

Abstract
Elucidation of biochemical, physiological, and cellular contributions to metabolic images of brain is important for interpretation of images of brain activation and disease. Discordant brain images obtained with [14C]deoxyglucose (DG) and [1- or 6-14C]glucose were previously ascribed to increased glycolysis and rapid [14C]lactate release from tissue, but direct proof of [14C]lactate release from activated brain structures is lacking. Analysis of factors contributing to images of focal metabolic activity evoked by monotonic acoustic stimulation of conscious rats reveals that labeled metabolites of [1- or 6-14C]glucose are quickly released from activated cells due to decarboxylation reactions, spreading via gap junctions, and efflux via lactate transporters. Label release from activated tissue accounts for most of the additional [14C]glucose consumed during activation compared to rest. Metabolism of [3,4-14C]glucose generates about four times more [14C]lactate compared to 14CO2 in extracellular fluid suggesting that most lactate is not locally oxidized. In brain slices, direct assays of lactate uptake from extracellular fluid demonstrate that astrocytes have faster influx and higher transport capacity than neurons. Also, lactate transfer from a single astrocyte to other gap junction-coupled astrocytes exceeds astrocyte-to-neuron lactate shuttling. Astrocytes and neurons have excess capacities for glycolysis, and oxidative metabolism in both cell types rises during sensory stimulation. The energetics of brain activation is quite complex and the proportion of glucose consumed by astrocytes and neurons, lactate generation by either cell type, and the contributions of both cell types to brain images during brain activation are likely to vary with the stimulus paradigm and activated pathways.
Keywords: Functional brain imaging, brain activation, glucose utilization, deoxyglucose, glucose, lactate, astrocyte, neuron, gap junction, lactate trafficking
IN VIVO METABOLIC BRAIN IMAGING
Glucose utilization tracers
Because mammalian brain is dependent on glucose as its primary fuel and changes in functional activity of brain alter local energy demand, development of the [14C]deoxyglucose ([14C]DG) method in the 1970's by Louis Sokoloff and colleagues1 to determine local rates of glucose utilization in living brain opened a new window to visualize and quantify brain activities in health and disease2. During the past three decades, brain imaging with [14C]DG and [18F]fluorodeoxyglucose (FDG) has enabled a wide variety of studies to evaluate brain functions in experimental animals and human subjects, as well as effects of pharmaceutical agents and the progression and treatment of neurological disorders. The contributions of different cell types to brain images and the downstream fate of the additional glucose consumed during brain activation over and above that utilized during ‘rest’ (i.e., when no specific stimulus is given to the subject) have been very important issues for decades due to technical difficulties associated with (i) determination of co-registration of metabolic tracers and cell-type markers and (ii) accounting for all of the label derived from labeled glucose after its metabolism at the hexokinase step, but considerable progress has been made towards these goals.
Metabolite trapping
Functional studies of brain activation using autoradiographic, positron emission tomographic (PET), magnetic resonance spectroscopic (MRS), or fluorescence microscopic technologies all rely on changes in metabolism of exogenous labeled tracers or of endogenous compounds to generate metabolites that are registered as signals, and all of these methodologies are, therefore, dependent on local trapping of metabolic products in activated cells. For example, conversion of [14C]DG or [18F]FDG by hexokinase to the corresponding hexose-6-phosphate (hexose-6-P) produces a ‘dead end’ metabolite (Fig. 1) that is not further metabolized by the glycolytic pathway1. Although some conversion of DG-6-P to other compounds can occur in brain, the labeled products are either phosphorylated or are incorporated into macromolecules3-6. These compounds are also trapped in the cell where the precursor was metabolized by hexokinase for the duration of the routine experimental period which is typically 45 min, but can range from about 30-60 min for fully quantitative studies1,2. Thus, the [14C]DG method has the advantage that hexokinase activity can be accurately measured in vivo in the midst of all other biochemical reactions in the brain. Under steady state conditions the rate of any step in a multi-step pathway is equal to the flux through the entire pathway, and determination of the rate of phosphorylation of glucose by hexokinase measures the rate of glucose utilization. The 45 min experimental period for [14C]DG takes advantage of the relative stability of [14C]DG-6-P in tissue to minimize the effects of uncertainties in the true values of rate constants for transport and metabolism on calculated rates of glucose utilization1,2. For autoradiographic assays with labeled glucose and acetate, short experimental periods are necessary to minimize loss of labeled products that would occur with longer experimental intervals. However, the metabolic calculated rates obtained from brief, 5-10 min assays with any tracer are not as accurate as desired due to the higher impact of errors in the estimates of the amount of unmetabolized precursor in tissue at the end of the experiment (this value is subtracted from the total 14C concentration to obtain concentration of labeled metabolites) and the integrated specific activity of the precursor pool in brain calculated from that in arterial plasma1, 2, 14, 15.
Fig. 1. Metabolic labeling by deoxyglucose and glucose.

The schematic illustrates the major pathways for metabolism of 2-deoxy-D-glucose (DG), 2-deoxy-2-fluoro-D-glucose (FDG), and glucose, plus sites of 14CO2 release from 14C-labeled glucose.
Sites of release of labeled carbon from glucose metabolites
Assays of rates of glucose utilization with DG or FDG eliminates the influence of rapid loss of diffusible products of glucose, but does not provide information about the downstream fate of the glucose, which is of particular interest during brain activation. Variously-labeled glucose can be used to track glucose utilization, but rapid formation and release from activated cells of labeled diffusible metabolites (e.g., lactate, CO2, glutamine, or other compounds) can limit the use of labeled glucose for determination of overall glucose utilization in brain in vivo (Fig. 1). Nevertheless, careful design of autoradiographic, biochemical, and MRS assays can take advantage of release or retention of specific carbon atoms from metabolites of labeled glucose at specific steps of glucose metabolism to evaluate aspects of the pathway. For example, entry of glucose into the pentose phosphate shunt pathway results in decarboxylation of carbon one (C1) so that differential labeling by [1-14C]- and [6-14C]glucose is a measure of pentose phosphate shunt activity7. Note that lactate retains all 14C label in glucose except that lost via the pentose phosphate shunt (Fig. 1) and if lactate or other metabolites are quickly released from tissue, calculated rates of glucose utilization (estimated as total 14C-metabolites retained in tissue by autoradiographic analysis divided by the precursor time-activity integral) would be underestimated by an amount proportional to the quantity of labeled metabolites lost from tissue. Release of 14CO2 from [3,4-14C]glucose at the pyruvate dehydrogenase step can be used to evaluate pyruvate entry into the tricarboxylic acid (TCA) cycle (Fig.1). Decarboxylation reactions in the TCA cycle release label from the C2 and C5 positions more rapidly than from C1 and C6, but label loss is retarded due to transamination reactions that catalyze exchange of label between α-ketoglutarate and glutamate and between oxaloacetate and aspartate, thereby diluting label into the large unlabeled amino acid pools (Fig. 1). Metabolic modeling in conjunction with MRS assays of the time courses of labeling of specific carbon atoms in TCA cycle-derived amino acids in each subject is used to calculate rates of glucose oxidation in neurons and astrocytes, glutamate-glutamine cycling, and anaplerosis72-76. Finally, changes in tissue NAD(P)H level can be detected and localized by fluorescence microscopy and provide information related to shifts in the net balance between glycolytic and oxidative metabolic pathways under various conditions77-81. Thus, different imaging and spectroscopic techniques can be used to probe specific aspects of glucose utilization and multiple approaches are required to evaluate metabolic changes during activation compared to rest, as well as under abnormal or disease conditions.
Comparison of labeling patterns obtained in parallel studies with labeled DG and labeled glucose during resting conditions and brain activation induced by a various stimuli can be a useful approach to help ‘dissect out’ changes in the metabolic fate of glucose during brain activation. This information, in turn, can help identify cellular processes that are amplified when information processing is stimulated. In the following sections, we summarize studies in our laboratory using imaging approaches to better understand the biology of brain activation and the basis for discrepant images of activation obtained with metabolic tracers.
IMAGING FOCAL BRAIN ACTIVATION WITH DIFFERENT TRACERS
Discordant images
A number of studies in different laboratories compared metabolic brain images obtained in autoradiographic assays using [14C]DG and [1- or 6-14C]glucose as glucose utilization tracers in parallel experiments in brain of conscious subjects in vivo and found that the magnitude of brain activation was greatly underestimated with labeled glucose under normal8-10 and pathophysiological conditions9,11-15. Because autoradiography registers total 14C levels, all labeled metabolites that are retained within tissue would be registered, indicating that diffusible products corresponding to the most of the additional [6-14C]glucose consumed during activation over and above that during rest are released from the activated tissue within the 5 min experimental period; lactate generation and release was suggested as the basis for these discordant brain images8-13. Lactate is an important glucose intermediate because its formation by lactate dehydrogenase helps to sustain glycolytic rate when glycolytic flux exceeds the rate of pyruvate oxidation or the rate of NAD+ regeneration by redox shuttle systems. Lactate is diffusible and rapidly transported along with H+ via monocarboxylic acid transporters, so it can also be used to clear H+ from the cell and serve an oxidative fuel for other cells. Because brain lactate concentration (0.5-1 μmol/g) is higher than the levels of other glycolytic metabolites between glucose-6-phosphate and pyruvate (range: 0.01-0.15 μmol/g), and its specific activity equilibrates with that of blood and brain glucose within 5 min14, 15, 39, any loss of labeled lactate from activated tissue would have a high impact on calculated glucose utilization rates based on total product accumulation. For example, during spreading cortical depression net release of both labeled and unlabeled lactate to cerebral venous blood was equal to about 20% of the labeled and unlabeled glucose entering brain and lactate efflux to blood accounted for about half of the magnitude of underestimation of glucose utilization rate by [6-14C]glucose14,15. As described below, in vivo studies in normal, conscious rats using tracer amounts of labeled compounds that do not alter metabolic fluxes support the likelihood that substantial amounts of labeled lactate are produced during brain activation and quickly released from brain by various routes.
Tonotopic bands in the auditory pathway
To evaluate imaging of focally-activated brain cells, we used a unilateral single tone stimulus to increase signaling and metabolic demand in specific groups of cells in the inferior colliculus of conscious rats10. The inferior colliculus is a good model structure for analysis of metabolite spreading and efflux because it has the highest glucose utilization and blood flow rates in brain, and metabolite dispersal and release processes should be comparably high. A monotonic stimulus increases functional activity in specific regions of the inferior colliculus due to the tonotopic organization of the ascending auditory pathway, thereby raising glucose utilization rate in the activated cells that can be visualized with radiolabeled DG16-18. Auditory projections from the cochlea to cortex are mainly, but not exclusively, to the contralateral hemisphere, and relative activation induced by a unilateral stimulus can be evaluated by assays of left-right differences; this approach minimizes animal number but, due to ipsilateral projections, this hemisphere is not a true ‘resting’ control and the magnitude of activation will be underestimated. Metabolic activity was assayed with two 14C-labeled glucose utilization tracers plus [14C]acetate; glucose is utilized by all brain cells, whereas acetate is preferentially taken up and oxidized by astrocytes19.
Robust tonotopic activation bands are readily detected with [14C]DG using either the routine, fully-quantitative 45 min experimental period or a brief, semi-quantitative 5 min assay interval which was included for direct comparison to brief labeling by other tracers. In contrast, focal activation was only modestly detectable with [6-14C]glucose and was not evident with [1-14C]glucose or with [2-14C]acetate (Fig. 2). Because the overall rate of glucose uptake into brain cells and phosphorylation by hexokinase is about twice as fast as DG1,2, brief tracer studies with [1- or 6-14C]glucose with unilateral activation were predicted to show much larger left-right differences than those obtained with [14C]DG, yet the converse is true (Fig. 2). Ratios of labeling of the major tonotopic band by [14C]DG in the activated to that in the contralateral colliculus were 1.87 and 1.29 for 45 and 5 min experiments, respectively, exceeding the corresponding ratios for overall labeling of the entire inferior colliculus, 1.47 and 1.1810. The mean activated-to-contralateral ratio for the 5 min [14C]DG assay was similar to or exceeded those for [1- or 6-14C]glucose10 even though there would be a greater fraction of unmetabolized [14C]DG in the total label pool in both the activated and contralateral hemisphere compared to [14C]glucose; this precursor background would blunt detection of a stimulus-induced rise in metabolism in brief experiments compared to the routine 45 min assay period in which most of label is [14C]DG-6-P.
Fig. 2. Focal acoustic activation of the inferior colliculus in the conscious rat is underestimated with [1- or 6-14C]glucose compared to [14C]deoxyglucose.
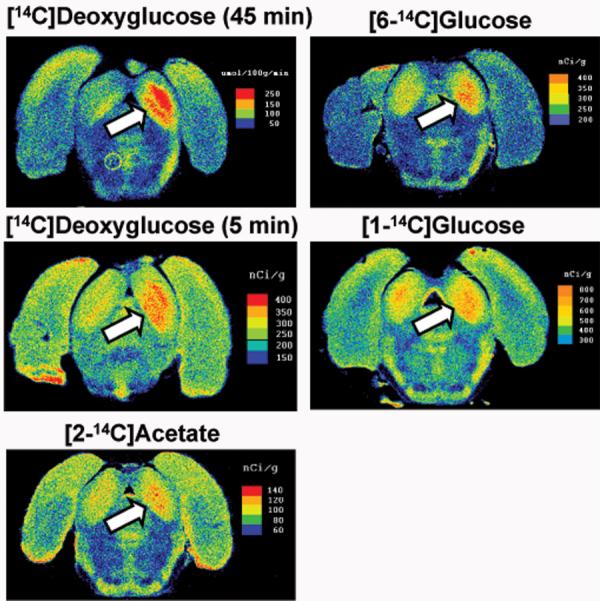
Functional metabolic activity was assayed during a unilateral monotonic (8 kHz tone at 103 dB) acoustic stimulation of conscious rats with the fully quantitative [14C]deoxyglucose (DG) method using the routine 45 experimental period, or using 5 min experimental periods and pulse-labeling with [14C]DG, [1- or 6-14C]glucose, or [2-14C]acetate. Autoradiographs were prepared from serial coronal sections of brain; the higher the optical density the greater the tissue 14C level, which is color coded in each panel as glucose utilization rate or tissue concentration10. Tonotopic bands of activated cells in the inferior colliculus (arrows) in the activated (right) hemisphere are most evident in rats pulse-labeled with [14C]DG for 45 or 5 min; crossover of fibers in the auditory pathway causes some activation of the contralateral inferior colliculus, which is most evident in the [14C]DG autoradiographs. Tonotopic bands are also detectable with [6-14C]glucose, but not with [1-14C]glucose or [2-14C]acetate (see text). DG and glucose are metabolized by all brain cells, whereas acetate is preferentially taken up and oxidized by astrocytes19.
Labeling of the tonotopic band regions in the activated inferior colliculus by 14C-labeled glucose or acetate was only about 13-26% higher than in the contralateral tissue. Greater labeling by [6-14C]glucose compared to [1-14C]glucose suggests increased flux of glucose into the pentose phosphate shunt pathway during acoustic stimulation because label is lost from [1-14C]glucose when 6-phosphogluconate is decarboxylated (Fig. 1). The flux through pentose phosphate shunt pathway is reported to be only ∼5% that of glucose utilization in adult rats, but this pathway has a high capacity that is revealed by inclusion of an artificial electron acceptor in the in vitro reaction mixture7 and detoxification of reactive oxygen species via the glutathione reductase-pentose shunt system may rise during brain activation in conscious rats. In vivo metabolic assays with [2-14C]acetate20,21 demonstrate that astrocytic oxidative metabolism does increase during sensory stimulation of conscious rats but does not progressively rise in proportion to stimulus magnitude during graded visual stimulation and does not generate tonotopic bands during acoustic stimulation. Glutamine is preferentially labeled by [14C]acetate, and glutamine is readily released to the medium during in vitro brain slice experiments (see discussion and cited references in referrence82), suggesting that dispersal of glutamine may contribute to efflux of labeled metabolites from activated astrocytes.
Two key findings were that visualization of tonotopic bands obtained with [1-14C]glucose could be amplified by either halothane anesthesia or by pre-treatment with probenecid, a lactate transport blocker10, implicating intracellular gap junctional trafficking and extracellular diffusion of lactate as important factors involved in dispersal of glucose-derived signals from focally-activated cells. In cultured astrocytes, 0.1-1 mM halothane reduced Lucifer yellow dye spread through gap junctions by about 75%83, whereas in cultured cardiac myocytes a higher halothane level was required to partially (1.5 mM) or fully (2 mM) block gap junctional current84. In our studies, the inhalation dose of halothane was ∼1% during the metabolic labeling assay, and in rats given a halothane inhalation dose of 0.5% or 1.5%, the brain halothane concentration is 40-50 μg/g85 (i.e., 0.2-0.25 mM) or 200 μg/g86 (i.e., 1 mM), respectively. Because the solubility of halothane in fat and brain tissue is higher than in 0.9% saline (about 125- and 3-to-8-fold, respectively87), the halothane concentration in brain membranes would be much greater than that in brain water (∼80% of brain weight) and anesthetic doses of halothane are likely to cause some inhibition of astrocytic gap junctional communication, thereby restricting intracellular spreading of metabolites among coupled astrocytes during activation. As described in more detail below, astrocytes in the inferior colliculus are highly coupled by gap junctions, and as many as 12,000 astrocytes are heterogeneously linked by gap junctional channels over a linear distance exceeding 1 mm28.
Total tissue lactate concentration in the inferior colliculus approximately doubles from the resting level of ∼0.5-1 μmol/g to ∼1-2 μmol/g during sensory stimulation of the conscious rat, and microdialysis studies show that extracellular lactate also rises 2-fold10. Probenecid blocks lactate transporters and amplifies the tonotopic bands10, but it does not inhibit gap junctional communication88, raising the intriguing possibility that neuronal generation and release of lactate to extracellular fluid via transporters contributes to degradation of tonotopic bands. If stimulus-induced increases in tissue lactate level were generated mainly by astrocytes and if this lactate were trapped within astrocytes by probenecid treatment, the lactate would still be able to spread widely within the large astrocytic syncytium of inferior colliculus, as shown for a larger fluorescent dye, Lucifer yellow28. Lucifer yellow can diffuse from the center of the slice of inferior colliculus to the collicular boundary within five min (the duration of glucose labeling) after microinjection into a single astrocyte28.
To summarize, metabolites of [14C]DG are trapped within activated cells and generate the best images of tonotopic bands evoked by acoustic stimulation. In contrast, two processes are associated with dispersion of metabolites of [14C]glucose from activated cells, (i) spreading within the activated structure, which blunts focal increases and peak-to-valley differences in metabolic activity in the tonotopic bands, and (ii) release of labeled metabolites from activated tissue, which reduces left-right differences. If local spreading were coupled to local metabolic trapping, spatial resolution of activation would be reduced but overall left-right differences for the activated structure would be similar to, or perhaps greater, than that obtained with [14C]DG, but this is not the case, consistent with substantial loss of label. Gap junctional trafficking in astrocytes, lactate transporters in astrocytes and neurons, and, perhaps, neuronal lactate production, are involved in metabolite dispersion, which was next examined in more detail.
Metabolite diffusion in vivo
To evaluate the relative spreading of glucose metabolites within the inferior colliculus of conscious rats during rest and acoustic activation, labeled compounds were micro-infused directly into the inferior colliculus and the volume of tissue labeled determined10. The volume labeled by an extracellular marker, [14C]inulin, was about twice that of [14C]DG, and labeling by both of these tracers was not altered by acoustic stimulation. The volume labeled by [U-14C]glucose and its metabolites was three times that of DG during rest, and it fell by half during activation, whereas labeling by U-14C-labeled lactate and glutamine (and their metabolites), two metabolites known to be released to extracellular fluid, were about 30-50% higher than DG during rest and increased by 23 and 49% (P<0.05), respectively, during acoustic stimulation. These results demonstrate that the volume of tissue that took up and metabolized the infused hexose (i.e., that registered by [14C]DG) was similar during rest and activation, and downstream metabolites of glucose-6-P spread into a 3-fold larger volume during rest. Because inulin spreading exceeded that of lactate and glutamine during rest and did not change during activation, the increased spreading of lactate and glutamine during acoustic stimulation may have involved intracellular routes. Also, the smaller volume of tissue labeled by glucose and its metabolites during activation is consistent with increased metabolite release from activated tissue, thereby reducing label spreading.
Metabolite spreading via the astrocytic syncytium
Amplification of tonotopic bands by halothane suggested that metabolite diffusion throughout astrocytic gap junctions reduces resolution of brain images of focal activation when labeled glucose is the tracer. This possibility is supported by our finding that two gap junction inhibitors, glycyrrhetinic acid and oleamide, reduced the volume of labeled by microinfusion of [1-14C]glucose tissue under resting conditions10. Because probenecid also enhanced the tonotopic bands, we anticipated that lactate spreading would contribute to the large tissue volume of labeled by microinfusion of [14C]glucose during rest. If this idea were correct, the tissue volume labeled by [3,4-14C]glucose should equal or exceed that labeled by [1-14C]glucose because lactate retains all of the label from both tracers, except that lost via the pentose phosphate shunt pathway (Fig. 1). Surprisingly, we found that infusion of [3,4-14C]glucose yielded a much lower labeled tissue volume, similar to that observed with [14C]DG10, indicating that spreading of TCA cycle-derived metabolites (e.g., glutamine) is predominant during resting conditions, and spreading of glutamine and lactate increases during acoustic activation.
Lactate labeling and oxidation to generate 14CO2
Low label trapping in [14C]glucose autoradiographs could arise from release of labeled lactate, 14CO2, or other diffusible compounds, and if glucose or lactate were quickly oxidized in small metabolic compartments (e.g., synaptosomes and astrocytic endfeet) that do not readily equilibrate with the large unlabeled amino acid pools (Figs. 1 and 3), 14CO2 production may be greater than expected. This possibility was tested by continuous microinfusion of [3,4-14C]glucose into interstitial fluid the inferior colliculus, and if the rate of lactate formation were matched by local lactate oxidation via pyruvate dehydrogenase in nearby cells, the levels of [14C]lactate and 14CO2 in extracellular fluid should be similar after a lag to label the metabolic and extracellular pools. However, this was not the case, acidic compounds (including lactate and pyruvate) accounted for 80% of the 14C in metabolites in the microdialysate whereas 14CO2 accounted for only 20% when sampled at 10 min intervals before, during, and after acoustic activation throughout the 60 min microinfusion interval (Cruz et al., unpublished). These results demonstrate that the intracellular metabolic pool of [14C]glucose-derived lactate/pyruvate exchanges with and labels the extracellular pool, as expected from in vitro studies showing that transmembrane lactate exchange is rapid compared to its metabolism29, 45. We did not assess the lag between pyruvate/lactate labeling compared to 14CO2 production, but previous studies by Cremer and colleagues89 showed that interconversion of label between pyruvate and lactate via the cytoplasmic lactate dehydrogenase reaction is faster than pyruvate transport into mitochondria, oxidative decarboxylation by the pyruvate dehydrogenase complex, and incorporation of its label into amino acids, but the lag time is short. At 10 sec after a bolus injection of [2-14C]pyruvate into the carotid artery of pentobarbital-anesthetized rats, 42% of the 14C in freeze-blown brain was recovered in lactate and 12% in glutamate, glutamine, and aspartate, and at 50 sec these amino acids accounted for 53% of the label compared to 23% for lactate89; oxidative metabolism of pyruvate/lactate was rapid in spite of potent metabolic depression by pentobarbital90, 91. Although the fate of labeled glucose inserted into interstitial fluid by microinfusion may differ somewhat from that taken up from blood by normal tissue, the differences in labeling of lactate and CO2 are large. Rapid pyruvate-lactate exchange labeling may amplify the initial labeling of lactate, but the lag between lactate labeling and generation of labeled CO2 in conscious stimulated rats is expected to be less than a minute. The 10 min sampling period for each microdialysate collection interval allows for brief lags in metabolic labeling of lactate and generation of CO2.
Fig. 3. Schematic illustrating pathways for label loss arising from glycolysis or oxidation of [14C]glucose or [14C]glucose-derived lactate to 14CO2 in small metabolic compartment(s).

Autoradiographic studies using [1- or 6-14C]glucose demonstrate that most of the label corresponding to the additional glucose consumed in focally-stimulated tissue is not retained in activated structures (see Fig. 2 and text). Diffusible and volatile compounds are most likely to be released after generation of pyruvate or lactate by the glycolytic pathway, followed by oxidation in the cell where formed or after its putative shuttling among neurons or astrocytes. If glucose or lactate were rapidly and locally oxidized in a small metabolic compartment that does not quickly equilibrate with the large amino acid pools that would facilitate dilution and trapping of label, then appearance of 14CO2 in the interstitial fluid would be expected to show a temporal profile similar to that of [14C]lactate when extracellular fluid is sampled by microdialysis (see text).
Constancy of the fraction of label recovered in extracellular acidic compounds suggests that lactate labeling tracked the 2-fold increases in total and extracellular unlabeled lactate during acoustic activation of conscious rats. In our studies, total lactate level rose from ∼1 to ∼2 μmol/g and, assuming similar intra- and extracellular concentrations at steady state and 20% extracellular fluid space, about 0.4 μmol lactate/g would be extracellular. The overall glucose utilization rate in the activated inferior colliculus10 is 1.08 μmol g−1 min−1 (it is 40% higher in the major tonotopic band) and, because two molecules of pyruvate are formed from each glucose, the rate of pyruvate formation would be about 2.2 μmol g−1 min−1. If the lactate pools mix quickly and completely, the extracellular lactate could turn over in about 0.2 min compared to a minute for the total lactate pool; brain lactate is a significant fuel reservoir.
To sum up, total tissue and extracellular unlabeled lactate rise by a similar magnitude during acoustic activation. The lactate transport blocker probenecid enhances tonotopic bands but does not interfere with astrocytic gap junctional trafficking (suggesting neuronal lactate release), and labeling of extracellular lactate was much higher than that of CO2. These findings, along with poor registration of metabolic activation by [1- and 6-14C]glucose, are consistent with substantial efflux of labeled lactate to blood. In normal human subjects given [1-11C]glucose PET scans, efflux of 11C-labeled acidic metabolites from brain to blood precedes 11CO2 release22.
Release of labeled lactate via exchange-mediated efflux across the blood-brain barrier
Rapid exchange of labeled lactate generated in brain from [14C]glucose for unlabeled lactate in blood could contribute to net label release from brain without net lactate transport across the blood-brain barrier. If blood-brain lactate exchange reactions are substantial, they would reduce the specific activity of the brain lactate/pyruvate pools, thereby reducing the flux of label into the TCA cycle-derived amino acids. Carrier-mediated uptake of lactate from blood to brain takes place in immature and adult brain, but suckling rats have much higher levels of monocarboxylic acid transporters in the cerebral vasculature than adult rats and calculated rates of lactate transport across the blood-brain barrier when the lactate level is 1-2 mM are 8-10-fold greater in 15-18 day-old compared to adult rats92, 93. In suckling rats, lactate exchange does markedly reduce the specific activity of brain lactate well below that of brain glucose after metabolic labeling with [14C]glucose94. However, in adult rat brain the rates of unidirectional lactate influx reported by three laboratories, 0.0692, 0.02-0.0495, and 0.1496 μmol g−1 min−1, are low compared to measured rates of pyruvate formation (i.e., twice the rate of glucose utilization) in the entire resting and acoustic-activated inferior colliculus (1.5 and 2.2 μmol g−1 min−1, respectively) and in the major tonotopic band the rate of pyruvate generation is even higher (2.8 μmol g−1 min−1)10. Thus, highest unidirectional lactate influx rate is <6% of the rate of pyruvate formation from blood glucose in the activated inferior colliculus, and lactate dilution should be low.
Exchange-mediated loss of labeled lactate from the brain metabolic pool was an obvious concern in early studies to develop a method using [14C]glucose to assay rates of brain glucose utilization in vivo, and arteriovenous differences for [14C]lactate were assayed. 14C-lactate efflux was reported to be small or not detectable in paralyzed-ventilated rats sedated with N2O/O2 (70:30), with or without 0.5% halothane anesthesia100-102. In contrast, we observed net efflux of equivalent amounts of labeled and unlabeled lactate from brain to blood in conscious rats with cortical spreading depression14, 15. Lactate release was substantial, but it accounted for only about half the magnitude of the underestimation of glucose utilization rate assayed with [6-14C]glucose compared to [14C]DG14, 15. In these rats, the specific activity of brain lactate was about half that of [6-14C]glucose, which is close to the theoretical maximum of 0.5 ([6-14C]glucose generates two molecules of pyruvate, one that is labeled and one that is not labeled). The relative specific activity of brain lactate was also close to 0.5 in normal conscious adult rats during three activity stages, rest, somatosensory stimulation, and recovery from stimulation39. There is some evidence for segregation of the blood-brain-exchangeable lactate pool from the metabolic pool in the adult rat 97-99, but if substantial, compartmentation of lactate taken up into brain would reduce the specific activity of purified brain lactate due to mixing of all pools when tissue homogenates are prepared. Thus, in all of the above examples, brain lactate dilution appears to be negligible.
In pulse intravenous labeling studies, an apparently-high relative specific activity for brain lactate could arise from a faster fall in specific activity of brain glucose compared to brain lactate. The specific activity of plasma glucose falls after reaching its peak level shortly after the bolus injection, and brain glucose specific activity declines in parallel; the specific activities of glucose in arterial plasma and brain are similar at 1, 3, and 10 min103. Because the rate of decline of brain glucose specific activity is about 2-4% per min (calculated from data of Hawkins et al.100 and Cremer et al.89), the difference in brain glucose and lactate specific activities determined at 5-6 min after pulse labeling arising from a 2 min lag between glucose and lactate labeling is expected to be <8% (the half-life of brain glucose is ∼1 min in conscious resting rats104 and metabolic turnover would be faster during activation).
To sum up, two lines of evidence, low unidirectional lactate influx from blood to brain in the adult rat and the high relative specific activity of brain lactate in conscious adult rats, support the conclusion that the quantity of labeled lactate released by exchange-mediated efflux and pool dilution is small compared to net release in the conscious adult rat. Because net lactate release to blood did not explain the magnitude of underestimation of glucose utilization rate determined with [6-14C]glucose in our earlier studies of spreading depression14, 15, we next evaluated the possibility that there may be an unrecognized, alternative route for metabolite efflux from brain.
A novel route for metabolite dispersal and clearance
Cerebral venous blood is a major sink for brain metabolites, but compounds released from astrocytic endfeet may also removed from brain by flow of perivascular fluid that is powered by arterial pulsations and can disperse horseradish peroxidase throughout the brain within minutes23. There is no diffusion barrier between interstitial, perivascular, and cerebrospinal (CSF) fluids, and extracellular can be removed from the brain by both absorption via arachnoid membranes and transport across the cribriform plate to lymphatic system24,25.
To identify pathways linked to extracellular fluid in the inferior colliculus, Evans blue albumin was microinfused into the inferior colliculus of conscious rats for different time intervals. The procedure quickly labeled the meninges on the dorsal surface of the inferior colliculus, the vasculature on the ventral region of the brain (e.g., Circle of Willis), and perivascular space along the middle cerebral arteries, with higher labeling in the ipsilateral compared to contralateral hemisphere (Cruz et al., unpublished). HPLC analysis of extracts of the dissected meninges and vasculature after microinfusion of [1-14C]glucose into the inferior colliculus for 5 min revealed labeled glucose, lactate, and other compounds. Infusion of D-[14C]lactate, a non-metabolizable compound, also labeled the vasculature and meningial membranes. These findings demonstrate that perivascular fluid flow is a novel route for removal of lactate and other compounds from the inferior colliculus, and material cleared from brain via this pathway would not be taken into account in assays of arteriovenous differences.
Summary: Discordant focal activation images in conscious adult rats in vivo
Increased glucose utilization evoked by monotonic acoustic stimulus is substantially underestimated by autoradiographic assays using [1- and 6-14C]glucose due to decarboxylation reactions, transporter-mediated lactate release, and astrocytic gap junctional trafficking. Amplification of the tonotopic bands by probenecid, a lactate transport blocker that does not impair astrocytic gap junctional trafficking, raises the intriguing possibility that lactate may be generated in and released by neurons because lactate formed in astrocytes would still be able to spread within the extensive syncytium and degrade tonotopic bands when astrocytic lactate transporters are inhibited. During activation, total and extracellular levels of unlabeled and labeled lactate level increase in parallel, and labeling of extracellular lactate exceeds that 14CO2, consistent with net release of the high-specific activity lactate from brain tissue to blood and perivascular drainage to the lymphatic system. Because the astrocytic syncytium was implicated in trafficking of glucose metabolites, we next examined astrocytic gap junctional communication in slices of the inferior colliculus.
GAP JUNCTION-MEDIATED TRANSFER OF SMALL MOLECULES
Astrocytes are interconnected via gap junctions and these channels are widely assumed to facilitate intercellular transfer of molecules <1kDa, including nutrients and signaling compounds105. Our autoradiographic studies do not have cellular resolution, but if highly-coupled astrocytes metabolize a large fraction of the blood-borne glucose during brain activation and if gap junction-mediated transfer of DG-6-P were unrestricted, spatial resolution of focal brain activation in [14C]DG assays would be poor, yet this is not the case (Fig. 2). We, therefore, directly examined two possible explanations, astrocytes may be poorly coupled in the inferior colliculus and DG-6-P may not readily pass through gap junctional channels. A third possibility, suggested by the probenecid studies, is that neurons generate most of the diffusible lactate; this question has not been examined in the current studies due to technical difficulties in quantitative determination of cellular labeling by [14C]DG or [14C]glucose.
A large astrocytic syncytium in the inferior colliculus
We first determined the distributions of the major astrocytic connexin (Cx) proteins, Cx43, Cx30, and Cx26 in the inferior colliculus of the rat, and found widespread but heterogeneous localization of the immunoreactive connexin proteins; most of the Cx43 was phosphorylated during rest and acoustic stimulation28. Next, Lucifer yellow dye spreading was used to evaluate the extent of coupling of astrocytes, and an extensive syncytial network was revealed. Using a 5 min labeling period that corresponded to the brief glucose utilization assays (Fig. 2), an average of about 6,100 astrocytes (range: 2,068-11,939) located as far as 1-1.5 mm from the impaled cell were labeled by diffusion of Lucifer yellow from a micropipette inserted into a single astrocyte located near the center of a tissue slice of the inferior colliculus28. Astrocytic endfeet are known to be gap junction-coupled, and perivascular labeling by Lucifer yellow was also extensive; the dye concentration in perivascular regions often exceeded that of nearby neuropil by a factor of two. Thus, astrocytes have high capacity to quickly and heterogeneously distribute intracellular molecules into a relatively large volume of the inferior colliculus. Gap junction-mediated spreading throughout the large astrocytic syncytium could readily degrade resolution of brain images and contribute to failure of [14C]glucose to register tonotopic activation bands (Fig. 2).
DG-6-P is a poor substrate for gap junctional transfer
Next we tested the hypothesis that astrocytic gap junctional transfer of glucose metabolites is selective. Single astrocytes in culture or brain slices were impaled with a micropipette containing a test compound plus an internal standard, a gap junction-permeable anionic fluorescent dye, to quantify cellular coupling by determination of dye-labeled area. 2-NBDG, a fluorescent glucose analog, and [3H]DG readily spread within the astrocytic syncytium, whereas gap junctional transfer of glucose-6-P, DG-6-P, and NBDG-6-P was very low, labeling areas about 10% (p<0.05) of the dyes or parent hexoses; syncytial trafficking of hexose-6-phosphates is highly restricted (Gandhi et al., unpublished). These findings demonstrate that brain imaging with labeled DG reflects metabolism in the cell where DG-6-P was generated, and suggest that limited gap junctional permeability of glucose-6-P, a regulatory glycolytic metabolite, might be important for local control of glycolytic flux. Minimal transfer of hexose-6-phosphates through astrocytic gap junctions challenges the long-held notion of unrestricted passage of small metabolites < ∼1 kDa105, including glucose-6-phosphate106, 107, through these channels.
Astrocytes have high capability for lactate uptake and intra-syncytial lactate shuttling compared to neuronal uptake from extracellular fluid or from nearby astrocytes
Because our studies described above implicated astrocytes in metabolite dispersal, the next experiments were designed to compare directly (i) lactate uptake from extracellular space into astrocytes and neurons and (ii) transfer of lactate among gap junction-coupled astrocytes compared to shuttling of lactate from astrocytes to neurons. To achieve this goal, we devised a sensitive, micro-enzymatic assay for lactate to detect changes in lactate levels in single cells in slices of inferior colliculus from adult brain. Results of ongoing studies clearly demonstrate that astrocytes have a much higher capacity for uptake of extracellular lactate compared to neurons (1.5- and 2.8-fold at 2 and 10 mM lactate, respectively; Gandhi et al., unpublished), as predicted29 from the kinetic properties of astrocytic and neuronal monocarboxylic acid transporters. Furthermore, intracellular astrocytic lactate is more readily dispersed to other gap junction-coupled astrocytes than to neighboring neurons (2.4-fold; Gandhi et al., unpublished), as expected from the lack of a membrane barrier between coupled cells within the large astrocytic syncytium28. MCT2, the predominant neuronal monocarboxylic acid transporter, has a Km for lactate of ∼0.7 mM, and when the local lactate concentration reaches or exceeds the average tissue level of ∼2 mM in our activation studies, neuronal lactate transporters would approach saturation and attain maximal velocity in a direction governed by the relative intracellular and extracellular lactate concentrations and pH. Maximal transport of lactate to or from neurons could result in diffusion of lactate in the interstitial fluid and astrocytic lactate uptake and dispersion via the syncytium. In cultured neurons continuously exposed to 100 μM noradrenaline, expression of immunoreactive MCT2 rises by about 75% at 3 h and 4-fold at 9 hours108. If this immuno-expression response corresponds to linear increases in neuronal lactate transport capacity, the anticipated rise at 1h is 25%, which will not have any impact on lactate shuttling or metabolism in 5 min stimulation studies in which [14C]glucose label trapping is very low. On the other hand, MCT1 and MCT4 are predominant in astrocytes and have higher Km's for lactate (3-5 mM and 15-30 mM, respectively) than MCT2. These transporters would facilitate lactate trafficking to and from astrocytes at rates that rise with local lactate level29,30, and this is what we observed experimentally in direct assays of lactate uptake and shuttling in adult brain slices.
These studies identify three important factors relevant to metabolite trafficking during brain activation in adult brain tissue 31, 32 (i) greater lactate uptake into and diffusion among astrocytes compared to lactate shuttling from astrocytes to neurons, (ii) trafficking of lactate, glutamine, and other compounds from activated cells via the astrocytic syncytium and their extensively gap junction-coupled perivascular endfeet, and (iii) the possibility of metabolite release from endfeet into perivascular fluid, followed by clearance via transfer to blood or removal by perivascular flow. Our data do not rule out some utilization of lactate by either or both major cell types during brain activation, but they support the notion that metabolite dispersal from unidentified activated cells and label release from activated tissue greatly exceed local trapping of label from the additional glucose consumed during activation.
BRAIN ACTIVATION FREQUENTLY UP-REGULATES NON-OXIDATIVE METABOLISM OF GLUCOSE MORE THAN OXYGEN CONSUMPTION
Two sets of observations, (i) failure of [6-14C]glucose to appropriately register the magnitude of brain activation, and (ii) a greater increase in glucose consumption compared to oxygen utilization in many studies of brain activation, demonstrate that there are significant changes in the fate of labeled and unlabeled glucose when brain activity is stimulated by physiological stimuli in normal, conscious rats. As described below, in vivo studies indicate that glycolytic and oxidative pathway fluxes are increased in both astrocytes and neurons.
Oxygen-glucose stoichiometry
The ratio of the rate of oxygen to glucose utilization is denoted by CMRO2/CMRglc, where CMR indicates the cerebral metabolic rate for oxygen or glucose (abbreviated as glc). The theoretical maximum of this ratio is 6 because 6 O2 are required to oxidize 1 glucose to 6 CO2 + 6 H2O. A fall in this ratio from the normal value of close to 6 in resting brain indicates a relative increase in non-oxidative metabolism of glucose, which could represent greater lactate production, an increase in biosynthetic reactions (e.g., amino acid or glycogen synthesis), or both.
The CMRO2/CMRglc ratio falls in many, but not all, studies that measured the oxygen-to-glucose utilization ratio during brain activation in humans and experimental animals, and metabolic changes appear to depend on the stimulus paradigm, duration, intensity, the neural pathway and station in the pathway that is assayed, and physiological state of the subject33, 109. Global measurements of arteriovenous differences across the brain reveal substantial decreases in the CMRO2/CMRglc ratio (Table 1), but these values probably underestimate the magnitude of fall in the most highly-activated regions due to averaging of the contributions of venous blood from structures that are not equally activated. For example, ratio fell from about 6 to about 5 when arteriovenous differences were assayed across the brain during somatosensory stimulation of the rat38, but the magnitude of activation of cerebral cortex was quite heterogeneous; some structures had large (almost 2-fold) increases in CMRglc, whereas others had small or no metabolic changes41. Unfortunately, it is not possible to sample the venous drainage of small brain structures that can be more uniformly activated, with the exception of the eye. In fact, the lowest CMRO2/CMRglc ratios occur in the outer retina (0.5-1.4; Table 1) due to high glycolytic metabolism of glucose and release of most of the glucose carbon as lactate into the venous drainage of the eye. These findings are consistent with the eye being an extremely glycolytic organ, particularly in predatory avian species, presumably because high mitochondrial density reduces visual acuity110, 111. In the 19 activation studies in awake humans tabulated by Rothman et al.109, six showed a 15-46% greater rise in CMRglc compared to CMRO2, seven had a 5-11% greater increase in CMRglc, and five reported no difference in the percent rise in CMRglc and CMRO2; curiously, CMRO2 rose 24% more than CMRglc in one report, suggesting oxidation of alternative fuel or unidentified factors influenced the determinations.
Table 1.
Oxygen utilization does not match glucose or glucose plus lactate consumption during brain activation.
| A. Brain of normal conscious subjects | |||||
|---|---|---|---|---|---|
| Human | Rest | Activationc | Reference | ||
| Whole brain (A-V difference assays) | |||||
| CMRO2/CMRglc | 6.1 | 5.4 | 60 | ||
| CMRO2/CMRglc | 6.04a | 5.35a (ns) | 61 | ||
| CMRO2/CMR(glc+1/2 lactate) | 5.92a | 4.70a | 61 | ||
| CMRO2/CMRglc | 5.95a | 6.02a (ns) | 62 | ||
| CMRO2/CMR(glc+1/2 lactate) | 5.84a | 4.42a | 62 | ||
| CMRO2/CMRglc | 5.8a | 5.89a (ns) | 63 | ||
| CMRO2/CMR(glc+1/2 lactate) | 6.11a | 4.35a | 63 | ||
| Primary visual cortex (PET assays) | |||||
| CMRO2/CMRglc | 4.1b | 2.8 | 64 | ||
| CMRO2/CMRglc | 6.2 | 4.8 | 65 | ||
| Rat | |||||
| Cerebral cortex (A-V difference assays) | |||||
| CMRO2/CMRglc | 5.94 | 5.10 | 66 | ||
| CMRO2/CMRglc | 6.1 | 5.0 | 38 | ||
| CMRO2/CMRglc | 6.1 | 4.0 | 67 | ||
|
B. Eye of anesthetized subjects | |||||
| Light |
Dark (Activated) |
||||
| Cat (A-V differences: CMRO2/CMRglc) | |||||
| Outer retinad (choroidal vein) | 1.35 | 0.84 | 68 | ||
| Pig (A-V differences: CMRO2/CMRglc) | |||||
| Inner retina (retinal vascular bed) | 5.14 | 4.14 | 69 | ||
| Outer retinad | 0.55 | 0.51 | 70 | ||
| Whole uvea (vortex vein ) | 0.87 | 0.78 | 70 | ||
Substrate utilization ratios previously summarized in ref. 33 were calculated from (i) arteriovenous differences (A-V) for oxygen and glucose or glucose plus lactate (note: dividing the lactate uptake by two converts it to glucose equivalents) measured in paired samples of blood from the jugular bulb (human whole brain), sagittal sinus (rat cerebral cortex), or identified veins of the eye or (ii) metabolic rates for oxygen and glucose determined by positron emission tomography (PET).
Ratios during rest or physical exercise, which markedly increases blood lactate levels and lactate uptake into brain from blood (reviewed by Ide & Secher71 and Dalsgaard37).
Note that the baseline ‘resting’ ratios are, for unexplained reasons, much lower than the expected value of about 6 observed in most studies (6 moles of oxygen are required for complete oxidation of 1 mole of glucose), but a further decrease was observed during activation.
All values during activation or exercise were statistically significantly different from rest, except those indicated by ‘ns’, denoting ‘not significant’.
Outer retina includes retinal pigment epithelium and photoreceptors; the whole uvea also includes the anterior uvea and contribution of aqueous humor70.
The failure of CMRO2 to routinely match the rise of CMRglc is unexpected because the brain has a large excess capacity for oxygen consumption that is revealed by Siesjö's review36 of studies of global seizures. CMRO2 can readily increase by at least 50% even though it does not match the concomitant rise in CMRglc36 and, in one example, CMRO2 rose by about 2.5-fold after global seizure onset and was sustained for two hours129. Local oxygen availability due to proximity to the vasculature could be a factor during brain activation130, and, during spreading cortical depression, increases in NADH fluorescence reveal local zones of relative hypoxia between capillaries81. However, in activated brain, the blood-oxygen-level-dependent functional magnetic resonance imaging effect (BOLD fMRI) indicates that, after an initial lag, blood oxygen is usually delivered in excess of its consumption131. It is, therefore, likely that normal functional activity preferentially up-regulates glycolysis.
The CMRO2/CMRglc metabolic ratio is generally calculated using only oxygen and glucose as substrates, and does not include other minor oxidative substrates (e.g., lactate, glycogen, fatty acids, and amino acids) that can be metabolized by brain cells in varying amounts under normal activating conditions32,34 and under pathophysiological states35,36. Inclusion of net consumption of minor substrates would lower the true CMRO2/CMRtotal fuel below the value calculated CMRO2/CMRglc ratio, as demonstrated in studies in vigorously-exercising human subjects (reviewed by Dalsgaard37); the higher the blood lactate level during exercise, the greater its uptake into brain and the further the fall in the oxygen-to-carbohydrate utilization ratio, from a resting value of about 6 to as low as 3.5 (Table 1). Under these conditions, nearly half of the glucose plus lactate taken up into human brain from blood is not oxidized. Because lactate does not accumulate in brain of exercising humans and the only metabolic fate of lactate is via oxidation, much of the glucose taken up into brain from blood appears to have been diverted into non-oxidative pathways (e.g., glycogen turnover) during strenuous muscular activity37. These results illustrate the importance of taking into account the contributions of blood lactate and other endogenous metabolites to oxygen/glucose utilization ratios in working brain. Non-oxidative metabolism can be substantial even when cerebral blood flow rises and blood oxygen levels are normal, and, as discussed below, the biochemical and cellular basis of the flux changes are not trivial to elucidate in vivo.
Metabolic activation in conscious rats in vivo
Studies in experimental animals have the advantage that metabolite levels and labeling patterns can be directly measured and correlated with results obtained by determination of arteriovenous differences. Generalized sensory stimulation of normal conscious rats increases cerebral blood flow and oxygen consumption but the 30% rise in CMRO2 did not match the 44% increase in CMRglc38. Incorporation of 14C from [6-14C]glucose into the labeled metabolites retained within the brain at the end of the 5 min labeling period rose 26%, and large fraction of the additional 14C trapped during activation was recovered in purified glutamate and GABA39. These results suggest that neuronal glycolytic and TCA cycle fluxes were enhanced because the large glutamate pool is neuronal and glutamate labeling is interpreted in terms of neuronal oxidation of glucose in MRS studies. A maximum of 20-25% increase in total label trapping in activated structures after pulse labeling with [6-14C]glucose was also observed during graded visual8 and acoustic10 stimulation of the normal conscious rat. Astrocytic glycolytic, oxidative and anaplerotic metabolism also increases during in vivo sensory stimulation. Acetate oxidation by astrocytes rises 15-25% in structures activated by auditory and visual stimulation20,21, CO2 fixation is higher in awake compared to anesthetized rats112, and glycogen turnover, which is also mainly astrocytic, is heterogeneously enhanced during multimodal sensory stimulation; in fact, glycogen turnover appears to account for about half of the additional glucose consumed in the sensory and parietal cortex in response to whisker stimulation38,40,41. Net accumulation of unlabeled lactate in brain tissue during sensory stimulation only accounted for about half of the glucose consumed in excess of oxygen38. The cellular origin of this lactate is unknown, but, on the basis of its specific activity (about half that of [6-14C]glucose), it was ultimately derived mainly from blood-borne glucose39. Because the specific activity of lactate is much higher than those of other diffusible metabolites of [6-14C]glucose (e.g., 5.8 times that of glutamine and almost twice that of alanine, which is also derived from pyruvate39), lactate is likely to be the major contributor to 14C release from activated brain. To summarize, activity-induced changes in functional metabolism are complex and involve glycolytic and oxidative metabolism in neurons and astrocytes; the proportion of glucose consumed by each major cell type during brain activation remains to be established.
Activation-induced change in brain lactate level: a minor fraction of glucose utilization
An increase in brain lactate level is often interpreted as a reflection of increased glycolysis (i.e., the rate of pyruvate formation exceeds that of pyruvate oxidation), but additional information is needed to interpret lactate concentration changes. The net rise in total brain tissue and extracellular lactate levels during physiological stimulation paradigms in normal subjects is generally quite small and corresponds to a minor fraction of the pyruvate/lactate generated from glucose. For example, in our studies in conscious rats, the 2-fold rise in the tissue lactate from ∼0.5-1 μmol/g during rest to ∼1-2 μmol/g during brain activation10,38,39,41-44 corresponds to only approximately 5% of the quantity of glucose metabolized during a 5-10 min period of brain activation38,41. Similarly small proportions of labeled lactate in brain are obtained after pulse labeling with [6-14C]glucose; the 14C recovered in purified brain lactate accounts for only 4 or 8% of the total 14C retained in the brain metabolite pool during rest or sensory stimulation, respectively39. The quantity of net lactate accumulation in activated brain is equivalent to about 0.25-0.5 μmol glucose/g, which could be metabolized within about 30 sec in the activated cortex or inferior colliculus. Other studies in humans or anesthetized rats report even smaller changes in brain lactate concentration during activation44, 113. Some lactate may be metabolized in brain, but endogenous lactate is not an major fuel reservoir; the flux through the brain lactate pools in vivo and the ultimate fates of lactate remain to be established.
Equilibrative reactions mediate lactate formation and transport
Because the lactate dehydrogenase reaction is defined by the relationship, [NADH]/[NAD+] = [lactate]/[pyruvate] × K/[H+] (K is the equilibrium constant)114 and lactate is co-transported along with H+ by the MCT transporters26, local lactate levels are influenced by many factors, including the relative rates of pyruvate formation and oxidation, pH, other [NADH]/[NAD]-dependent metabolic and shuttling reactions, and lactate concentration gradients114, 115. The free cytoplasmic [NAD]/[NADH] ratio in freeze-blown rat brain tissue is calculated to be about 670114 and the measured brain lactate/pyruvate ratio is in the range of 10-1336, 114. In electrically-stimulated brain slices the changes in total NAD(P)H fluorescence (denoted as ΔF/F) are typically relatively small, in the range of ±4-8%77-80, in spite of substantial differences in the magnitude and duration of stimulation in these studies, as well as the pathways and cellular structures to which the metabolic changes are ascribed. These findings suggest that NADH turnover greatly exceeds its net concentration change (as is the case for brain lactate), underscoring the importance of obtaining additional information from a variety of experimental conditions to interpret the functional significance and cellular localization of changes in NAD(P)H fluorescence.
A rise in intracellular pyruvate level may be necessary to increase flux through the regulated pyruvate dehydrogenase reaction, and an increase in lactate level may simply reflect changes in pyruvate concentration. The Km of pyruvate dehydrogenase (PDH) for pyruvate (0.02-0.06 mM46) is similar to the pyruvate concentration in normal brain (range: ∼0.05-0.15 μmol/g36, 114), and increased glycolytic and glycogenolytic fluxes would be expected to raise intracellular pyruvate level and contribute to increasing the velocity of the pyruvate dehydrogenase reaction until the enzyme becomes saturated. Because maximal lactate dehydrogenase activity (Vmax = 60-70 μmol min−1 g wet wt−1 when assayed in extracts of mouse and human brain) is 15-70-fold higher than maximal pyruvate dehydrogenase activity (Vmax = 1-4 μmol min−1 g wet wt−1)47, pyruvate and lactate levels should quickly equilibrate. Thus, modest shifts in pyruvate concentration that arise from normal fluctuations in CMRglc should be accompanied by corresponding proportionate changes in tissue lactate level. In fact, in normal, control (‘resting’) brain tissue there is a linear relationship between measured lactate and pyruvate concentrations (Fig. 4, solid line), suggesting that “small” (i.e., <2-fold) increases in lactate level within the normal range may represent pyruvate-lactate equilibration. In contrast, when oxidative metabolism is compromised, e.g., during carbon monoxide poisoning and recovery from transient ischemia, ‘excess’ lactate is produced and the lactate/pyruvate ratio rises, as illustrated by the data points that fall above the dashed lines in Fig. 4; the dashed lines are extrapolated in both directions from the solid linear regression line for control values. Notably, the lactate/pyruvate ratio can also increase even when lactate and pyruvate levels are reduced (Fig. 4), such as during hypercapnia, which causes tissue acidification48, depresses the calculated rate of brain glucose utilization, and apparently stimulates oxidation of endogenous substrates35. Thus, a fall in lactate or pyruvate level may, but need not, reflect increased oxidation of these substrates; a decrease can arise from a lower production rate when its rate of consumption or diffusion away from the sampling site is constant.
Fig. 4. Brain lactate level as function of brain pyruvate concentration in normal and pathophysiological conditions.
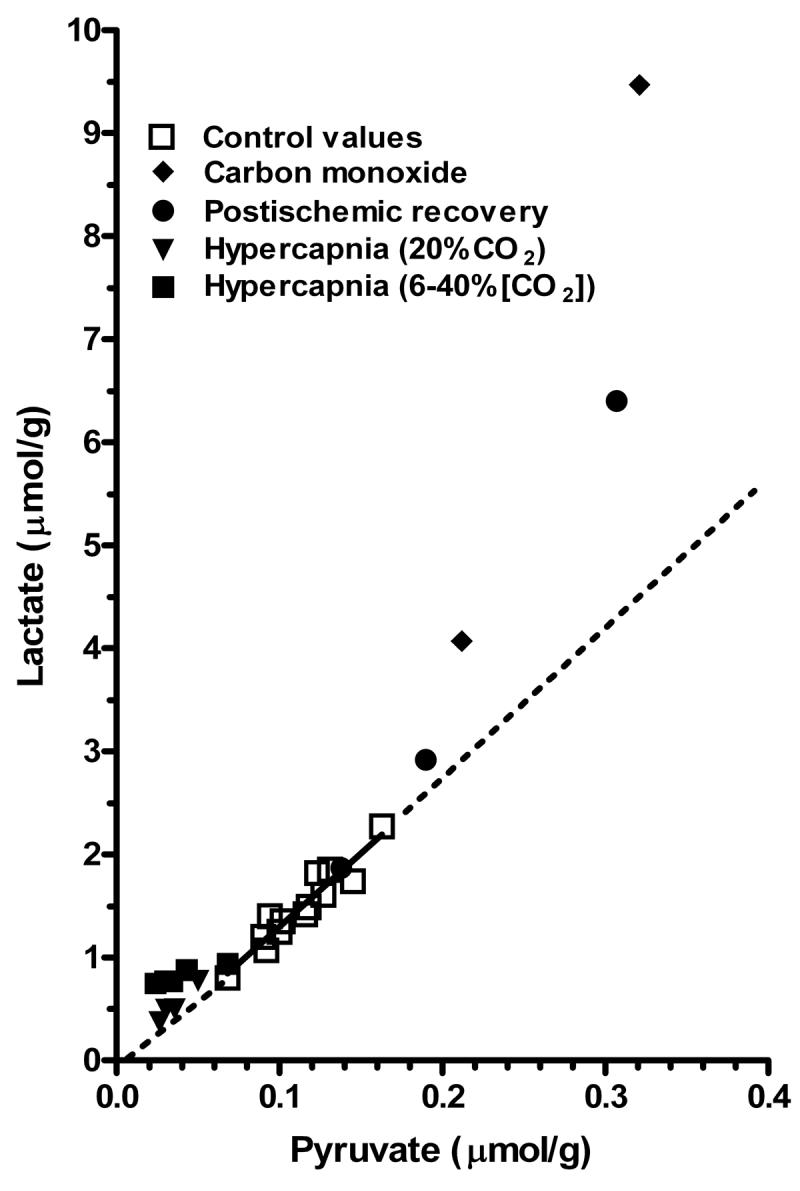
Brain lactate and pyruvate concentrations determined in brain of normal control rats were compiled from studies in different laboratories35,48,56-59. The solid line is the linear regression (r2 = 0.903) for the control values (open squares); the dashed lines denote extrapolation of the regression line above and below the normal range and they serve as references to easily identify levels of lactate formed in ‘excess’ of pyruvate under abnormal conditions. Data sets are also plotted for paired lactate-pyruvate levels determined in carbon monoxide-treated rats56, postischemic rats with different pre-ischemic glucose levels57, rats exposed to 20% CO2 for intervals up to 60 min35, and rats exposed to different levels of CO2 for a fixed time48.
To sum up, lactate is generated and transported by passive, concentration-driven reactions, contrasting the tightly-regulated metabolism of glucose via the glycolytic and oxidative pathways. Lactate levels are the net result of all input and output processes, including transport to and from blood, diffusion within brain, generation from glucose, glycogen or other precursors, and oxidative metabolism in all cell types29,45,49. As emphasized by Veech115, “measurements of lactate content per se are able to provide relatively little information other than the level of lactate itself.”
Low energetic cost of release of excess lactate from brain during activation
Co-transport of lactate and H+ out of glycolytically-active cellular structures and rapid removal of these by-products by various processes would serve two functions, (i) to ‘pull’ regeneration of NAD+ from NADH by product (lactate) removal and (ii) to help minimize intracellular acidosis. These functions may be more important to overall cellular energetics than release of a high-energy fuel because delivery of glucose from blood to brain is not limiting under normal physiological conditions. Brain glucose concentration is maintained within the normal range during sensory stimulation and brain-to-plasma ratio for glucose is close to its usual value of 0.228,38,41. Matching of glucose supply with local demand can be sustained over a large range of metabolic rates because the maximal capacity for glucose transport from blood to brain (Tmax) is about 3-fold higher than maximal brain hexokinase velocity (Vmax); unidirectional glucose influx into brain exceeds its phosphorylation rate55, 116. It is possible that transient reduction of local glucose level may occur just after onset of an intense stimulus, and in astrocytes fuel demand may be buffered by glycogen. More recent studies of glucose dynamics in vivo and in cultured brain cells show that this process is quite complex, involving regulation of glucose transporters at a cellular level according to demand117-119. To sum up, under normal conditions, glucose is delivered to brain in excess of its utilization, glucose and lactate transporters are equilibrative, and the lactate can be readily oxidized by other body tissues. If some glucose is metabolized only to the level of lactate in brain cells, then released from brain, there is little, if any, energetic cost. From the perspective of whole-body metabolism, fuel is not wasted.
CELLULAR BASIS OF BRAIN GLUCOSE UTILIZATION IN VIVO DURING REST AND ACTIVATION
Cellular localization of CMRglc
The fraction of glucose consumed by astrocytes and neurons has been of intense interest for more than 30 years. Routine 14C-autoradiographic assays do not have cellular resolution, and studies to identify metabolically-labeled cells have been hindered by a number of serious technical difficulties, particularly quantitative retention of labeled metabolites in cells and tissue during histological processing to identify cell type; losses can be as high as 90%120-123. Even in the most recent studies with about 50% label retention124 or in vivo assays using a fluorescent glucose analog125, the best estimates suggest that overall glucose utilization rates in astrocytes and neurons are similar under resting conditions. However, specific subsets neurons and astrocytes are more metabolically active than others during resting conditions and accumulate high levels of [3H]DG compared to other cells126, 127. During swimming or rotation paradigms increased neuronal trapping of labeled DG occurs in activated structures120, 121. Increased neuronal glucose utilization during in vivo activation is consistent with the large excess capacity of synaptosomes to carry out glycolysis and respiration. Synaptosomal glycolytic rate increased 10-fold for at least 30 min and respiration rose 6-fold after addition of an uncoupler (FCCP, carbonylcyanide p-trifluoromethoxyphenylhydrozone)128. Thus, neurons can upregulate glucose utilization to a greater extent than respiration and if this occurs during physiological stimulation, neurons can contribute to the fall in CMRO2/CMRglc.
Metabolic anatomy
In the late 1980's, Collins and co-workers8 and Lear and Ackermann9,11, 12, 13 suggested that substantial increases in glycolysis and lactate release during brain activation in vivo explained the differences in product trapping in autoradiographic studies using labeled glucose compared to DG. Analysis of local capillary density, glucose utilization rate, and activities of lactate dehydrogenase and cytochrome oxidase revealed distinct differences in local glycolytic and oxidative capacities that were partly associated with pathway specificity132. These and related studies of the metabolic architecture of brain led Collins133 to propose the notion of ‘red brain and white brain’; local differences in relative oxidative and glycolytic capacities were related to the type of synaptic input (excitatory or inhibitory), the type of physiological activity (tonic compared to brief phasic activity), and the history of the structure (metabolic re-modeling can take place), and compared to metabolic specialization of oxidative and glycolytic muscle fibers that underlies lactate shuttling among peripheral tissues134-136.
In cardiac muscle, disposal of NADH was identified as a rate-limiting factor that causes lactate production because utilization of glucose and lactate was not sustained under progressively-increasing work loads, whereas pyruvate oxidation and oxygen consumption increased linearly over the same range137; this limitation may also contribute to lactate production in brain cells (see below, ‘redox shuttles’). In smooth vascular muscle, the NADH/NAD redox state of glycolytic compartments and tissue lactate level remain relatively constant in spite of substantial differences in glycolytic flux and lactate production caused by varying Na+,-K+-ATPase activity138. These data support the notion that redox state, NAD(P)H fluorescence (ΔF/F), and lactate level do not provide sufficient information to evaluate changes in metabolic fluxes in muscle or brain. Finally, functional compartmentation of glycogen- and exogenous glucose-derived glycolytic fluxes in vascular smooth muscle is quite complex, with parallel non-mixing glycolytic pathways, one involving preferential oxidative metabolism of glycogen, the other, release of lactate derived from exogenous glucose in conjunction with pumping of sodium and potassium139-144; this degree of metabolic complexity has not yet been established in brain cells.
Cytoplasm-mitochondria redox shuttles
In brain, the malate-aspartate shuttle is the major redox shuttle that transfers reducing equivalents from glycolytically-produced NADH to the mitochondrial electron transport chain. The activity of this shuttle in adult brain astrocytes has been debated because the amount of detectable immunoreactive aspartate/glutamate carrier protein, a critical component of the malate-aspartate shuttle, is low in adult astrocytes compared to adult neurons, whereas this carrier is detectable in astrocytes in immature brain tissue and cultured astrocytes145-147. The apparently-low capacity of adult astrocytes for the malate-aspartate shuttle would be a key contributory factor for predominance of glycolysis and lactate production in astrocytes. However, the above studies did not appear to use antigen retrieval techniques to improve detection of the aspartate/glutamate carrier protein; retrieval procedures substantially increase the detectable amount of immunoreactive heat shock protein 70 in brain tissue148.
Because astrocytes require pyruvate for oxidation and anaplerotic reactions, an alternative mechanism for adult brain astrocytes to obtain pyruvate has been proposed by Cerdán and colleagues149, 150. Their redox switch/redox coupling hypothesis involves a transcellular redox system in which lactate generated in astrocytes is released and taken up by neurons, then converted to pyruvate, which is then released and taken up and oxidized by astrocytes. Thus, redox limitations and shuttling could modulate cellular glucose fluxes in astrocytes and neurons, provide energy to neurons, and return oxidizable pyruvate to astrocytes; many complex processes may contribute to astrocyte-neuron metabolic interactions.
Oxidative capabilities of adult astrocytes
Astrocytes have glycolytic, glycogenolytic, and oxidative capacities32, 152, 153, and their highly-specialized subcellular structures may use glycolytic or oxidative pathways to sustain specific functions. For example, the filopodial processes of astrocytes surround and interact with synapses; filopodia are very thin and were reported to have a paucity of mitochondria, leading to our previous suggestion that these structures may be highly glycolytic31,32. However, identification of cellular processes in electron microscopic studies is difficult due to lack of appropriate markers, and a recent study by Lovatt, Nedergaard, and colleagues151 using transgenic mice that express green fluorescent protein in astrocytes clearly demonstrates the abundance of mitochondria in perisynaptic astrocytic processes. In fact, the relative density of cortical astrocytic mitochondria in green fluorescent protein-positive processes was about 66% greater than in the surrounding neuropil, indicating a higher oxidative capacity in perisynaptic astrocytic processes than nearby neuronal elements151. Furthermore, the astrocytes acutely isolated from adult mice express transcripts for NAD/NADH mitochondrial shuttle systems and they are capable of oxidative metabolism of glucose (i.e., incorporation of label from [13C]glucose into TCA cycle intermediates and TCA cycle-derived amino acids), pyruvate recycling (i.e., the ability to completely degrade glutamate and other compounds by exit of malate from the TCA cycle, conversion of malate to pyruvate, and oxidation of pyruvate), and pyruvate carboxylation (the anaplerotic process necessary for de novo biosynthesis of TCA cycle-derived amino acids)151.
The ability of isolated adult murine astrocytes to oxidize [13C]glucose infers active redox shuttling to transfer reducing equivalents from the NADH generated via glycolysis to the electron transport chain in mitochondria. This capacity is consistent with astrocytic oxidation of interstitial [14C]lactate and [14C]glucose to 14CO2 when the labeled substrates were delivered into brain of normal, conscious adult rats by microdialysis54. Astrocytic oxidation was inferred from inclusion of fluorocitrate in the perfusate to inhibit astrocytic aconitase54, and interpretation of these results depends on the specificity of blockade of the astrocytic compared to the neuronal TCA cycle; cell-selective action is dose dependent. Although there is some uncertainty of the actual drug dose delivered to tissue, the extent of transfer of various compounds across microdialysis membranes is typically ∼10-15%, so tissue inhibitor levels are anticipated to be about 7-10 times lower than the 5, 20, or 100 μM fluorocitrate perfused through the dialysis probe54. Inhibitor specificity for astrocytes is suggested by assays in guinea pig hippocampal synaptosomes; a 10-fold higher fluoroacetate concentration did not deplete ATP levels154, 155. When slices were incubated in artificial cerebrospinal fluid containing 10 mM glucose plus 1 mM fluoroacetate for 30 min, synaptic transmission was impaired and the slice ATP level was reduced 35%; similar incubations did not alter the ATP content of the synaptosomal preparation but did reduce the ATP level in glioma cells by 65%154, 155. In these studies, synaptosomal glycolysis is unlikely to compensate for severe impairment of mitochondrial ATP production because synaptosomal ATP concentration fell by about 70% after treatment of with cyanide or the uncoupler FCCP128. To summarize, adult brain astrocytes can oxidize glucose, and although glucose oxidation is calculated to be much lower than in neurons in MRS studies75, it is likely that oxidative metabolism plays important roles in the perisynaptic processes, soma, and endfeet of astrocytes.
Glycolysis in neurons
Neuronal functions have a critical but unidentified dependence on glucose that has been repeatedly demonstrated in studies in brain slices from adult rodents since the 1970-1980's. For example, replacement of glucose by various alternative substrates, such as mannose, fructose, lactate, and pyruvate, can maintain slice ATP concentrations within 80-90% of control but, depending on the specific procedure used for slice preparation and its duration, these alternative substrates may or may not sustain evoked population spikes even though energy levels are adequate50,51. Thus, glucose has a critical role in maintaining synaptic transmission that is independent of energy generation, and this specific requirement for glucose is removed after about 30 min of metabolic stress by an unidentified process that depends on calcium entry into the cell51. Conceivably, activation of the AMP-dependent protein kinase system, a ‘metabolic sensor’ that can be activated by calcium and serve neuroprotective functions156-158, may participate in adaptive processes in postischemic brain slices. Critical roles of glucose are also evident in other experimental systems, and cultured glutamatergic neurons show a dependence on glucose to maintain neurotransmitter homeostasis during synaptic activity52. The specific neuronal functions that rely on glycolytic metabolism remain to be established, but may include generation of NADPH by the pentose phosphate shunt pathway and packaging of glutamate into synaptic vesicles. Preferential utilization of glycolytic ATP for glutamate transport into synaptic vesicles is conferred by binding of glyceraldehyde phosphate dehydrogenase and 3-phosphoglycerate kinase to vesicular membranes53, and vesicular glutamate loading directly ties neuronal glycolysis to excitatory neurotransmission.
Cell-to-cell lactate shuttling
The concept of the astrocyte-to-neuron lactate shuttle was formulated in 1994 by Pellerin and Magistretti159, who observed that sodium-dependent glutamate uptake into cultured astrocytes stimulated glucose utilization and lactate release to the culture medium; the glycolytic stimulation by glutamate was not inhibited by glutamate receptor antagonists (D-AP5, CNQX, L-AP3, or L-AP4) and glutamate receptor agonists did not mimic the glutamate effects, whereas a glutamate transport inhibitor and a Na+,-K+-ATPase blocker did abolish the metabolic stimulation. Based on these in vitro findings, excitatory glutamatergic neurotransmission was proposed to increase astrocytic glycolysis, followed by release of lactate that may be taken up and oxidized in neurons. Aspects of this concept have been modified and refined in the intervening years since the initial report, and interested readers are referred to more recent reviews75, 160, 161 and references cited therein for the details and proposed circumstances of its applicability. Calculated rates of glucose oxidation in neurons determined by MRS are considerably higher than in astrocytes, and these findings have been incorporated into a model supporting lactate shuttling from astrocytes to neurons, while allowing for some lactate efflux from activated tissue75. Further development of metabolic modeling approaches75, 76, 162, 163 in conjunction with greater sensitivity of MRS assays is anticipated to lead more accurate determination cellular metabolic fluxes in brain in vivo.
The lactate shuttle hypothesis has brought a lot of attention to astrocytic energetics, to potential contributions of astrocytes to brain imaging, and to neuron-astrocyte metabolic interactions, but it is also controversial30, 33, 49, 164 for a number of reasons, including (i) glutamate-induced stimulation of glucose utilization and lactate production in cultured astrocytes is not a universal finding, (ii) the proportion of glucose consumed by astrocytes and neurons in brain in vivo during activation in normal conscious subjects is not known, (iii) the cellular origin and fate of lactate produced in vivo in normal conscious subjects must be established under various paradigms of activation involving different neural pathways, and (iv) the quantity of net cell-to-cell shuttling and oxidation of lactate compared to lactate release must be determined.
Glutamate-induced glycolysis in vitro
Some laboratories consistently observe the in vitro glutamate-induced glycolytic response, whereas others do not (summarized in Tables 5 and 6 in ref.33 and Figs 2 and 3 in ref.165). The reasons for this apparent discrepancy have not been elucidated, but a key issue is oxidative capacities of different astrocyte culture preparations and whether oxidation of glucose, glutamate, or other substrates also supports the energetics sodium extrusion and glutamate conversion to glutamine in astrocytes in vitro and in vivo. For example, glutamate markedly stimulates oxygen consumption by cultured astrocytes166, it is well established that astrocytes oxidize glutamate and the fraction oxidized compared to that converted to glutamine rises as extracellular glutamate concentration is increased167, and an astrocyte preparation that does not increase CMRglc during exposure L-glutamate does show an increase in CMRglc when glutamate is replaced with D-aspartate, a transportable, non-metabolizable analog168. Culture conditions and developmental plasticity can influence oxidative capability, and, for example, oxidation of glucose and lactate by astrocytes grown in a culture medium containing 20-25 mM glucose is half that of cultures grown in low glucose169. However, astrocytes grown in parallel in low or high glucose and assayed under different conditions showed no increase in glucose utilization or lactate release during exposure to 0.2 or 1 mM glutamate (see Table 6 in ref. 33). Thus, unidentified factors have a high impact on the astrocytic glutamate-response phenotype, and extrapolation of data from in vitro studies to in vivo activities must be made with caution. The recent discovery of mitochondrial abundance in perisynaptic astrocytic processes in adult mice151 strengthens the likelihood that oxidative metabolism contributes to the astrocytic energetics of excitatory neurotransmission32.
Neuronal roles in lactate generation during activation
As noted above, synaptosomes have considerable excess glycolytic capacity128, and metabolic modeling of energy supply and demand based on kinetic properties, distributions, and levels of glucose and lactate transporters led Simpson and colleagues to conclude that glucose uptake and metabolism and generation of lactate transients is mainly neuronal27. This conclusion is strongly supported by a recent study by Caesar et al.113 who showed that postsynaptic neuronal activation governs the activity-dependent increases in CMRglc and lactate concentration. The stimulus-induced rise in glucose consumption and the increase in extracellular lactate level in cerebellum of anesthetized rats were both blocked by CNQX113, a glutamate (AMPA) receptor antagonist that does not to inhibit the glutamate-induced rise in CMRglc in cultured astrocytes159. Because glutamate will be continuously taken up into cerebellar astrocytes during CNQX treatment, the in vivo stimulus-induced glycolytic activity is not related to glutamate uptake. Activity-dependent stimulation of blood flow and CMRO2 in various rat brain structures is dependent on postsynaptic glutamate AMPA or NMDA receptor activity (see studies cited by Caesar et al.113), and postsynaptic neuronal activation involving mitochondrial dynamics has been linked to NAD(P)H transients in hippocampal slices by Shuttleworth and colleagues77, 79, 80. These findings emphasize the importance of inclusion of neuronal glucose utilization and lactate production into modeling of functional metabolism; astrocytic glutamate uptake is not necessarily the major or the only factor governing lactate generation during brain activation in vivo.
Quantification of lactate utilization and release
Functional activity alters fluxes in many pathways, and large fraction of the labeled metabolites of [1- and 6-14C]glucose consumed during activation over and above that utilized during rest is quickly released from activated tissue, probably mainly as lactate (Fig. 5). Lactate release from activated cells may be necessary to sustain high, local glycolytic activity, but the energetic cost of lactate release would be negligible during most normal stimulatory conditions. This lactate may, in part, be consumed by other brain regions, but labeling gradients from focally-activated areas into adjacent non-activated tissue are not obvious in autoradiographic studies, suggesting that widespread distribution may slightly increase overall labeling throughout the brain, along with substantial release to blood via multiple routes. Quantification of the fate of lactate in brain in vivo is a major challenge to neurochemists.
Fig. 5. Summary of key aspects of glucose metabolism during brain activation and probable roles of astrocytes in metabolite spreading and release from brain.

Increased functional activity stimulates fluxes of glucose into different pathways, leading to discordant brain images obtained with [1- and 6-14C]glucose compared to [14C]deoxyglucose; the magnitude of glucose utilization is greatly underestimated with [14C]glucose. Astrocytes have specialized metabolic compartments, and metabolites generated from labeled glucose can spread in neurons or astrocytes via intra- and extracellular routes, thereby influencing local levels of labeled metabolites. Gap junction-coupled astrocytes form a large syncytium and the endfeet interface with the major fluid transport systems in brain: blood, interstitial fluid, and cerebrospinal fluid.
The functional anatomy of astrocytes suggests that they have major roles in metabolite trafficking involving rapid distribution within large, heterogeneous syncytia (Fig. 5). Astrocytes have a higher capacity than neurons for lactate uptake from extracellular fluid of brain slices and for lactate dispersal to other coupled astrocytes compared to shuttling to nearby neurons. Astrocytes are poised to interact with local and distant major fluid pathways in brain because their processes and endfeet are highly coupled by gap junctions and the endfeet are in close proximity to the cerebral vasculature, perivascular fluid, and cerebrospinal fluid. Thus, astrocytic gap junction-coupled syncytium can be used to transport nutrients from blood to inter-capillary sites, as well as rapidly disperse metabolic by-products from activated cells to blood, CSF, and perivascular fluid drainage systems. Because astrocytes have close apposition to synapses, the concept of the tripartite synapse comprised of pre- and post-synaptic neuronal elements plus synaptically-associated astrocytes170 should be expanded to include the large astrocytic syncytium linked to each perisynaptic astrocyte.
COMPLEX BIOLOGY UNDERLYING BRAIN IMAGES: CLINICAL IMPLICATIONS
Brain images reflect the biochemical reactions and cellular physiology of the living brain and may provide deceptively simple pictures of brain activation and neurological disease (Fig. 6). In the simplest case, [14C]DG and 18F[FDG] images represent hexokinase activity, and glucose utilization rates calculated from DG or FDG phosphorylation correspond to glucose utilization under steady state conditions. Under pathophysiological conditions in which glucose levels are abnormal, especially during hypoglycemia, or when the properties of hexokinase differ (e.g., in tumors), appropriate corrections to the usual calculation factors must be made. PET images derived from [11C]glucose and MRS studies using [13C]glucose are much more complex because the quantities and identities of the labeled compounds released from the brain are not known unless additional studies are carried out. Thus, increased glycolysis associated with massive lactate release during an energy crisis would be registered by FDG-PET images but not by [11C]glucose-PET or [13C]glucose-MRS assays. Many neurodegenerative diseases are associated with progressive mitochondrial dysfunction, and the temporal changes in functional activity of brain structures affected by these diseases are often tracked by FDG-PET. In these cases, caution must be applied to interpretation of results, since oxidative deficits would probably lead to compensatory increases in glycolysis. Thus, FDG-PET images may appear to be normal or, perhaps, somewhat higher than normal until irreversible damage ensues and metabolic rates fall as cells die. In these cases, the earliest phases of the disease would not be detected and appropriately treated in a timely manner; treatments may, in fact, be initiated during the irreversible stages of cellular damage. Also, many long-term diseases with cardiovascular complications involve changes in the basement membranes of cerebral vessels. Vascular membrane thickening has the potential to alter pulsatile perivascular flow and impair nutrient trafficking to and from brain, and abnormal delivery and clearance of imaging tracers and their metabolites could affect PET and MRS studies of brain disease. A better understanding the biology of interactions among neurons, glia, and the vasculature is necessary to improve interpretation of images of brain activation and disease.
Fig. 6. Imaging neurodegenerative diseases.

Slow, progressive evolution of mitochondrial damage may lead to compensatory increases in glycolysis. Increased glycolytic utilization of glucose would be registered by [18F]fluorodeoxyglucose (FDG)-positron emission tomographic (PET) studies, but not by use of labeled glucose if lactate efflux from brain is increased. Glucose utilization rates that are within or above the normal range during early, potentially-treatable stages of disease may not be appropriately interpreted unless lactate generation and release is also assessed when imaging of diseased brain is carried out. Pathophysiological changes involving the vasculature could affect nutrient and by-product trafficking, as well as transport of tracers used for brain imaging and clearance of their metabolites.
ACKNOWLEDGEMENTS
This work was supported, in part, by NIH grant NS36728.
REFERENCES
- 1.Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]deoxyglucose method for the measurement of local glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- 2.Sokoloff L. Cerebral circulation, energy metabolism, and protein synthesis: General characteristics and principles of measurement. In: Phelps M, Mazziotta J, Schelbert H, editors. Positron Emission Tomography and Autoradiography: Principles and Applications for the Brain and Heart. Raven Press; New York: 1986. pp. 1–71. [Google Scholar]
- 3.Nelson T, Sokoloff L. 2-Deoxyglucose incorporation into rat brain glycogen during measurement of local cerebral glucose utilization by the 2-deoxyglucose method. In: Kaufman EE, editor. J. Neurochem. Vol. 43. 1984. pp. 949–956. [DOI] [PubMed] [Google Scholar]
- 4.Dienel GA, Cruz NF, Mori K, Sokoloff L. Acid lability of metabolites of 2-deoxyglucose in rat brain: Implications for estimates of kinetic parameters of deoxyglucose phosphorylation and transport between blood and brain. J. Neurochem. 1990;54:1440–1448. doi: 10.1111/j.1471-4159.1990.tb01981.x. [DOI] [PubMed] [Google Scholar]
- 5.Dienel GA, Cruz NF, Sokoloff L. Metabolites of 2-deoxy-[14C]glucose in plasma and brain: Influence on rate of glucose utilization determined with deoxyglucose in rat brain. J. Cereb. Blood Flow Metab. 1993;13:315–327. doi: 10.1038/jcbfm.1993.40. [DOI] [PubMed] [Google Scholar]
- 6.Dienel GA, Cruz NF. Synthesis of deoxyglucose-1-phosphate, deoxyglucose-1,6-bisphosphate, and other metabolites of 2-deoxy-D-[14C]glucose in rat brain in vivo: influence of time and tissue glucose level. J. Neurochem. 1993;60:2217–2231. doi: 10.1111/j.1471-4159.1993.tb03508.x. [DOI] [PubMed] [Google Scholar]
- 7.Baquer NZ, Hothersall JS, McLean P. Function and regulation of the pentose phosphate pathway in brain. Curr. Top. Cell. Regul. 1988;29:265–289. [PubMed] [Google Scholar]
- 8.Collins RC, McCandless DW, Wagman IL. Cerebral glucose utilization: Comparison of [14C]deoxyglucose and [6-14C]glucose quantitative autoradiography. J. Neurochem. 1987;49:1564–1570. doi: 10.1111/j.1471-4159.1987.tb01028.x. [DOI] [PubMed] [Google Scholar]
- 9.Ackermann RF, Lear JL. Glycolysis-induced discordance between glucose metabolic rates measured with radiolabeled fluorodeoxyglucose and glucose. J. Cereb. Blood Flow Metab. 1989;9:774–785. doi: 10.1038/jcbfm.1989.111. [DOI] [PubMed] [Google Scholar]
- 10.Cruz NF, Ball KK, Dienel GA. Functional imaging of focal brain activation in conscious rats: Impact of [(14)C]glucose metabolite spreading and release. J. Neurosci. Res. 2007;85:3254–3266. doi: 10.1002/jnr.21193. [DOI] [PubMed] [Google Scholar]
- 11.Lear JL, Ackermann RF. Comparison of cerebral glucose metabolic rates measured with fluorodeoxyglucose and glucose labeled in the 1, 2, 3-4, and 6 positions using double label quantitative digital autoradiography. J. Cereb. Blood Flow Metab. 1988;8:575–585. doi: 10.1038/jcbfm.1988.99. [DOI] [PubMed] [Google Scholar]
- 12.Lear J, Ackermann RF. Why the deoxyglucose method has proven so useful in cerebral activation studies: The unappreciated prevalence of stimulation-induced glycolysis. J. Cereb. Blood Flow Metab. 1989;9:911–913. doi: 10.1038/jcbfm.1989.128. [DOI] [PubMed] [Google Scholar]
- 13.Lear J, Ackermann R. Autoradiographic comparison of FDG-based and GLU-based measurements of cerebral glucose transport and metabolism: Normal and activated conditions. In: Lassen N, Ingvar D, Raichle M, Friberg L, editors. Brain work and mental activity, Alfred Benzon Symposium 31. Copenhagen, Munskgaard: 1991. pp. 142–152. [Google Scholar]
- 14.Adachi K, Cruz NF, Sokoloff L, Dienel GA. Labeling of metabolic pools by [6-14C]glucose during K+-induced stimulation of glucose utilization in rat brain. J. Cereb. Blood Flow Metab. 1995;15:97–110. doi: 10.1038/jcbfm.1995.11. [DOI] [PubMed] [Google Scholar]
- 15.Cruz NF, Adachi K, Dienel GA. Metabolite trafficking during K+-induced spreading cortical depression: Rapid efflux of lactate from cerebral cortex. J. Cereb. Blood Flow Metab. 1999;19:380–392. doi: 10.1097/00004647-199904000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Serviere J, Webster WR. A combined electrophysiological and [14C] 2-deoxyglucose study of the frequency organization of the inferior colliculus of the cat. Neurosci Lett. 1981;27:113–118. doi: 10.1016/0304-3940(81)90253-6. [DOI] [PubMed] [Google Scholar]
- 17.Ryan AF, Woolf NK, Sharp FR. Tonotopic organization in the central auditory pathway of the Mongolian gerbil: a 2-deoxyglucose study. J. Comp. Neurol. 1982;207:369–380. doi: 10.1002/cne.902070408. [DOI] [PubMed] [Google Scholar]
- 18.Webster WR, Serviere J, Crewther D, Crewther S. Iso-frequency 2-DG contours in the inferior colliculus of the awake monkey. Exp. Brain Res. 1984;56:425–437. doi: 10.1007/BF00237983. [DOI] [PubMed] [Google Scholar]
- 19.Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J. Neurosci. 1998;18:5225–5233. doi: 10.1523/JNEUROSCI.18-14-05225.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruz NF, Lasater A, Zielke HR, Dienel GA. Activation of astrocytes in brain of conscious rats during acoustic stimulation: acetate utilization in working brain. J. Neurochem. 2005;92:934–947. doi: 10.1111/j.1471-4159.2004.02935.x. [DOI] [PubMed] [Google Scholar]
- 21.Dienel GA, Schmidt KC, Cruz NF. Astrocyte activation in vivo during graded photic stimulation. J Neurochem. 2007;103:1506–1522. doi: 10.1111/j.1471-4159.2007.04859.x. [DOI] [PubMed] [Google Scholar]
- 22.Blomqvist G, Stone-Elander S, Halldin C, Roland PE, Widen L, Lindqvist M, Swahn CG, Langstrom B, Wiesel FA. Positron emission tomographic measurements of cerebral glucose utilization using [1-11C]D-glucose. J. Cereb. Blood Flow Metab. 1990;10:467–483. doi: 10.1038/jcbfm.1990.89. [DOI] [PubMed] [Google Scholar]
- 23.Rennels ML, Gregory TF, Blaumanis OR, Fujimoto K, Grady PA. Evidence for a ‘paravascular’ fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res. 1985;326:47–63. doi: 10.1016/0006-8993(85)91383-6. [DOI] [PubMed] [Google Scholar]
- 24.Koh L, Zakharov A, Johnston M. Integration of the subarachnoid space and lymphatics: is it time to embrace a new concept of cerebrospinal fluid absorption? Cerebrospinal Fluid Research. 2005;2:6. doi: 10.1186/1743-8454-2-6. ( http://www.cerebrospinalfluidresearch.com/content/2/1/6) [DOI] [PMC free article] [PubMed]
- 25.Nagra G, Koh L, Zakharov A, Armstrong D, Johnston M. Quantification of cerebrospinal fluid transport across the cribriform plate into lymphatics in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006;291:R1383–R1389. doi: 10.1152/ajpregu.00235.2006. [DOI] [PubMed] [Google Scholar]
- 26.Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J. 1999;343:281–299. [PMC free article] [PubMed] [Google Scholar]
- 27.Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007;27:1766–1791. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ball KK, Gandhi GK, Thrash J, Cruz NF, Dienel GA. Astrocytic connexin distributions and rapid, extensive dye transfer via gap junctions in the inferior colliculus: Implications for [(14)C]glucose metabolite trafficking. J. Neurosci. Res. 2007;85:3267–3283. doi: 10.1002/jnr.21376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hertz L, Dienel GA. Lactate transport and transporters: general principles and functional roles in brain cells. J. Neurosci. Res. 2005;79:11–18. doi: 10.1002/jnr.20294. [DOI] [PubMed] [Google Scholar]
- 30.Chih CP, Roberts EL., Jr. Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J. Cereb. Blood Flow Metab. 2003;23:1263–1281. doi: 10.1097/01.WCB.0000081369.51727.6F. [DOI] [PubMed] [Google Scholar]
- 31.Dienel GA, Cruz NF. Neighborly interactions of metabolically-activated astrocytes in vivo. Neurochem. Int. 2003;43:339–354. doi: 10.1016/s0197-0186(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 32.Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- 33.Dienel GA, Cruz NF. Nutrition during brain activation: does cell-to-cell lactate shuttling contribute significantly to sweet and sour food for thought? Neurochem. Int. 2004;45:321–351. doi: 10.1016/j.neuint.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 34.Zielke HR, Zielke CL, Baab PJ. Oxidation of (14)C-labeled compounds perfused by microdialysis in the brains of free-moving rats. J. Neurosci. Res. 2007;85:3145–3149. doi: 10.1002/jnr.21424. [DOI] [PubMed] [Google Scholar]
- 35.Miller AL, Hawkins RA, Veech RL. Decreased rate of glucose utilization by rat brain in vivo after exposure to atmospheres containing high concentrations of CO2. J. Neurochem. 1975;25:553–558. doi: 10.1111/j.1471-4159.1975.tb04367.x. [DOI] [PubMed] [Google Scholar]
- 36.Siesjö BK. Brain energy metabolism. John Wiley & Sons; Chicester: 1978. [Google Scholar]
- 37.Dalsgaard MK. Fuelling cerebral activity in exercising man. J. Cereb. Blood Flow Metab. 2006;26:731–750. doi: 10.1038/sj.jcbfm.9600256. [DOI] [PubMed] [Google Scholar]
- 38.Madsen PL, Cruz N,F, Sokoloff L, Dienel GA. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J. Cereb. Blood Flow Metab. 1999;19:393–400. doi: 10.1097/00004647-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Dienel GA, Wang RY, Cruz NF. Generalized sensory stimulation of conscious rats increases labeling of oxidative pathways of glucose metabolism when the brain glucose-oxygen uptake ratio rises. J. Cereb. Blood Flow Metab. 2002;22:1490–1502. doi: 10.1097/01.WCB.0000034363.37277.89. [DOI] [PubMed] [Google Scholar]
- 40.Cruz NF, Dienel GA. High brain glycogen levels in brains of rats with minimal environmental stimuli: Implications for metabolic contributions of working astrocytes. J. Cereb. Blood Flow Metab. 2002;22:1476–1489. doi: 10.1097/01.WCB.0000034362.37277.C0. [DOI] [PubMed] [Google Scholar]
- 41.Dienel GA, Ball KK, Cruz NF. A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J. Neurochem. 2007;102:466–478. doi: 10.1111/j.1471-4159.2007.04595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schasfoort EM, De Bruin LA, Korf J. Mild stress stimulates rat hippocampal glucose utilization transiently via NMDA receptors, as assessed by lactography. Brain Res. 1988;475:58–63. doi: 10.1016/0006-8993(88)90198-9. [DOI] [PubMed] [Google Scholar]
- 43.De Bruin LA, Schasfoort EM, Steffens AB, Korf J. Effects of stress and exercise on rat hippocampus and striatum extracellular lactate. Am. J. Physiol. 1990;259:R773–R779. doi: 10.1152/ajpregu.1990.259.4.R773. [DOI] [PubMed] [Google Scholar]
- 44.Mangia S, Tkac I, Logothetis NK, Gruetter R, Van de Moortele PF, Ugurbil K. Dynamics of lactate concentration and blood oxygen level-dependent effect in the human visual cortex during repeated identical stimuli. J. Neurosci. Res. 2007;85:3340–3346. doi: 10.1002/jnr.21371. [DOI] [PubMed] [Google Scholar]
- 45.Dienel GA, Hertz L. Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 2001;66:824–838. doi: 10.1002/jnr.10079. [DOI] [PubMed] [Google Scholar]
- 46.Ksiezak-Reding H, Blass JP, Gibson GE. Studies on the pyruvate dehydrogenase complex in brain with the arylamine acetyltransferase-coupled assay. J. Neurochem. 1982;38:1627–1636. doi: 10.1111/j.1471-4159.1982.tb06643.x. [DOI] [PubMed] [Google Scholar]
- 47.Dienel GA. Energy Generation in the Central Nervous System. In: Edvinsson L, Krause D, editors. Cerebral Blood Flow and Metabolism. 2nd Ed. Lippincott, Williams & Wilkins; Phila: 2002. pp. 140–161. [Google Scholar]
- 48.Folbergrova J, MacMillan V, Siesjö BK. The effect of moderate and marked hypercapnia upon the energy state and upon the cytoplasmic NADH-NAD+ ratio of the rat brain. J. Neurochem. 1972;19:2497–2505. doi: 10.1111/j.1471-4159.1972.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 49.Korf J. Is brain lactate metabolized immediately after neuronal activity through the oxidative pathway? J. Cereb. Blood Flow Metab. 2006;26:1584–1586. doi: 10.1038/sj.jcbfm.9600321. [DOI] [PubMed] [Google Scholar]
- 50.Dienel GA, Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50:362–388. doi: 10.1002/glia.20157. [DOI] [PubMed] [Google Scholar]
- 51.Okada Y, Lipton P. Glucose, oxidative energy metabolism, and neural function in brain slices—Glycolysis plays a key role in neural activity. In: Gibson GE, Dienel GA, editors. Handbook of Neurochemistry and Molecular Neurobiology. Brain Energetics. Integration of Molecular and Cellular Processes. Springer-Verlag; Berlin: 2007. pp. 17–39. [Google Scholar]
- 52.Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS. Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J. Cereb. Blood Flow Metab. 2006;26:1285–1297. doi: 10.1038/sj.jcbfm.9600281. [DOI] [PubMed] [Google Scholar]
- 53.Ikemoto A, Bole DG, Ueda T. Glycolysis and glutamate accumulation into synaptic vesicles. Role of glyceraldehyde phosphate dehydrogenase and 3-phosphoglycerate kinase. J. Biol. Chem. 2003;278:5929–5940. doi: 10.1074/jbc.M211617200. [DOI] [PubMed] [Google Scholar]
- 54.Zielke HR, Zielke CL, Baab PJ, Tildon JT. Effect of fluorocitrate on cerebral oxidation of lactate and glucose in freely moving rats. J. Neurochem. 2007;101:9–16. doi: 10.1111/j.1471-4159.2006.04335.x. [DOI] [PubMed] [Google Scholar]
- 55.Holden JE, Mori K, Dienel GA, Cruz NF, Nelson T, Sokoloff L. Modeling the dependence of hexose distribution volumes in brain on plasma glucose concentration: implications for estimation of the local 2-deoxyglucose lumped constant. J. Cereb Blood Flow Metab. 1991;11:171–182. doi: 10.1038/jcbfm.1991.50. [DOI] [PubMed] [Google Scholar]
- 56.MacMillan V. The effects of acute carbon monoxide intoxication on the cerebral energy metabolism of the rat. Can. J. Physiol. Pharmacol. 1975;53:354–362. doi: 10.1139/y75-051. [DOI] [PubMed] [Google Scholar]
- 57.Ljunggren B, Norberg K, Siesjö BK. Influence of tissue acidosis upon restitution of brain energy metabolism following total ischemia. Brain Res. 1974;77:173–186. doi: 10.1016/0006-8993(74)90782-3. [DOI] [PubMed] [Google Scholar]
- 58.Duffy TE, Howse DC, Plum F. Cerebral energy metabolism during experimental status epilepticus. J. Neurochem. 1975;24:925–934. doi: 10.1111/j.1471-4159.1975.tb03657.x. [DOI] [PubMed] [Google Scholar]
- 59.Ponten U, Ratcheson RA, Salford LG, Siesjö BK. Optimal freezing conditions for cerebral metabolites in rats. J. Neurochem. 1973;21:1127–1138. doi: 10.1111/j.1471-4159.1973.tb07567.x. [DOI] [PubMed] [Google Scholar]
- 60.Madsen PL, Hasselbalch SG, Hagemann LP, Olsen KS, Bülow J, Holm S, Wildschiodtz G, Paulson OB, Lassen NA. Persistent resetting of the cerebral oxygen/glucose uptake ratio by brain activation: Evidence obtained with the Kety-Schmidt technique. J. Cereb. Blood Flow Metab. 1995;15:485–491. doi: 10.1038/jcbfm.1995.60. [DOI] [PubMed] [Google Scholar]
- 61.Ide K, Horn A, Secher NH. Cerebral metabolic response to submaximal exercise. J. Appl. Physiol. 1999;87:1604–1608. doi: 10.1152/jappl.1999.87.5.1604. [DOI] [PubMed] [Google Scholar]
- 62.Ide K, Schmalbruch IK, Quistorff B, Horn A, Secher NH. Lactate, glucose and O2 uptake in human brain during recovery from maximal exercise. J. Physiol. 2000;522:159–164. doi: 10.1111/j.1469-7793.2000.t01-2-00159.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dalsgaard MK, Ide K, Cai Y, Quistorff B, Secher NH. The intent to exercise influences the cerebral O(2)/carbohydrate uptake ratio in humans. J. Physiol. 2002;540:681–689. doi: 10.1113/jphysiol.2001.013062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- 65.Ribeiro L, Kuwabara H, Meyer H, Fujita H, Marrett S, Evans A, Gjedde A. Cerebral blood flow and metabolism during nonspecific bilateral visual stimulation in normal subjects. In: Uemura K, Lassen NA, Jones T, Kanno I, editors. Quantification of brain function: Tracer kinetics and image analysis in brain PET. Elsevier; Amsterdam: 1993. pp. 229–234. [Google Scholar]
- 66.Madsen PL, Linde R, Hasselbalch SG, Paulson OB, Lassen NA. Activation-induced resetting of cerebral oxygen and glucose uptake in the rat. J. Cereb. Blood Flow Metab. 1998;18:742–748. doi: 10.1097/00004647-199807000-00005. [DOI] [PubMed] [Google Scholar]
- 67.Schmalbruch IK, Linde R, Paulson OB, Madsen PL. Activation-induced resetting of cerebral metabolism and flow is abolished by beta-adrenergic blockade with propranolol. Stroke. 2002;33:251–255. doi: 10.1161/hs0102.101233. [DOI] [PubMed] [Google Scholar]
- 68.Wang L, Kondo M, Bill A. Glucose metabolism in cat outer retina. Effects of light and hyperoxia. Invest. Ophthalmol. Vis. Sci. 1997;38:48–55. [PubMed] [Google Scholar]
- 69.Wang L, Tornquist P, Bill A. Glucose metabolism of the inner retina in pigs in darkness and light. Acta Physiol. Scand. 1997;160:71–74. doi: 10.1046/j.1365-201X.1997.00131.x. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Tornquist P, Bill A. Glucose metabolism in pig outer retina in light and darkness. Acta Physiol. Scand. 1997;160:75–81. doi: 10.1046/j.1365-201X.1997.00030.x. [DOI] [PubMed] [Google Scholar]
- 71.Ide K, Secher NH. Cerebral blood flow and metabolism during exercise. Prog. Neurobiol. 2000;61:397–414. doi: 10.1016/s0301-0082(99)00057-x. [DOI] [PubMed] [Google Scholar]
- 72.Mason GF, Rothman DL, Behar KL, Shulman RG. NMR determination of the TCA cycle rate and alpha-ketoglutarate/glutamate exchange rate in rat brain. J. Cereb. Blood Flow Metab. 1992;12:434–447. doi: 10.1038/jcbfm.1992.61. [DOI] [PubMed] [Google Scholar]
- 73.Mason GF, Gruetter R, Rothman DL, Behar KL, Shulman RG, Novotny EJ. Simultaneous determination of the rates of the TCA cycle, glucose utilization, alpha-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J. Cereb Blood Flow Metab. 1995;15:12–25. doi: 10.1038/jcbfm.1995.2. [DOI] [PubMed] [Google Scholar]
- 74.Gruetter R, Seaquist ER, Ugurbil K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am. J. Physiol. Endocrinol. Metab. 2001;281:E100–112. doi: 10.1152/ajpendo.2001.281.1.E100. [DOI] [PubMed] [Google Scholar]
- 75.Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J. Cereb. Blood Flow Metab. 2006;26:865–877. doi: 10.1038/sj.jcbfm.9600263. [DOI] [PubMed] [Google Scholar]
- 76.Shestov AA, Valette J, Uğurbil K, Henry PG. On the reliability of (13)C metabolic modeling with two-compartment neuronal-glial models. J/ Neurosci Res. 2007;85:3294–3303. doi: 10.1002/jnr.21269. [DOI] [PubMed] [Google Scholar]
- 77.Shuttleworth CW, Brennan AM, Connor JA. NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J. Neurosci. 2003;23:3196–3208. doi: 10.1523/JNEUROSCI.23-08-03196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- 79.Brennan AM, Connor JA, Shuttleworth CW. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J. Cereb. Blood Flow Metab. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- 80.Brennan AM, Connor JA, Shuttleworth CW. Modulation of the amplitude of NAD(P)H fluorescence transients after synaptic stimulation. J. Neurosci. Res. 2007;85:3233–3243. doi: 10.1002/jnr.21288. [DOI] [PubMed] [Google Scholar]
- 81.Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- 82.Badar-Goffer RS, Ben-Yoseph O, Bachelard HS, Morris PG. Neuronal-glial metabolism under depolarizing conditions. A 13C-n.m.r. study. Biochem. J. 1992;282:225–230. doi: 10.1042/bj2820225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mantz J, Cordier J, Giaume C. Effects of general anesthetics on intercellular communications mediated by gap junctions between astrocytes in primary culture. Anesthesiology. 1993;78:892–901. doi: 10.1097/00000542-199305000-00014. [DOI] [PubMed] [Google Scholar]
- 84.Burt JM, Spray DC. Volatile anesthetics block intercellular communication between neonatal rat myocardial cells. Circ. Res. 1989;65:829–837. doi: 10.1161/01.res.65.3.829. [DOI] [PubMed] [Google Scholar]
- 85.Divakaran P, Joiner F, Rigor BM, Wiggins RC. Accumulation and persistence of halothane in adult and fetal rat brain as a result of subanesthetic exposure. J. Neurochem. 1980;34:1543–1546. doi: 10.1111/j.1471-4159.1980.tb11239.x. [DOI] [PubMed] [Google Scholar]
- 86.Duncan WA, Raventós J. The pharmacokinetics of halothane (fluothane) anaesthesia. Br. J. Anaesth. 1959;31:302–315. doi: 10.1093/bja/31.7.302. [DOI] [PubMed] [Google Scholar]
- 87.Larson CP, Jr, Eger EI, 2nd, Severinghaus JW. The solubility of halothane in blood and tissue homogenates. Anesthesiology. 1962;23:349–355. doi: 10.1097/00000542-196205000-00009. [DOI] [PubMed] [Google Scholar]
- 88.Eugenín EA, Brañes MC, Berman JW, Sáez JC. TNF-alpha plus IFN-gamma induce connexin43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses. J. Immunol. 2003;170:1320–1328. doi: 10.4049/jimmunol.170.3.1320. [DOI] [PubMed] [Google Scholar]
- 89.Cremer JE, Sarna CS, Teal HM, Cunningham VJ. Amino acid precursors: Their transport into brain and initial metabolism. In: Fonnum F, editor. Amino acids as chemical transmitters (Proceedings of the NATO Advanced Study Institute on Amino Acids as Chemical Transmitters; New York, NY: Plenum Press; 1977. pp. 669–689. [Google Scholar]
- 90.Crosby G, Crane AM, Sokoloff L. A comparison of local rates of glucose utilization in spinal cord and brain in conscious and nitrous oxide- or pentobarbital-treated rats. Anesthesiology. 1984;61:434–438. doi: 10.1097/00000542-198410000-00012. [DOI] [PubMed] [Google Scholar]
- 91.Sakabe T, Tsutsui T, Maekawa T, Ishikawa T, Takeshita H. Local cerebral glucose utilization during nitrous oxide and pentobarbital anesthesia in rats. Anesthesiology. 1985;63:262–266. doi: 10.1097/00000542-198509000-00004. [DOI] [PubMed] [Google Scholar]
- 92.Cremer JE, Cunningham VJ, Pardridge WM, Braun LD, Oldendorf WH. Kinetics of blood-brain barrier transport of pyruvate, lactate and glucose in suckling, weanling and adult rats. J. Neurochem. 1979;33:439–445. doi: 10.1111/j.1471-4159.1979.tb05173.x. [DOI] [PubMed] [Google Scholar]
- 93.Vannucci SJ, Simpson IA. Developmental switch in brain nutrient transporter expression in the rat. Am. J. Physiol. Endocrinol. Metab. 2003;285:E1127–1134. doi: 10.1152/ajpendo.00187.2003. [DOI] [PubMed] [Google Scholar]
- 94.Cremer JE, Heath DF. The estimation of rates of utilization of glucose and ketone bodies in the brain of the suckling rat using compartmental analysis of isotopic data. Biochem. J. 1974;142:527–544. doi: 10.1042/bj1420527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.LaManna JC, Harrington JF, Vendel LM, Abi-Saleh K, Lust WD, Harik SI. Regional blood-brain lactate influx. Brain Res. 1993;614:164–170. doi: 10.1016/0006-8993(93)91030-v. [DOI] [PubMed] [Google Scholar]
- 96.Lear JL, Kasliwal RK. Autoradiographic measurement of cerebral lactate transport rate constants in normal and activated conditions. J. Cereb. Blood Flow Metab. 1991;11:576–580. doi: 10.1038/jcbfm.1991.106. [DOI] [PubMed] [Google Scholar]
- 97.Konitzer K, Voigt S. Metabolism of blood-borne lactate in rat brain in vivo. Acta Biol. Med. Ger. 1977;36:1049–1059. [PubMed] [Google Scholar]
- 98.Leegsma-Vogt G, Venema K, Korf J. Evidence for a lactate pool in the rat brain that is not used as an energy supply under normoglycemic conditions. J. Cereb. Blood Flow Metab. 2003;23:933–941. doi: 10.1097/01.WCB.0000080650.64357.8F. [DOI] [PubMed] [Google Scholar]
- 99.Leegsma-Vogt G, van der Werf S, Venema K, Korf J. Modeling cerebral arteriovenous lactate kinetics after intravenous lactate infusion in the rat. J. Cereb. Blood Flow Metab. 2004;24:1071–1080. doi: 10.1097/01.WCB.0000129417.80261.A6. [DOI] [PubMed] [Google Scholar]
- 100.Hawkins RA, Miller AL, Cremer JE, Veech RL. Measurement of the rate of glucose utilization by rat brain in vivo. J. Neurochem. 1974;23:917–923. doi: 10.1111/j.1471-4159.1974.tb10743.x. [DOI] [PubMed] [Google Scholar]
- 101.Hawkins RA, Hawkins PA, Mans AM, Viña JR, DeJoseph MR. Optimizing the measurement of regional cerebral glucose consumption with [6-14C]glucose. J. Neurosci. Methods. 1994;54:49–62. doi: 10.1016/0165-0270(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 102.Mans AM, Davis DW, Hawkins RA. Regional brain glucose use in unstressed rats after two days of starvation. Metab. Brain Dis. 1987;2:213–221. doi: 10.1007/BF00999693. [DOI] [PubMed] [Google Scholar]
- 103.Hawkins R, Hass WK, Ransohoff J. Measurement of regional brain glucose utilization in vivo using [2(−14)C] glucose. Stroke. 1979;10:690–703. doi: 10.1161/01.str.10.6.690. [DOI] [PubMed] [Google Scholar]
- 104.Savaki HE, Davidsen L, Smith C, Sokoloff L. Measurement of free glucose turnover in brain. J. Neurochem. 1980;35:495–502. doi: 10.1111/j.1471-4159.1980.tb06293.x. [DOI] [PubMed] [Google Scholar]
- 105.Loewenstein WR. Junctional intercellular communication: the cell-to-cell membrane channel. Physiol. Rev. 1981;61:829–913. doi: 10.1152/physrev.1981.61.4.829. [DOI] [PubMed] [Google Scholar]
- 106.Giaume C, Tabernero A, Medina JM. Metabolic trafficking through astrocytic gap junctions. Glia. 1997;21:114–123. [PubMed] [Google Scholar]
- 107.Tabernero A, Medina JM, Giaume C. Glucose metabolism and proliferation in glia: role of astrocytic gap junctions. J. Neurochem. 2006;99:1049–1061. doi: 10.1111/j.1471-4159.2006.04088.x. [DOI] [PubMed] [Google Scholar]
- 108.Pierre K, Debernardi R, Magistretti PJ, Pellerin L. Noradrenaline enhances monocarboxylate transporter 2 expression in cultured mouse cortical neurons via a translational regulation. J. Neurochem. 2003;86:1468–1476. doi: 10.1046/j.1471-4159.2003.01964.x. [DOI] [PubMed] [Google Scholar]
- 109.Rothman DL, Behar KL, Hyder F, Shulman RG. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu. Rev. Physiol. 2003;65:401–427. doi: 10.1146/annurev.physiol.65.092101.142131. [DOI] [PubMed] [Google Scholar]
- 110.Krebs HA. The Pasteur effect and the relations between respiration and fermentation. Essays Biochem. 1972;8:1–34. [PubMed] [Google Scholar]
- 111.Winkler BS, Starnes CA, Sauer MW, Firouzgan Z, Chen SC. Cultured retinal neuronal cells and Müller cells both show net production of lactate. Neurochem. Int. 2004;45:311–320. doi: 10.1016/j.neuint.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 112.Öz G, Berkich DA, Henry PG, Xu Y, LaNoue K, Hutson SM, Gruetter R. Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J. Neurosci. 2004;24:11273–11279. doi: 10.1523/JNEUROSCI.3564-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Caesar K, Hashemi P, Douhou A, Bonvento G, Boutelle MG, Walls AB, Lauritzen M. Glutamate receptor dependent increments in lactate, glucose and oxygen metabolism evoked in rat cerebellum in vivo. J. Physiol. 2008 Jan 10; doi: 10.1113/jphysiol.2007.144154. In press. DOI: 10.1113/jphysiol.2007.144154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Veech RL, Harris RL, Veloso D, Veech EH. Freeze-blowing: a new technique for the study of brain in vivo. J. Neurochem. 1973;20:183–188. doi: 10.1111/j.1471-4159.1973.tb12115.x. [DOI] [PubMed] [Google Scholar]
- 115.Veech RL. The metabolism of lactate. NMR in Biomedicine. 1991;4:53–58. doi: 10.1002/nbm.1940040204. [DOI] [PubMed] [Google Scholar]
- 116.Hargreaves RJ, Planas AM, Cremer JE, Cunningham VJ. Studies on the relationship between cerebral glucose transport and phosphorylation using 2-deoxyglucose. J. Cereb. Blood Flow Metab. 1986;6:708–716. doi: 10.1038/jcbfm.1986.127. [DOI] [PubMed] [Google Scholar]
- 117.Barros LF, Bittner CX, Loaiza A, Porras OH. A quantitative overview of glucose dynamics in the gliovascular unit. Glia. 2007;55:1222–12237. doi: 10.1002/glia.20375. [DOI] [PubMed] [Google Scholar]
- 118.Porras OH, Ruminot I, Loaiza A, Barros LF. Na(+)-Ca(2+) cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia. 2008;56:59–68. doi: 10.1002/glia.20589. [DOI] [PubMed] [Google Scholar]
- 119.Nehlig A, Coles JA. Cellular pathways of energy metabolism in the brain: is glucose used by neurons or astrocytes? Glia. 2007;55:1238–1250. doi: 10.1002/glia.20376. [DOI] [PubMed] [Google Scholar]
- 120.Sharp FR. Relative cerebral glucose uptake of neuronal perikarya and neuropil determined with 2-deoxyglucose in resting and swimming rat. Brain Res. 1976;110:127–139. doi: 10.1016/0006-8993(76)90213-4. [DOI] [PubMed] [Google Scholar]
- 121.Sharp FR. Rotation induced increases of glucose uptake in rat vestibular nuclei and vestibulocerebellum. Brain Res. 1976;110:141–151. doi: 10.1016/0006-8993(76)90214-6. [DOI] [PubMed] [Google Scholar]
- 122.Durham D, Woolsey TA, Kruger L. Cellular localization of 2-[3H]deoxy-D-glucose from paraffin-embedded brains. J. Neurosci. 1981;1:519–526. doi: 10.1523/JNEUROSCI.01-05-00519.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Duncan GE, Stumpf WE, Pilgrim C, Breese GR. High resolution autoradiography at the regional topographic level with [14C]2-deoxyglucose and [3H]2-deoxyglucose. J. Neurosci. Methods. 1987;20:105–113. doi: 10.1016/0165-0270(87)90043-4. [DOI] [PubMed] [Google Scholar]
- 124.Nehlig A, Wittendorp-Rechenmann E, Lam CD. Selective uptake of [14C]2-deoxyglucose by neurons and astrocytes: high-resolution microautoradiographic imaging by cellular 14C-trajectography combined with immunohistochemistry. J. Cereb. Blood Flow Metab. 2004;24:1004–1014. doi: 10.1097/01.WCB.0000128533.84196.D8. [DOI] [PubMed] [Google Scholar]
- 125.Itoh Y, Abe T, Takaoka R, Tanahashi N. Fluorometric determination of glucose utilization in neurons in vitro and in vivo. J. Cereb. Blood Flow Metab. 2004;24:993–1003. doi: 10.1097/01.WCB.0000127661.07591.DE. [DOI] [PubMed] [Google Scholar]
- 126.Duncan GE, Stumpf WE, Pilgrim C. Cerebral metabolic mapping at the cellular level with dry-mount autoradiography of [3H]2-deoxyglucose. Brain Res. 1987;401:43–49. doi: 10.1016/0006-8993(87)91161-9. [DOI] [PubMed] [Google Scholar]
- 127.Duncan GE, Kaldas RG, Mitra KE, Breese GR, Stumpf WE. High activity neurons in the reticular formation of the medulla oblongata: a high-resolution autoradiographic 2-deoxyglucose study. Neuroscience. 1990;35:593–600. doi: 10.1016/0306-4522(90)90331-w. [DOI] [PubMed] [Google Scholar]
- 128.Kauppinen RA, Nicholls DG. Synaptosomal bioenergetics. The role of glycolysis, pyruvate oxidation and responses to hypoglycaemia. Eur. J. Biochem. 1986;158:159–165. doi: 10.1111/j.1432-1033.1986.tb09733.x. [DOI] [PubMed] [Google Scholar]
- 129.Meldrum BS, Nilsson B. Cerebral blood flow and metabolic rate early and late in prolonged epileptic seizures induced in rats by bicuculline. Brain. 1976;99:523–542. doi: 10.1093/brain/99.3.523. [DOI] [PubMed] [Google Scholar]
- 130.Ndubuizu O, LaManna JC. Brain tissue oxygen concentration measurements. Antioxid. Redox Signal. 2007;9:1207–1219. doi: 10.1089/ars.2007.1634. [DOI] [PubMed] [Google Scholar]
- 131.Steinbrink J, Villringer A, Kempf F, Haux D, Boden S, Obrig H. Illuminating the BOLD signal: combined fMRI-fNIRS studies. Magn. Reson. Imaging. 2006;24:495–505. doi: 10.1016/j.mri.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 132.Borowsky IW, Collins RC. Metabolic anatomy of brain: a comparison of regional capillary density, glucose metabolism, and enzyme activities. J. Comp. Neurol. 1989;288:401–413. doi: 10.1002/cne.902880304. [DOI] [PubMed] [Google Scholar]
- 133.Collins RC. Red and white brain. Ann. N. Y. Acad. Sci. 1997;835:250–254. doi: 10.1111/j.1749-6632.1997.tb48635.x. [DOI] [PubMed] [Google Scholar]
- 134.Baldwin KM, Hooker AM, Herrick RE. Lactate oxidative capacity in different types of muscle. Biochem. Biophys. Res. Commun. 1978;83:151–157. doi: 10.1016/0006-291x(78)90410-2. [DOI] [PubMed] [Google Scholar]
- 135.Brooks GA. Lactate production under fully aerobic conditions: the lactate shuttle during rest and exercise. Fed. Proc. 1986;45:2924–2929. [PubMed] [Google Scholar]
- 136.Gladden LB. Perspectives: Is there an intracellular lactate shuttle in skeletal muscle? J. Physiol. 2007;582:899. doi: 10.1113/jphysiol.2007.138487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kobayashi K, Neely JR. Control of maximum rates of glycolysis in rat cardiac muscle. Circ. Res. 1979;44:166–175. doi: 10.1161/01.res.44.2.166. [DOI] [PubMed] [Google Scholar]
- 138.Barron JT, Gu L, Parrillo JE. NADH/NAD redox state of cytoplasmic glycolytic compartments in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H2872–2878. doi: 10.1152/ajpheart.2000.279.6.H2872. [DOI] [PubMed] [Google Scholar]
- 139.Paul RJ. Functional compartmentalization of oxidative and glycolytic metabolism in vascular smooth muscle. Am. J. Physiol. 1983;244:C399–409. doi: 10.1152/ajpcell.1983.244.5.C399. [DOI] [PubMed] [Google Scholar]
- 140.Paul RJ, Bauer M, Pease W. Vascular smooth muscle: aerobic glycolysis linked to sodium and potassium transport processes. Science. 1979;206:1414–6. doi: 10.1126/science.505014. [DOI] [PubMed] [Google Scholar]
- 141.Lynch RM, Paul RJ. Compartmentation of glycolytic and glycogenolytic metabolism in vascular smooth muscle. Science. 1983;222:1344–1346. doi: 10.1126/science.6658455. [DOI] [PubMed] [Google Scholar]
- 142.Lynch RM, Paul RJ. Compartmentation of carbohydrate metabolism in vascular smooth muscle: evidence for at least two functionally independent pools of glucose 6-phosphate. Biochim. Biophys. Acta. 1986;887:315–318. doi: 10.1016/0167-4889(86)90159-x. [DOI] [PubMed] [Google Scholar]
- 143.Lynch RM, Paul RJ. Compartmentation of carbohydrate metabolism in vascular smooth muscle. Am. J. Physiol. 1987;252:C328–334. doi: 10.1152/ajpcell.1987.252.3.C328. [DOI] [PubMed] [Google Scholar]
- 144.Hardin CD, Kushmerick MJ. Simultaneous and separable flux of pathways for glucose and glycogen utilization studied by 13C-NMR. J. Mol. Cell. Cardiol. 1994;26:1197–1210. doi: 10.1006/jmcc.1994.1138. [DOI] [PubMed] [Google Scholar]
- 145.Ramos M, del Arco A, Pardo B, Martinez-Serrano A, Martinez-Morales JR, Kobayashi K, Yasuda T, Bogonez E, Bovolenta P, Saheki T, Satrustegui J. Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res. Dev. Brain Res. 2003;143:33–46. doi: 10.1016/s0165-3806(03)00097-x. [DOI] [PubMed] [Google Scholar]
- 146.Berkich DA, Ola MS, Cole J, Sweatt AJ, Hutson SM, LaNoue KF. Mitochondrial transport proteins of the brain. J. Neurosci. Res. 2007;85:3367–3377. doi: 10.1002/jnr.21500. [DOI] [PubMed] [Google Scholar]
- 147.Xu Y, Ola MS, Berkich DA, Gardner TW, Barber AJ, Palmieri F, Hutson SM, LaNoue KF. Energy sources for glutamate neurotransmission in the retina: absence of the aspartate/glutamate carrier produces reliance on glycolysis in glia. J. Neurochem. 2007;101:120–131. doi: 10.1111/j.1471-4159.2006.04349.x. [DOI] [PubMed] [Google Scholar]
- 148.Nishino K, Nowak TS., Jr Time course and cellular distribution of hsp27 and hsp72 stress protein expression in a quantitative gerbil model of ischemic injury and tolerance: thresholds for hsp72 induction and hilar lesioning in the context of ischemic preconditioning. J. Cereb. Blood Flow Metab. 2004;24:167–178. doi: 10.1097/01.WCB.0000100853.67976.8B. [DOI] [PubMed] [Google Scholar]
- 149.Cerdán S, Rodrigues TB, Sierra A, Benito M, Fonseca LL, Fonseca CP, García-Martín ML. The redox switch/redox coupling hypothesis. Neurochem. Int. 2006;48:523–530. doi: 10.1016/j.neuint.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 150.Ramírez BG, Rodrigues TB, Violante IR, Cruz F, Fonseca LL, Ballesteros P, Castro MM, García-Martín ML, Cerdán S. Kinetic properties of the redox switch/redox coupling mechanism as determined in primary cultures of cortical neurons and astrocytes from rat brain. J. Neurosci. Res. 2007;85:3244–3253. doi: 10.1002/jnr.21386. [DOI] [PubMed] [Google Scholar]
- 151.Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hertz L, Peng L. Energy metabolism at the cellular level of the CNS. Can. J. Physiol. Pharmacol. 1992;70(Suppl):S145–157. doi: 10.1139/y92-256. [DOI] [PubMed] [Google Scholar]
- 153.Peng L, Zhang X, Hertz L. High extracellular potassium concentrations stimulate oxidative metabolism in a glutamatergic neuronal culture and glycolysis in cultured astrocytes but have no stimulatory effect in a GABAergic neuronal culture. Brain Res. 1994;663:168–172. doi: 10.1016/0006-8993(94)90475-8. [DOI] [PubMed] [Google Scholar]
- 154.Keyser DO, Pellmar TC. Synaptic transmission in the hippocampus: critical role for glial cells. Glia. 1994;10:237–243. doi: 10.1002/glia.440100402. [DOI] [PubMed] [Google Scholar]
- 155.Keyser DO, Pellmar TC. Regional differences in glial cell modulation of synaptic transmission. Hippocampus. 1997;7:73–77. doi: 10.1002/(SICI)1098-1063(1997)7:1<73::AID-HIPO7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 156.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J. Biol. Chem. 2005;280:20493–520402. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 157.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J. Physiol. 2006;574:85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calve A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U. S. A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- 161.Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- 162.Henry PG, Oz G, Provencher S, Gruetter R. Toward dynamic isotopomer analysis in the rat brain in vivo: automatic quantitation of 13C NMR spectra using LC Model. NMR Biomed. 2003;16:400–412. doi: 10.1002/nbm.840. [DOI] [PubMed] [Google Scholar]
- 163.Uffmann K, Gruetter R. Mathematical modeling of (13)C label incorporation of the TCA cycle: the concept of composite precursor function. J. Neurosci. Res. 2007;85:3304–3317. doi: 10.1002/jnr.21392. [DOI] [PubMed] [Google Scholar]
- 164.Chih CP, Lipton P, Roberts EL., Jr Do active cerebral neurons really use lactate rather than glucose? Trends Neurosci. 2001;24:573–578. doi: 10.1016/s0166-2236(00)01920-2. [DOI] [PubMed] [Google Scholar]
- 165.Dienel GA, Cruz NF. Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem Int. 2006;48:586–595. doi: 10.1016/j.neuint.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 166.Eriksson G, Peterson A, Iverfeldt K, Walum E. Sodium-dependent glutamate uptake as an activator of oxidative metabolism in primary astrocyte cultures from newborn rat. Glia. 1995;15:152–156. doi: 10.1002/glia.440150207. [DOI] [PubMed] [Google Scholar]
- 167.McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 1996;66:386–393. doi: 10.1046/j.1471-4159.1996.66010386.x. [DOI] [PubMed] [Google Scholar]
- 168.Peng L, Swanson RA, Hertz L. Effects of L-glutamate, D-aspartate, and monensin on glycolytic and oxidative glucose metabolism in mouse astrocyte cultures: further evidence that glutamate uptake is metabolically driven by oxidative metabolism. Neurochem. Int. 2001;38:437–443. doi: 10.1016/s0197-0186(00)00104-2. [DOI] [PubMed] [Google Scholar]
- 169.Abe T, Takahashi S, Suzuki N. Oxidative metabolism in cultured rat astroglia: effects of reducing the glucose concentration in the culture medium and of D-aspartate or potassium stimulation. J. Cereb. Blood Flow Metab. 2006;26:153–160. doi: 10.1038/sj.jcbfm.9600175. [DOI] [PubMed] [Google Scholar]
- 170.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]