

Abstract
Background
There is growing recognition that oxidative stress plays a role in the pathogeneses of myocardial repair/remodeling following infarction (MI). NADPH oxidase is a major source for cardiac reactive oxygen species production. Herein, we studied the importance of NADPH oxidase in development of cardiac oxidative stress and its induced molecular and cellular changes related to myocardial repair/remodeling.
Methods
MI was created by coronary artery ligation in C57/BL (wild type) and NADPH oxidase (gp91phox) knockout mice. Cardiac oxidative stress, inflammatory/fibrogenic responses, apoptosis and hypertrophy were detected by in situ hybridization, immunohistochemistry, TUNEL, picrosirius red staining and image analysis, respectively, at different stages postMI.
Results
In wild type mice with MI and compared to sham-operated animals, we observed significantly increased gp91phox and 3-nitrotyrosine, a marker of oxidative stress, in the infarcted myocardium; accumulated macrophages and myofibroblasts at the infarct site; abundant apoptotic myocytes primarily at border zones on day 3 and numerous apoptotic inflammatory/myofibroblasts in the later stages. In addition, we detected significantly increased TGF-β1, TIMP-2 and type 1 collagen gene expression and continuously increasing collagen volume in the infarcted myocardium; and hypertrophy in noninfarcted myocardium. Compared to wild type mice with MI, we did not observe significant difference in infarct size/thickness, cardiac hypertrophy, myocyte apoptosis, inflammatory/fibrogenic responses as well as cardiac oxidative stress in gp91phox knockout mice.
Conclusion
Our findings indicate that during NADPH oxidase deficiency, superoxide production can be compensated by other sources, which leads to cardiac oxidative stress and its related molecular/cellular events in the infarcted heart.
Keywords: Myocardial infarction, cardiac remodeling, oxidative stress, NADPH oxidase, mice
INTRODUCTION
Heart failure has emerged as a major health problem during the past two decades. It appears most commonly in patients with previous MI. Myocardial remodeling, which occurs in both infarcted and non-infarcted myocardium, contributes significantly to the development of heart failure (1-3). This leads to ventricular remodeling characterized by alterations in left ventricular size, shape, and wall thickness (4-6). Multiple factors may, in fact, contribute to left ventricular remodeling at different stages postMI. There is experimental evidence to suggest that oxidative stress mediated by reactive oxygen species (ROS) plays a role in the pathogeneses of myocardial repair/remodeling after MI (7-11). Oxidative stress results from an oxidant/antioxidant imbalance: an excess of oxidants relative to antioxidant capacity. The heart with acute MI experiences increased ROS production as well as an antioxidant deficit. Chronic antioxidant treatment suppresses cardiac oxidative stress and attenuates ventricular remodeling, partially preserving left ventricular function and improving survival in rats or mice with experimental MI (12-14). Experimental studies have also demonstrated that oxidative stress can induce most, if not all, of the changes that are thought to contribute to myocardial remodeling including proinflammatory cytokine release, cardiomyocyte apoptosis (15), fibrogenesis (16), cell proliferation (17-18), and hypertrophy (19).
NADPH oxidase is one of the major sources of superoxide, O2-, in the heart (20). Following MI, NADPH oxidase expression is significantly increased in the infarcted myocardium (21), suggesting that ROS production is enhanced. However, the importance of NADPH oxidase expression on cardiac repair and remodeling remain to be elucidated. In the current study by using experimental MI model created by coronary artery ligation in wild type and NADPH oxidase (gp91phox) gene knockout mice, we sought to determine the role of NADPH oxidase on development of cardiac oxidative stress and its related molecular and cellular events including hypertrophy, myocyte apoptosis, cell proliferation, inflammatory and fibrogenic responses in the infarcted heart.
MATERIAL AND METHODS
Animal Model
Left ventricular anterior transmural MI was created in 8-week-old male wild-type C57BL/6J mice and gp91phox knockout mice by permanent ligation of the left coronary artery with silk ligature (22). Mice were anesthetized, intubated and ventilated with a rodent respirator. After left thoracotomy, the heart was exposed and 7-0 silk suture placed around the left coronary artery. The vessel was ligated, the chest closed and lungs reinflated using positive end-expiratory pressure. Animals were sacrificed on day 3, 7, 14 and 28 (n=10/time point/group). Sham-operated mice served as controls. Hearts were removed, rinsed in cold normal saline, weighed, frozen in isopentane with dry ice, and kept at −80°C. Serial cryostat coronal sections were prepared for the following studies. This study was approved by the University of Tennessee Health Science Center Animal Care and Use Committee.
In Situ Hybridization
The localization and optical density of cardiac gp91phox, transforming growth factor (TGF)-β1, type I collagen and tissue inhibitor of metalloprotease (TIMP)- 2 mRNAs were detected by quantitative in situ hybridization. In brief, cardiac sections (16μm) were fixed in 4% formaldehyde for 10 min, washed with phosphate-buffered saline (PBS, pH 7.4), and incubated in 0.25% acetic anhydride in 0.1M TE-HCl for 10 min. Sections were then hybridized overnight with [35S]dATP-labeled DNA probes for gp91phox, TGF-β1, TIMP-2, and type I collagen at 45°C. The hybridized sections were then washed, dried, and subsequently exposed to Kodak Biomax X-ray film. After exposure, the film was developed and sections were stained with hematoxylin and eosin. Quantitation of mRNA optical density was performed using a computer image analysis system (NIH Image, 1.60) (23).
Immunohistochemistry
The appearance, population and localization of macrophages, lymphocytes, myofibroblasts and proliferating cells as well as expression of 3-nitrotyrosine in the mouse heart were detected by immunohistochemistry. Cardiac sections (6μm) were air-dried, fixed in 10% buffered formalin for 5 min, and washed in PBS for 10 min. Sections were then incubated with the primary antibody against ED1 (macrophages), CD4 (lymphocytes), α–smooth muscle actin (SMA) (myofibroblasts) (Sigma, St Louis, MO), and Ki67 (proliferating cells) (ID Labs, London, ON Canada) and 3-nitrotyrosine (Cayman Chemical Co, Ann Arbor, MI) for 1 h at room temperature. Sections were then incubated with IgG-peroxidase conjugated secondary antibody (Sigma, St. Louis, MO) for 1 h at room temperature, washed in PBS for 10 min, and incubated with 0.5 mg/mL diaminobenzidine tetrahydrochloride 2-hydrate + 0.05% H2O2 for 5 min. Negative control sections were incubated with secondary antibody alone. All sections were counterstained with hematoxylin, dehydrated, mounted, and viewed by light microscopy (24).
TUNEL
Apoptosis was detected by TUNEL technique using an Apop Tag Fluorescein kit (Intergen, Norcross, GA). In brief, cardiac sections (6μm) were fixed in 1% faraformaldehyde, followed by incubation with TdT enzyme. Sections were then incubated with antidigoxigenin conjugated with fluorescein. Sections were counterstained with Propidium iodide (PI), mounted and viewed by fluorescence microscopy (25). TUNEL-positive nuclei were distinguished from the TUNEL-negative nuclei by counterstaining with PI and were counted after being photographed. The percentage of nuclei labeled by TUNEL per unit of cells stained with PI nuclear dye reflected the apoptotic index.
Cardiac Morphology
Cardiac sections (6μm) were prepared to determine the fibrillar collagen accumulation by collagen-specific picrosirius red staining and observed by light microscopy as previously reported (26). Collagen volume fraction of each section was determined using a computer image analysis system (NIH image, 1.60) as previously reported (26).
Infarction Size/Thickness
Cardiac sections (6μm) were taken at the apex, mid-cavity, and base and stained with H&E. Infarct size were determined as the mean percentage of epicardial and endocardial circumference occupied by infarct tissue for the three sections as previously described by Pfeffer et al (27). Infarcted myocardium thickness was measured using a computer image analysis system (NIH Image, 1.60).
Statistical Analysis
Statistical analysis of ratio of heart weight, infarct size, wall thickness, gene expression and apoptosis findings was performed using analysis of variance. Values are expressed as mean±SEM with P<0.05 considered significant. Multiple group comparisons among controls and each group were made by Scheffé’s F-test.
RESULTS
Cardiac Gene Expression of gp91phox following MI
Low gp91phox mRNA levels were observed in the normal heart of wild type mice (Figure 1, panel A). Following MI, gp91phox mRNA levels were significantly increased in the infarcted myocardium on day 3, reached peak on day 7 (Figure 1, panel B) and gradually declined thereafter. However, gp91phox mRNA remained unchanged in noninfarcted myocardium compared to controls. Cardiac gp91phox mRNA was not detectable in gp91phox knockout mice (panel C). The quantitative gp91phox mRNA data in the infarcted myocardium of wild type mice are shown in Figure 1, panel D.
Figure 1.
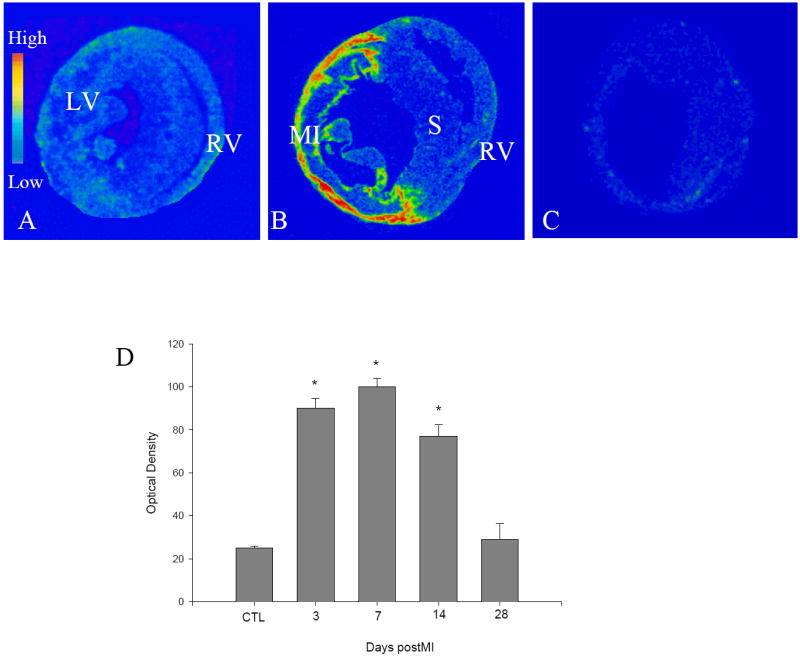
Cardiac gp91phox gene expression in wild type mice. In sham-operated heart, low levels of gp91phox mRNA were present in both left and right ventricles (LV, RV) (panel A). On day 7 postMI, gp91phox mRNA levels were largely increased at the site of MI, but not in septum (S) and right ventricle (panel B). Cardiac gp91phox mRNA was not detectable in gp91phox knockout mice (panel C). Panel D shows temporal response of gp91phox mRNA in the infarcted myocardium. * p<0.05 vs controls (CTL).
Heart Weight, Infarct Size/Thickness and Myocyte Hypertrophy
Compared to controls, heart weight was continuously increased in both wild type and gp91phox knockout mice following MI, indicating the development of hypertrophy in noninfarcted myocardium. However, there was no significant difference in the heart weight between wild type and gp91phox knockout mice (Figure 2). Myocyte hypertrophy became evident in the noninfarcted left ventricle (septum) in both groups at week 2 and 4 postMI. Left ventricle infarct size and thickness (Figure 2) were similar between wild type and gp91phox knockout mice at all studied time points.
Figure 2.

Heart weight, infarct size and infarct thickness in wild type and gp91phox knockout (gp91phox-/-) mice.
Cells Involved in Cardiac Repair/Remodeling and Proliferation
By immunohistochemistry, we detected cells involved in cardiac inflammatory/fibrogenic responses following MI. In wild type mice with MI, we observed accumulated ED-1 positive macrophages at the site of infarction on day 3. The population of macrophages reached peak on day 7 (Figure 3, panel B) and gradually declined thereafter. A small population of lymphocytes was present at the site of MI (Figure 3, panel D). Inflammatory cell accumulation was not seen in noninfarcted myocardium.
Figure 3.

Cells involved in cardiac repair following MI in wild type mice. ED-1+ macrophages, CD3+ lymphocytes and Ki67+ proliferating cells were rarely seen in the normal heart (panels A, C, and E, respectively). On day 7 postMI, macrophages were accumulated at the site of MI (panel B, brown), while only a few lymphocytes were seen in the same site (panel D, arrows). Abundant proliferating cells (brown) including fibroblast-like cells (arrows), vascular endothelial and smooth muscle cells (V), were present in the infarcted myocardium (panel F). α-SMA+ myofibroblasts were not present in the normal heart (panel G, arrows: α-SMA+ vascular smooth muscle cells). On day 7 postMI, myofibroblasts appeared and accumulated in the infarcted myocardium (panel H, brown).
Cell proliferation was detected by immunohistochemical Ki67 staining. Proliferating cells were rarely seen in the normal heart (Figure 3, panel E). Following MI, abundant proliferating cells appeared at infarct site, particularly on day 7 (Figure 3, panel F). The majority of these proliferating cells are fibroblast-like cells and newly-formed vascular smooth muscle cells and endothelial cells.
Myofibroblasts are phenotypically transformed fibroblasts and play a major role on collagen synthesis in the injured tissue. A hallmark of these cells is the expression of α-smooth muscle actin (SMA). By immunohistochemical α-SMA labeling, we detected involvement of myofibroblasts on cardiac repair in mice following MI. Normal myocardium does not contain myofibroblasts (Figure 3, panel G). Following MI, myofibroblasts began to appear in the infarcted myocardium on day 3, became abundant on day 7 (Figure 3, panel H) and 14 and gradually declined thereafter. Myofibroblasts were not seen in noninfarcted myocardium.
Compared to wild type mice with MI, inflammatory cell response, cell proliferation and appearance of myofibroblasts were similar to those found in gp91phox knockout mice.
Cardiac Apoptosis
By using TUNEL technique, we detected apoptosis in the infarcted heart. Apoptotic cells were rarely seen in the normal mouse heart (Figure 4, panel A). On day 3 postMI, we observed abundant apoptotic cells in the infarcted myocardium, particularly at the border zones, the regions between infarcted and noninfarcted myocardium in both wild-type (Figure 4, panel B) and gp91phox knockout mice (not shown). The majority of cells undergoing apoptosis at this time point were myocytes. The number of apoptotic myocytes was largely reduced in the infarcted myocardium in the later time points, while most apoptotic cells were inflammatory cells on day 7 (Figure 4, panel C) and 14 and myofibroblasts on day 28 (Figure 4, panel D). There was, however, no significant difference in apoptotic rate in the infarcted myocardium between wild type and gp91phox knockout mice (Figure 4, panel E).
Figure 4.
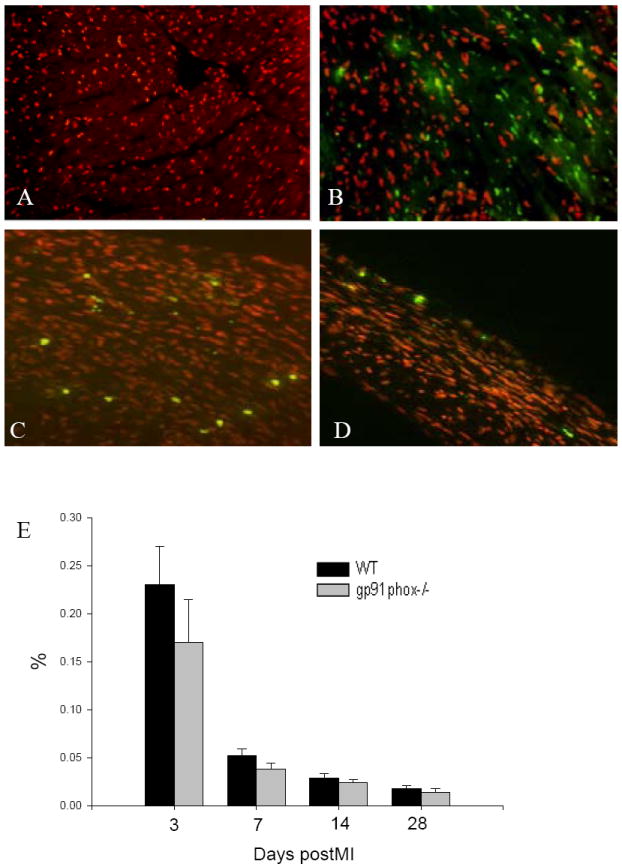
Apoptosis in the infarcted mouse heart. Apoptotic cells were not observed in the normal mouse heart (panel A). Following MI, numerous apoptotic myocytes (bright spots) were seen in the infarcted myocardium on day 3 (panel B). Apoptotic inflammatory cells (panel C) and myofibroblasts (panel D) became evident in the infarcted myocardium of wild type mice on day 7 and 28, respectively. Temporal cardiac apoptosis data in both wild type and gp91phox knockout mice are shown in panel E.
Cardiac Gene Expression of TGF-β1, TIMP-2 and Type I Collagen
As detected by quantitative in situ hybridization, low levels of TGF-β1 (Figure 5, panel A), TIMP-2 (Figure 5, panel C) and type I collagen (Figure 5, panel E) mRNAs were observed in the heart of both wild type and gp91phox knockout mice. On day 3 and 7 following MI, TGF-β1 mRNA was significantly increased at the site of MI in both wild type (Figure 5, panel B) and gp91phox knockout mice and then declined to normal levels on day 14 and 28. TIMP-2 mRNA was significantly elevated in the infarcted myocardium on day 3, peaked on day 7 (Figure 5, panel D) and declined thereafter, but still remained significantly higher than controls in both wild-type and gp91phox knockout mice. Compared to controls, type I collagen mRNA was significantly increased in the infarcted myocardium on day 3, reached peak on day 7 (Figure 5, panel F), declined but remained significantly higher on day 14 and returned to normal levels on day 28 in both wild type and gp91phox gene knockout mice. However, there is no significant difference in cardiac TGF-β1, TIMP-2 and type I collagen mRNA levels between wild type and gp91phox gene knockout mice at any studied time point. The quantitative data on TGF-β1, TIMP-2 and type I collagen mRNAs in the infarcted myocardium are shown in Figure 6.
Figure 5.
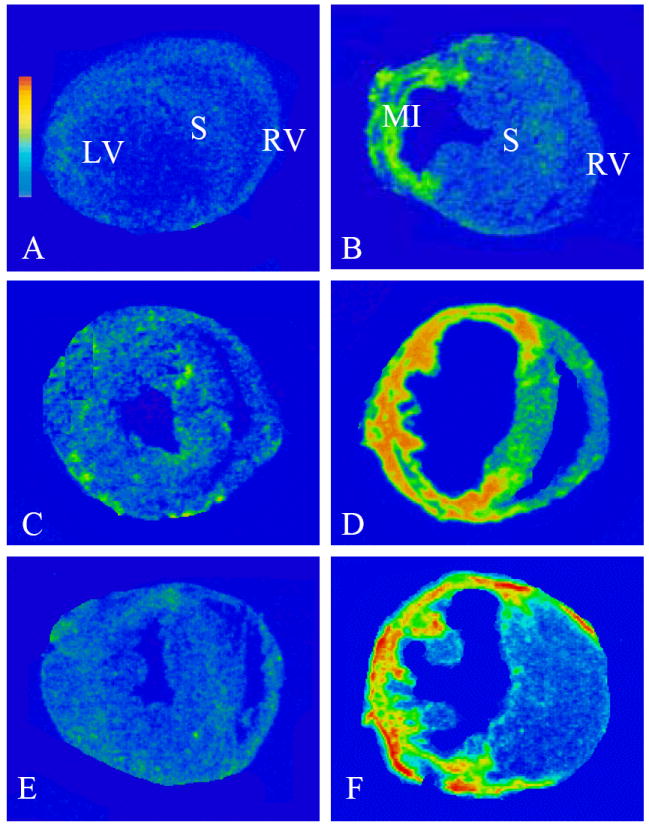
Gene expression of profibrogenic mediators in the infarcted wild type mouse heart. Detected by in situ hybridization, low levels of TGF-β1 (panel A), TIMP-2 (panel C), and type I collagen (panel E) mRNAs are located in both left and right ventricles (LV, RV). On day 7 postMI, TGF-β1 (panel B), TIMP-2 (panel D), and type I collagen (panel F) mRNAs were largely increased in the infarcted myocardium.
Figure 6.
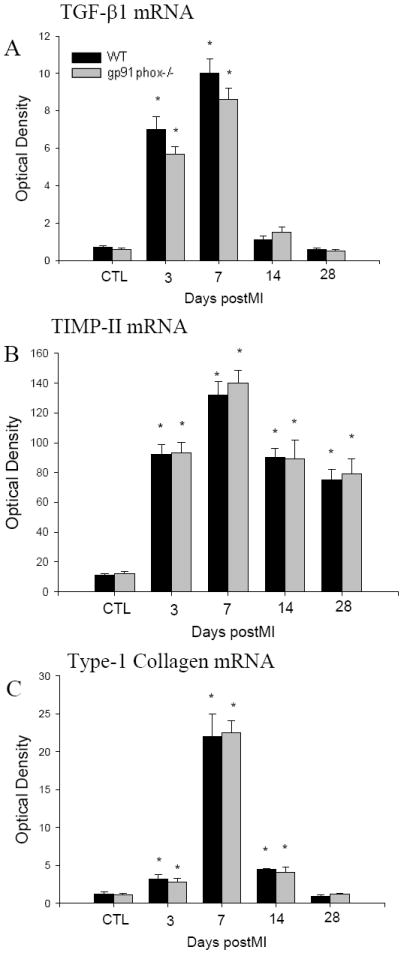
Temporal response of cardiac TGF-β1, TIMP-2 and type I collagen gene expression in the infarcted heart of wild type and gp91phox knockout mice.
Cardiac Collagen Volume
By collagen-specific picrosirius red staining, we observed a small amount of collagen in the cardiac interstitial space in both wild type (Figure 7, panel A) and gp91phox knockout mice (not shown). Following MI, collagen started to accumulate at the infarcted myocardium on day 3 and continuously increased over the course of 4 weeks in both wild type and gp91phox knockout mice. However, collagen volume in the infarcted myocardium was not significantly different between wild type and gp91phox knockout mice (Figure 7). Collagen volume was not significantly elevated in noninfarcted myocardium at any time point.
Figure 7.

Cardiac collagen volume and 3-nitrotyrosine expression. A small amount of collagen (red) is present in the interstitial space (pane A, arrows). Following MI, collagen volume is largely increased at the site of MI on day 28 (pane B). Temporal response of collagen volume fraction at the site of MI in both wild type and gp91phox knockout mice is shown in panel C. On day 7 postMI, 3-nitrotyrosine is similarly expressed in the inflammatory cells at the site of MI in wild type and gp91phox knockout mice (panels D and E, respectively).
Expression of 3-nitrotyrosine
Superoxide can react with nitric oxide to form short-lived peroxynitrite. Peroxynitrite forms stable 3-nitrotyrosine, which serves as a marker of oxidative stress (28). By immunohistochemistry, 3-nitrotyrosine is negatively labeled in the normal myocardium of both wild type and gp91phox knockout mice. Following MI, 3-nitrotyrosine is extensively expressed in the infarcted myocardium in both wild type and gp91phox knockout mice, which is most evident on day 7 and cells expressing 3-nitrotyrosine were primarily inflammatory cells (Figure 7).
DISCUSSION
Ventricular structural remodeling after MI is characterized by cardiac repair in the infarcted myocardium as well as interstitial fibrosis and hypertrophy in noninfarcted sites (4-6). Increasing evidence from both animal and human studies has demonstrated that cardiac oxidative stress appearing following MI plays an important role in the pathogenesis of cardiac remodeling and progression of cardiac dysfunction (7, 29-30). NADPH oxidase is a major source of superoxide production in the heart. Herein we addressed the importance of NADPH oxidase on cardiac molecular and cellular events related to cardiac repair/remodeling that appear within the infarcted heart.
Firstly, this study has demonstrated that in wild type mice, gp91phox expression was significantly increased in the infarcted myocardium, particularly in the early stage of MI. The elevation of NADPH oxidase in the infarcted heart may result in enhanced ROS production. The occurrence of cardiac oxidative stress has also been demonstrated in rats and mice with MI (13-14). ROS in low concentrations serve as signaling molecules (31). However, these agents elicit harmful effects when produced in excess (32). Experimental studies have also demonstrated that oxidative stress can induce most of the changes that contribute to myocardial remodeling. Chronic antioxidant treatment has been shown to suppress cardiac oxidative stress, attenuate ventricular remodeling, and improve left ventricular function in rats or mice with experimental MI (12-14).
Secondly, we addressed oxidative stress related molecular and cellular events that appear within the infarcted mouse heart. We explored cells involved in cardiac repair/remodeling and the expression of profibrogenic mediators. Following MI, monocyte-derived macrophages were found to be the major inflammatory cells in the infarcted myocardium. Macrophages are responsible for cytokine release, phagocytosis and induction of oxidative stress in the repairing tissue. Enhanced cardiac gp91phox in wild type mice is primarily expressed by macrophages. After week 2 postMI, macrophages gradually disappeared from the infarcted heart, which is coincident with the decline in gp91phox expression in the late stage of MI. Mediated by ROS, macrophages have been demonstrated to stimulate proinflammatory/profibrogenic cytokine release in the repairing tissue, that include tissue necrosis factor (TNF)-α and TGF-β. This study has shown enhanced cardiac TGF-β1 expression, which is spatially coincident with macrophages in the infarcted myocardium. TGF-β1 is the key stimulator on the differentiation/proliferation of myofibroblasts and their collagen synthesis in the repairing tissue. Myofibroblasts are phenotypically transformed fibroblasts, which are not present in the normal tissue, but appear in the repairing tissue, where they play a key role on fibrous tissue formation (33). Following MI, abundant myofibroblasts were observed at the site of infarction and are co-localized with enhanced TGF-β1 and collagen expression. Myofibroblasts are, however, not seen in noninfarcted myocardium. TGF-β also stimulates the synthesis of TIMPs. TIMPs primarily inhibit collagen degradation by matrix metalloproteinases and therefore facilitate collagen accumulation (34). TIMP expression is tightly controlled at the transcription level. In the current study, we observed significantly elevated TIMP-2 gene expression at the site of infarction. Thus, the imbalance of cardiac collagen synthesis and degradation that appears in the infarcted heart results in fibrous tissue accumulation.
We then detected cardiac apoptosis and hypertrophy following MI. Loss of myocytes is an important mechanism in the development of myocardial remodeling and cardiac failure. Myocyte apoptosis has been shown to occur in the infarcted myocardium in rats (24). In the current study, we observed abundant apoptotic myocytes at the site of MI, particularly at day 3 and 7 postMI. However, myocyte apoptosis was not evident in noninfarcted myocardium. In the late stage of MI, apoptotic macrophages and myofibroblasts became evident in the infarcted myocardium. These repairing cells are transient cells in the infarcted heart and are finally eliminated through apoptosis when cardiac repair is completed. The regulation of myocyte apoptosis involves multiple mechanisms. ROS have been proven to be one of the stimulators of myocyte apoptosis (35-37). Treatment with the antioxidants has been indicated to attenuate oxidative stress and cardiomyocyte apoptosis (38-40). Following MI, myocardial hypertrophy is developed in non-infarcted left ventricle, which is most evident at week 4. Extracellular stimuli such as mechanical strain, neurohormones or cytokines have been well recognized to promote myocyte hypertrophy. Recent studies have further demonstrated that these extracellular stimuli may mediate myocyte hypertrophy via oxidative stress.
Finally, we studied the effect of NADPH oxidase on cardiac oxidative stress and myocardial repair/remodeling following MI in mice. Frantz et al have recently reported that lack of gp91phox did not improve cardiac function, infarct size and systemic oxidative stress (41). In the current study, we further examined the potential effect of gp91phox on cardiac oxidative stress and its related inflammatory response, cell proliferation, fibrogenic response, hypertrophy and apoptosis in the infarcted heart. We found that gp91phox expression was significantly elevated with oxidative stress occurring at the site of infarction in wild type mice, particularly in the first week postMI. However, in gp91phox deficiency mice, cardiac oxidative stress was also developed and oxidative stress related cardiac inflammatory/fibrogenic responses, myocyte apoptosis and hypertrophy as well as infarct size were similar to those found in wild-type mice. The protective effects of gp91phox deficiency on cardiac repair/remodeling were therefore not evident. Even though NADPH oxidase is a major source of superoxide, it has been reported that oxidative stress in the injured heart can be derived from other sources. In addition to NADPH oxidase, ROS can also be produced intracellularly through electron leakage from the mitochondria during oxidative phosphorylation and through the activation of several cellular enzymes, including xanthine oxidase and nitric oxide synthase. Therefore, during gp91phox deficiency, appearance of oxidative stress in the infarcted heart might be due to compensating ROS production by other sources. Furthermore, Frantz et al’s study showed that systemic oxidative stress was also not reduced in gp91phox gene knockout mice with MI (41). In addition to gp91phox, NADPH oxidase is composed of several other subunits including p22phox, p40phox, p47phox, p67phox and racl. Doerries et al have recently indicated that lack of p47phox improved cardiac remodeling and ventricular function, indicating the importance of p47phox in cardiac remodeling following MI (42). This observation suggests that the individual subunits of NADPH oxidase may differ in the effects they have on cardiac remodeling postMI. The potential role of gp91phox in the heart has been studied in other models. Maytin et al have indicated that myocardial hypertrophy induced by chronic pressure overload after aortic constriction was also not affected in gp91phox knockout mice (43). However, another study has shown the reduced cardiotoxicity following doxorubicin treatment in gp91phox knockout mice (44). Taking together, these studies suggest that gp91phox has different functions in cardiac remodeling depending on the nature of cardiac damage. In addition to the heart, it has also been reported that in intrapulmonary arteries, endothelial dysfunction induced by chronic hypoxia depends on gp91phox (45).
In summary, the current study has examined the spatial and temporal responses related to cardiac remodeling following MI including cells responsible for cardiac repair, cardiac apoptosis, inflammatory/fibrogenic responses and hypertrophy in mice. By using gp91phox knockout mice, we detected the importance of NADPH oxidase on cardiac oxidative stress and repair/remodeling following MI. Our results have indicated that NADPH oxidase deficiency does not suppress cardiac oxidative stress and is not protective to cardiac remodeling postMI. Our findings indicate that during NADPH oxidase deficiency, superoxide production can be compensated by other sources, leading to cardiac oxidative stress and its related molecular/cellular events in the infarcted heart.
Acknowledgments
The authors wish to gratefully acknowledge the skillful technical assistance of Yuanjian Chen and Youde Jiang.
This work was supported by the National Heart, Lung, and Blood Institute (RO1-HL077668 to Yao Sun).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olivetti G, Capasso JM, Meggs LG, Sonnenblick EH, Anversa P. Cellular basis of chronic ventricular remodeling after myocardial infarction in rats. Circ Res. 1991;68:856–869. doi: 10.1161/01.res.68.3.856. [DOI] [PubMed] [Google Scholar]
- 2.Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395–403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 3.Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res. 2005;66:22–32. doi: 10.1016/j.cardiores.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 4.Anversa P, Li P, Zhang X. Ischemic myocardial injury and ventricular remodeling. Cardiovasc Res. 1993;27:145–157. doi: 10.1093/cvr/27.2.145. [DOI] [PubMed] [Google Scholar]
- 5.Francis GS, Mcdonald K, Chu G, Cohn JN. Pathophysiologic aspects of end-stage heart failure. Am J Cardio. 1995;75:11A–16A. doi: 10.1016/s0002-9149(99)80378-6. [DOI] [PubMed] [Google Scholar]
- 6.Weber KT. Extracellular matrix remodeling in heart failure. Circulation. 1997;96:4065–4082. doi: 10.1161/01.cir.96.11.4065. [DOI] [PubMed] [Google Scholar]
- 7.Hill MF, Singal PK. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol. 1996;148:291–300. [PMC free article] [PubMed] [Google Scholar]
- 8.Fukui T, Yoshiyama M, Hanatani A, Omura T, Yoshikawa J, Abe Y. Expression of p22-phox an dgp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem Biophys Res Commun. 2001;281:1200–1206. doi: 10.1006/bbrc.2001.4493. [DOI] [PubMed] [Google Scholar]
- 9.Lu L, Quinn MT, Sun Y. Oxidative stress in the infarcted heart: role of de novo angiotensin II production. Biochem Biophys Res Commun. 2004;325:943–951. doi: 10.1016/j.bbrc.2004.10.106. [DOI] [PubMed] [Google Scholar]
- 10.Usal A, Acarturk E, Yuregir GT, Unlukurt I, Demirci C, Kurt GI, Birand A. Decreased glutathione levels in acute myocardial infarction. Jpn Heart J. 1996;37:177–182. doi: 10.1536/ihj.37.177. [DOI] [PubMed] [Google Scholar]
- 11.Khaper N, Kaur K, Li T, Farahmand F, Singal PK. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol Cell Biochem. 2003;251:9–15. [PubMed] [Google Scholar]
- 12.Sia YT, Parker TG, Liu P, Tsoporis JN, Adam A, Rouleau JL. Imporved post-myocardial infarction survival with probucol in rats: effects on left ventricular function, morphology, cardiac oxidative stress and cytokine expression. J Am Col Cardiol. 2002;39:148–156. doi: 10.1016/s0735-1097(01)01709-0. [DOI] [PubMed] [Google Scholar]
- 13.Sia YT, Lapointe N, Parker T, Tsoporis JN, Deschepper CF, Calderone A, Pourdjabbar A, Jasmin JF, Sarrazin JF, Liu P, Adam A, Butany J, Rouleau JL. Beneficial effects of long-term use of the antioxidant probucol in heart failure in the rat. Circulation. 2002;105:2549–2555. doi: 10.1161/01.cir.0000016721.84535.00. [DOI] [PubMed] [Google Scholar]
- 14.Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S, Utsumi H, Takeshita A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res. 2000;87:392–398. doi: 10.1161/01.res.87.5.392. [DOI] [PubMed] [Google Scholar]
- 15.Hare JM. Oxidative stress and apoptosis in heart failure progression. Circ Res. 2001;89:198–201. [PubMed] [Google Scholar]
- 16.Poli G, Parola M. Oxidative damage and fibrogenesis. Free Radical Biol Med. 1997;22:287–305. doi: 10.1016/s0891-5849(96)00327-9. [DOI] [PubMed] [Google Scholar]
- 17.Gorlach A, Kietzmann T, Hess J. Redox signaling through NADPH oxidases: involvement in vascular proliferation and coagulation. Ann N Y Acad Sci. 2002;973:505–507. doi: 10.1111/j.1749-6632.2002.tb04691.x. [DOI] [PubMed] [Google Scholar]
- 18.Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Whorton AR, Hoidal JR. A NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am J Physiol Cell Physiol. 2002;282:C1212–C1224. doi: 10.1152/ajpcell.00496.2001. [DOI] [PubMed] [Google Scholar]
- 19.Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:851–859. doi: 10.1016/s0022-2828(03)00145-7. [DOI] [PubMed] [Google Scholar]
- 20.Mohazzab-H KM, Kaminski PM, Wolin MS. Lactate and PO2 modulate superoxide anion production in bovine cardiac myocytes: potential role of NADH oxidase. Circulation. 1997;15:614–620. doi: 10.1161/01.cir.96.2.614. [DOI] [PubMed] [Google Scholar]
- 21.Fukui T, Yoshiyama M, Hanatani A, Omura T, Yoshikawa J, Abe Y. Expression of p22-phox and gp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem Biophys Res Commun. 2001;281:1200–1206. doi: 10.1006/bbrc.2001.4493. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y, Zhang JQ, Zhang J, Ramires FJ. Angiotensin II, transforming growth factor-beta1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–1569. doi: 10.1006/jmcc.1998.0721. [DOI] [PubMed] [Google Scholar]
- 23.Zhao W, Ahokas RA, Weber KT, Sun Y. ANG II-induced cardiac molecular and cellular events: role of aldosterone. Am J Physiol Heart Circ Physiol. 2006;291(1):H336–43. doi: 10.1152/ajpheart.01307.2005. [DOI] [PubMed] [Google Scholar]
- 24.Zhao W, Lu L, Chen SS, Sun Y. Temporal and spatial characteristics of apoptosis in the infarcted rat heart. Biochem Biophys Res Commun. 2004;325(2):605–11. doi: 10.1016/j.bbrc.2004.10.064. [DOI] [PubMed] [Google Scholar]
- 25.Hunter AL, Choy JC, Granville DJ. Detection of apoptosis in cardiovascular diseases. Methods Mol Med. 2005;112:277–289. doi: 10.1385/1-59259-879-x:277. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y, Ratjaska A, Weber KT. Inhibition of angiotensin-converting enzyme and attenuation of myocardial fibrosis by lisinopril in rats receiving angiotensin II. J Lab Clin Med. 2005;126:95–101. [PubMed] [Google Scholar]
- 27.Pfeffer MA, Pfeffer Jam Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, Brounwald E. Myocardial infarct size and ventricular function in rats Circ Res. 1979;44:503–512. doi: 10.1161/01.res.44.4.503. [DOI] [PubMed] [Google Scholar]
- 28.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 29.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Hadal-Ginard B, Kajstura J, Leri A, Anversa P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89(3):279–86. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 30.Hill MF, Singal PK. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. 1997;96:2414–2420. doi: 10.1161/01.cir.96.7.2414. [DOI] [PubMed] [Google Scholar]
- 31.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–52. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 32.Desmouliere A, Gabbiani G. The role of the myofibroblast in wound healing and fibrocontractive diseases. In: Richard AF, editor. The molecular and cellular biology of wound repair. New York, NY: Plenum Press; 1996. pp. 391–423. [Google Scholar]
- 33.Docherty AJ, Murphy G. The tissue metalloproteinase family and the inhibitor TIMP: a study using cDNAs and recombinant proteins. Ann Rheum Dis. 1990;49:469–479. [PubMed] [Google Scholar]
- 34.Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–1292. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 35.Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 36.Kumar D, Kirshenbaum LA, Li T, Danelisen I, Singal PK. Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Antioxid Redox Signal. 2001;3:135–145. doi: 10.1089/152308601750100641. [DOI] [PubMed] [Google Scholar]
- 37.von Harsdorf R, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation. 1998;99:2934–2941. doi: 10.1161/01.cir.99.22.2934. [DOI] [PubMed] [Google Scholar]
- 38.Li WG, Coppey L, Weiss RM, Oskarsson HJ. Antioxidant therapy attenuates JNK activation and apoptosis in the remote noninfarcted myocardium after large myocardial infarction. Biochem Biophys Res Commun. 2001;280:353–357. doi: 10.1006/bbrc.2000.4134. [DOI] [PubMed] [Google Scholar]
- 39.Zeng H, Liu X, Zhao H. Effects of carvedilol on cardiomyocyte apoptosis and gene expression in vivo after ischemia-reperfusion in rats. J Huazhong Univ Sci Technolog Med Sci. 2003;23:127–130. doi: 10.1007/BF02859934. [DOI] [PubMed] [Google Scholar]
- 40.Kumar D, Jugdutt BI. Apoptosis and oxidants in the heart. J Lab Clin Med. 2003;142:288–297. doi: 10.1016/S0022-2143(03)00148-3. [DOI] [PubMed] [Google Scholar]
- 41.Frantz S, Brandes RP, Hu K, Rammelt K, Wolf J, Scheuermann H, Ertl G, Bauersachs J. Left ventricular remodeling after myocardial infarction in mice with targeted deletion of the NADPH oxidase subunit gp91PHOX. Basic Res Cardiol. 2006;101:127–132. doi: 10.1007/s00395-005-0568-x. [DOI] [PubMed] [Google Scholar]
- 42.Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, Sorrentino S, Manes C, Schieffer B, Drexler H, Landmesser U. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res. 2007;100:894–903. doi: 10.1161/01.RES.0000261657.76299.ff. [DOI] [PubMed] [Google Scholar]
- 43.Maytin M, Siwik DA, Ito M, Xiao L, Sawyer DB, Liao R, Colucci WS. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox. Circulation. 2004;109:1168–1171. doi: 10.1161/01.CIR.0000117229.60628.2F. [DOI] [PubMed] [Google Scholar]
- 44.Deng S, Kruger A, Kleschyov AL, Kalinowski L, Daiber A, Wojnowski L. Gp91phox-containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Fee Radic Biol Med. 2007;42:466–473. doi: 10.1016/j.freeradbiomed.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 45.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol. 2006;148:714–723. doi: 10.1038/sj.bjp.0706779. [DOI] [PMC free article] [PubMed] [Google Scholar]