitochondrial length
To control for potential effects of dMiro on the size of transported mitochondria, we analyzed their length in larval motor axons (Fig. 8A). Control AM mitochondria exhibited a mean length of 1.3 ± 0.1 μm, whereas RM mitochondria were a little shorter, averaging 1.2 ± 0.1 μm. The difference in mean length of AM and RM control mitochondria was not significant (p > 0.5) (Fig. 8B–F). However, the average length of AM and RM mitochondria in dmiro null mutant axons was significantly reduced by 59 ± 6% to 0.6 ± 0.1 μm and by 64 ± 4% to 0.4 ± 0.1 μm of control, respectively (p < 0.001) (Fig. 8B–E). Motile null mutant mitochondria were on average shorter than immobile ones, but this difference was statistically indistinguishable (p > 0.05). The length defect was restored by expression of normal dMiro protein in dmiro null mutant motor neurons (Fig. 8B–E).
Figure 8.
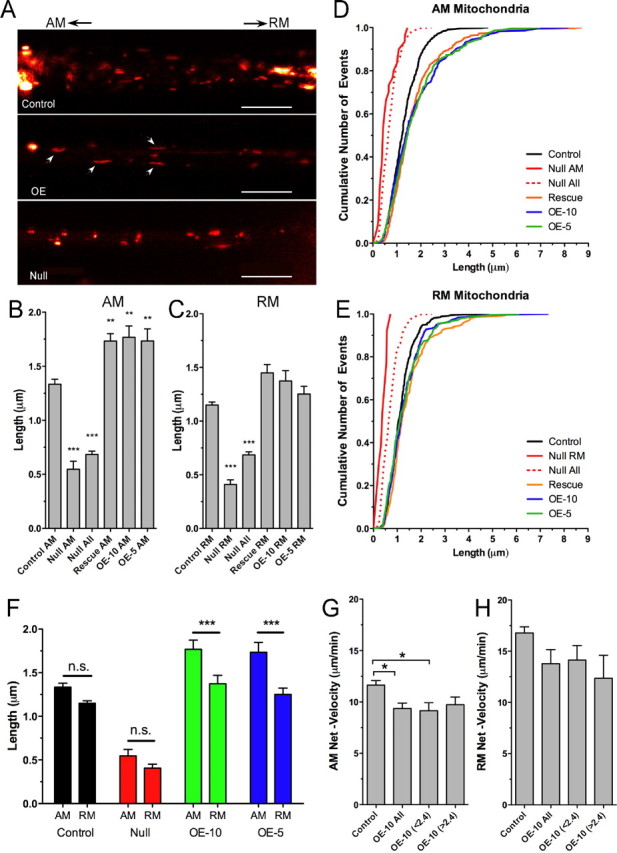
Mutations in dMiro alter mitochondrial length. A, Confocal images of GFP-tagged mitochondria in larval motor axons of control, dmiro null mutants (Null) and during OE of dMiro. Note the altered length of mitochondria during loss and OE of dMiro. Scale bars, 10 μm. B, C, Average length of motile AM (B) and RM (C) mitochondria in larval motor axons of control, dmiro null mutants, dmiro null mutants expressing normal dMiro protein, and during OE of dMiro. OE-10 and OE-5 indicate independently derived transgenic strains. “Null All” combines the average length of motile and stationary mitochondria in dmiro null mutant axons. Asterisks indicate significant differences to control (p < 0.01; N > 20 one-way ANOVA). Error bars represent SEM. D, E, Cumulative frequency distribution for the length of AM (D) and RM mitochondria (E). Genotypes as in B. F, OE of dMiro causes a significant difference in the length of AM and RM mitochondria (p < 0.001; N > 20; two-way ANOVA). Error bars represent SEM. G, H, Average net velocity of differentially sized AM (G) and RM mitochondria (H). Note the similar velocity of normal-sized (<2.4 μm) and elongated mitochondria (>2.4 μm) during OE of dMiro (p < 0.05; N > 7; one-way ANOVA). Error bars represent SEM.
OE of dMiro in an otherwise wild-type background significantly increased the average length of AM mitochondria to >130 ± 6% of control (p < 0.01) (Fig. 8B–F). Interestingly, the increase in length of RM mitochondria was much less pronounced and not significant (p > 0.05). Consequently, the size of AM and RM mitochondria was significantly different during dMiro OE (p < 0.01) (Fig. 8F). This phenomenon was not observed in control and dmiro null mutants, although a trend was indicated (p > 0.05) (Fig. 8F).
It seemed possible that the observed alterations in mitochondrial size could have been caused by a homeostatic mechanism compensating for a potentially slower transport of normal-sized mitochondria. To investigate this possibility, we compared the transport characteristics of normal and elongated mitochondria during dMiro OE. We used 2.4 μm as the threshold since 95% of control mitochondria were shorter (n = 558). Upon dMiro OE, antero- and retrograde net velocities of normal-sized and elongated mitochondria were similar (p > 0.05) (Fig. 8G–H). Most surprisingly, run kinetics of normal and elongated AM and RM mitochondria were essentially indistinguishable (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), suggesting that changes in mitochondrial length have little effect on mitochondrial transport kinetics. Since the effects of dmiro null mutations and dMiro OE on mitochondrial transport and length do not correlate, we suggest that dMiro may modulate the machineries of mitochondrial transport and fusion or fission through mechanistically separate pathways.
Discussion
dMiro executes directional programming of mitochondrial transport
Effective net antero- and retrograde transport of bidirectionally moving mitochondria is achieved by a directional program that favors one motor activity over the other (Morris and Hollenbeck, 1993). The nature of this mechanism is poorly understood but likely requires coordination of motor activities ensuring that when plus end-directed motors (kinesin) are active, minus end-directed motors (dynein) are not, and vice versa (Gross, 2003; Welte, 2004; Hollenbeck and Saxton, 2005). Evidence supporting the existence of motor coordination comes from several transport systems (Brady et al., 1990; Waterman-Storer et al., 1995; Stenoien and Brady, 1997; Waterman-Storer et al., 1997; Martin et al., 1999; Valetti et al., 1999; Deacon et al., 2003).
Bidirectional motility may be coordinated by an interaction between KIF5 and dynein that is mediated through dynein intermediate chain 1 and kinesin light chains (Ligon et al., 2004). In addition, the dynactin complex, specifically the subunit p150Glued, has been implicated in coordinating bidirectional transport of vesicles by preventing opposing force production (Martin et al., 1999; Gross et al., 2002, 2003; Deacon et al., 2003). Genetic interactions between Drosophila KIF5, dynein, and dynactin mutations support interactions among all three proteins, and loss of Drosophila dynein heavy chain activity reduces the velocity of kinesin movements (Martin et al., 1999; Pilling et al., 2006). However, loss of p150Glued enhanced, rather than reduced, kinesin and dynein movements (Pilling et al., 2006). Hence, the molecular nature of motor coordination remains unclear.
In addition to motor coordination, effective transport of bidirectionally moving cargo also requires control over the processivity of motors (Welte et al., 1998; Suomalainen et al., 1999; Gross et al., 2000, 2002; Smith et al., 2001; Rodionov et al., 2003). It is unclear whether the mechanisms controlling motor coordination and processivity act hierarchically or in parallel, but it is likely that their actions are coordinated. Loss of control over either motor coordination or processivity should impair the bias for one motor movement, although in different ways: impairing the activity of a coordinated on/off switch essentially causes a “tug-of-war” where the more powerful motor dictates the main direction of transport. Accordingly, it is expected that changes in the activity of one motor are inversely correlated to changes in the activity of the opposing motor. In contrast, impairing a mechanism that controls motor processivity is expected to alter only the properties of the targeted motor, which is the motor that directs movement in the programmed direction of net transport. Hence, it is expected that this motor will lose its dominance, which then increases the time allocated to stationary phases but not to movements in the opposite direction.
Loss of dMiro mirrored the expected effects during loss of control over motor processivity, as it predominantly reduced the distance and duration of movements in the main direction of net transport and increased the duration of stops accordingly. Hence, we suggest that dMiro selectively promotes the processivity of the motor that facilitates movement in the programmed direction of transport.
Potential mechanisms of dMiro signaling
We suggest that dMiro ensures effective mitochondrial transport by selectively promoting either kinesin- or dynein-mediated movements during a signal that confers the direction of net transport. This role requires control over both motors but also integration of signals that activate mitochondria for either anterograde or retrograde transport. Little is currently known about such potentially long-range signals but they could include NGF, phosphatidylinositols, PKC, and signals on the functional state of mitochondria (Chada and Hollenbeck, 2003; De Vos et al., 2003; Chada and Hollenbeck, 2004; Miller and Sheetz, 2004; Morfini et al., 2007; Kang et al., 2008).
How Miro achieves control over the processivity of kinesin and dynein motors remains to be seen but could be facilitated by its known interactions with KIF5 and Milton (Stowers et al., 2002; Fransson et al., 2006; Glater et al., 2006; MacAskill et al., 2009a,b). Ca2+ binding by Miro's EF-hands disrupts binding of Miro1 to KIF5, inhibiting antero- and retrograde transport of mitochondria (Saotome et al., 2008; MacAskill et al., 2009b; Wang and Schwarz, 2009). Accordingly, Miro could increase the processivity of kinesin-mediated movements through a direct interaction with KIF5 in the absence of Ca2+. Alternatively, Miro may control the processivity of kinesin and potentially dynein motors through its interaction with Milton/GRIF1/OIP106.
Initially, it has been suggested that Milton/GRIF1/OIP106 and Miro form an adaptor complex that couples KIF5 to mitochondria (Fransson et al., 2006; Glater et al., 2006; MacAskill et al., 2009a), but the recent finding that mammalian Miro1 binds directly to KIF5 challenges this view (MacAskill et al., 2009b). Our findings do not exclude the possibility that Miro/Milton couple mitochondria to kinesin motors. However, dMiro is clearly not the only membrane anchor for kinesin since its loss did not abolish kinesin movements. Instead, lack of dMiro only impaired the processivity (duration and distance) of either kinesin- or dynein-mediated mitochondrial movements depending on the direction of net transport. Hence, dMiro plays a more complex role than that of a membrane anchor. The same may not be the case for Milton since it binds to mitochondria not only through Miro but also through a second unknown mitochondrial protein (Glater et al., 2006). Accordingly, Milton may couple KIF5 to mitochondria through protein X and act as a scaffolding protein for Miro's actions upregulating the processivity of kinesin and dynein movements.
Consistent with a “bidirectional” role of Milton is the finding that Milton is required for dynein movements during the formation of the “mitochondrial cloud” in fly oocytes (Cox and Spradling, 2006). Mechanistically, the nonkinesin-associated Milton isoform (Glater et al., 2006) may promote these dynein movements in cooperation with Miro. Evidence that specific Milton/GRIF1/OIP106 isoforms directly bind to dynein subunits is lacking, although the HAP-1 domain, which differs between Milton isoforms, has been predicted to mediate interactions with dynactin (Li et al., 1998). Interestingly, dynactin increases the processivity of dynein-driven movements in vitro (Karki and Holzbaur, 1995; King and Schroer, 2000). However, loss of the dynactin subunit p150Glued enhanced kinesin- and dynein-driven movements of mitochondria in Drosophila motor axons (Pilling et al., 2006). Alternatively, Miro's direct or indirect interactions with KIF5 could alter the known interaction of KIF5 with dynein (Ligon et al., 2004), achieving control over the processivity of the dynein motor, and vice versa.
Miro's influence on mitochondrial fusion and fission
Mitochondrial fusion and fission are critical for maintaining a functional population of mitochondria. They control the shape, length, and number of mitochondria and allow the exchange of lipids and proteins (Detmer and Chan, 2007; Twig et al., 2008a). Fusion and fission may be critical for a quality control mechanism that governs mitochondrial turnover, since repetitive rounds of selective fusion and fission segregate severely dysfunctional mitochondria and target them for autophagy (Twig et al., 2008b). Especially in neurons, abnormal fusion and fission often leads to abnormal intracellular distributions of mitochondria and may underlie a number of neurodegenerative diseases (Li et al., 2004; Verstreken et al., 2005; Chan, 2006; Chen et al., 2007; Detmer and Chan, 2007).
The phenotypic effects of loss and OE of dMiro on the length of mitochondria suggest a role for dMiro in mitochondrial fusion or fission, consistent with phenotypic effects of mutations in yeast and human Miro (Frederick et al., 2004, 2008; Fransson et al., 2006). However, Miro is unlikely directly involved in fusion or fission since mitochondria in yeast null mutants are competent of fusion and fission per se (Frederick et al., 2004, 2008). For several reasons, the mitochondrial transport defects of dmiro mutants are unlikely caused by abnormal mitochondrial fusion or fission and vice versa: First, phenotypic effects during loss and OE of dMiro on mitochondrial transport and length did not correlate with each other. Second, mitochondrial length did not influence transport kinetics of mitochondria during dMiro OE. Third, the effects on mitochondrial length were not a consequence of a homeostatic mechanism that compensates for deficits in mitochondrial transport. Therefore, it is likely that dMiro modulates mitochondrial transport and fusion or fission through mechanistically separate pathways.
The selective effect of dMiro OE on the length of AM but not RM mitochondria indicates that dMiro may be part of a mechanism that coordinates fusion or fission events with transport. In principle, such a link should be biologically relevant. Since repeatedly occurring fusion and fission events seem to ensure mitochondrial health (Twig et al., 2008b), an “aging” mitochondrion in need of fusion is expected to actively approach other mitochondria, whereas two freshly separated mitochondria may have a need for being actively transported away from each other. Supporting anecdotal evidence for a need of coordinating mitochondrial fission and trafficking comes from the recent finding that reducing mitochondrial size by fission is presumably important before actin-based mitochondrial trafficking into dendritic spines can occur (Sung et al., 2008). In analogy, dMiro could be part of a similar mechanism coordinating fission and/or fusion of mitochondria with MT-based transport.
Footnotes
This work was supported by a grant to K.E.Z from National Institute of Neurological Disorders and Stroke (R01 NS052664). G.J.R was supported by a National Institutes of Health training grant (T32 AG007434). We thank Dan Burhans for early contributions, Becky Emerson and Jinhui Zhang for technical support, and Mays Imad and Milosh Babic for critical comments on this manuscript.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophot Internat. 2004;11:36–42. [Google Scholar]
- Brady ST, Pfister KK, Bloom GS. A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci U S A. 1990;87:1061–1065. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol. 2003;206:1985–1992. doi: 10.1242/jeb.00263. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14:1272–1276. doi: 10.1016/j.cub.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cox RT, Spradling AC. Milton controls the early acquisition of mitochondria by Drosophila oocytes. Development. 2006;133:3371–3377. doi: 10.1242/dev.02514. [DOI] [PubMed] [Google Scholar]
- Deacon SW, Serpinskaya AS, Vaughan PS, Lopez Fanarraga M, Vernos I, Vaughan KT, Gelfand VI. Dynactin is required for bidirectional organelle transport. J Cell Biol. 2003;160:297–301. doi: 10.1083/jcb.200210066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Sable J, Miller KE, Sheetz MP. Expression of phosphatidylinositol (4,5) bisphosphate-specific pleckstrin homology domains alters direction but not the level of axonal transport of mitochondria. Mol Biol Cell. 2003;14:3636–3649. doi: 10.1091/mbc.E02-10-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy JB. GAL4 system in Drosophila: a fly geneticist's Swiss army knife. Genesis. 2002;34:1–15. doi: 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, Aspenström P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–6502. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- Fransson S, Ruusala A, Aspenström P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Com. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol. 2004;167:87–98. doi: 10.1083/jcb.200405100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick RL, Okamoto K, Shaw JM. Multiple pathways influence mitochondrial inheritance in budding yeast. Genetics. 2008;178:825–837. doi: 10.1534/genetics.107.083055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SP. Dynactin: coordinating motors with opposite inclinations. Curr Biol. 2003;13:R320–R322. [PubMed] [Google Scholar]
- Gross SP, Welte MA, Block SM, Wieschaus EF. Dynein-mediated cargo transport in vivo. A switch controls travel distance. J Cell Biol. 2000;148:945–956. doi: 10.1083/jcb.148.5.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SP, Welte MA, Block SM, Wieschaus EF. Coordination of opposite-polarity microtubule motors. J Cell Biol. 2002;156:715–724. doi: 10.1083/jcb.200109047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SP, Vershinin M, Shubeita GT. Cargo transport: two motors are sometimes better than one. Curr Biol. 2007;17:R478–R486. doi: 10.1016/j.cub.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. The pattern and mechanisms of mitochondrial transport in axons. Front Biosci. 1996;1:d91–d102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, Kovács R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292:C641–C657. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem. 1995;270:28806–28811. doi: 10.1074/jbc.270.48.28806. [DOI] [PubMed] [Google Scholar]
- King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2:20–24. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- Li SH, Gutekunst CA, Hersch SM, Li XJ. Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci. 1998;18:1261–1269. doi: 10.1523/JNEUROSCI.18-04-01261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dentritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL. A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J Biol Chem. 2004;279:19201–19208. doi: 10.1074/jbc.M313472200. [DOI] [PubMed] [Google Scholar]
- Louie K, Russo GJ, Salkoff DB, Wellington A, Zinsmaier KE. Effects of imaging conditions on mitochondrial transport and length in larval motor axons of Drosophila. Comp Biochem Physiol A Mol Integr Physiol. 2008;151:159–172. doi: 10.1016/j.cbpa.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Brickley K, Stephenson FA, Kittler JT. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol Cell Neurosci. 2009a;40:301–312. doi: 10.1016/j.mcn.2008.10.016. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009b;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallik R, Gross SP. Molecular motors: strategies to get along. Curr Biol. 2004;14:R971–R982. doi: 10.1016/j.cub.2004.10.046. [DOI] [PubMed] [Google Scholar]
- Maly IV. A stochastic model for patterning of the cytoplasm by the saltatory movement. J Theor Biol. 2002;216:59–71. doi: 10.1006/jtbi.2002.2531. [DOI] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Mitochondrial regulation of neuronal plasticity. Neurochem Res. 2007;32:707–715. doi: 10.1007/s11064-006-9170-3. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Opalach K, Serulle Y, Moreira JE, Sugimori M, Llinas RR, Brady ST. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci U S A. 2007;104:2442–2447. doi: 10.1073/pnas.0611231104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov V, Yi J, Kashina A, Oladipo A, Gross SP. Switching between microtubule- and actin-based transport systems in melanophores is controlled by cAMP levels. Curr Biol. 2003;13:1837–1847. doi: 10.1016/j.cub.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DA, Simmons RM. Models of motor-assisted transport of intracellular particles. Biophys J. 2001;80:45–68. doi: 10.1016/S0006-3495(01)75994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Gross SP, Enquist LW. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A. 2001;98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Ranjan R, Dawson-Scully K, Bronk P, Marin L, Seroude L, Lin YJ, Nie Z, Atwood HL, Benzer S, Zinsmaier KE. Presynaptic regulation of neurotransmission in Drosophila by the G protein-coupled receptor Methuselah. Neuron. 2002;36:105–119. doi: 10.1016/s0896-6273(02)00932-7. [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Brady ST. Immunochemical analysis of kinesin light chain function. Mol Biol Cell. 1997;8:675–689. doi: 10.1091/mbc.8.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Roegiers F, Rolls MM. Microtubules have opposite orientation in axons and dendrites of Drosophila neurons. Mol Biol Cell. 2008;19:4122–4129. doi: 10.1091/mbc.E07-10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Sung JY, Engmann O, Teylan MA, Nairn AC, Greengard P, Kim Y. WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc Natl Acad Sci U S A. 2008;105:3112–3116. doi: 10.1073/pnas.0712180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen M, Nakano MY, Keller S, Boucke K, Stidwill RP, Greber UF. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008a;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008b;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD. The molecular toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Valetti C, Wetzel DM, Schrader M, Hasbani MJ, Gill SR, Kreis TE, Schroer TA. Role of dynactin in endocytic traffic: effects of dynamitin overexpression and colocalization with CLIP-170. Mol Biol Cell. 1999;10:4107–4120. doi: 10.1091/mbc.10.12.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Karki S, Holzbaur EL. The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1) Proc Natl Acad Sci U S A. 1995;92:1634–1638. doi: 10.1073/pnas.92.5.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Karki SB, Kuznetsov SA, Tabb JS, Weiss DG, Langford GM, Holzbaur EL. The interaction between cytoplasmic dynein and dynactin is required for fast axonal transport. Proc Natl Acad Sci U S A. 1997;94:12180–12185. doi: 10.1073/pnas.94.22.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA. Bidirectional transport along microtubules. Curr Biol. 2004;14:13. doi: 10.1016/j.cub.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Welte MA, Gross SP, Postner M, Block SM, Wieschaus EF. Developmental regulation of vesicle transport in Drosophila embryos: forces and kinetics. Cell. 1998;92:547–557. doi: 10.1016/s0092-8674(00)80947-2. [DOI] [PubMed] [Google Scholar]