

Abstract
The enzymes that are involved in the elongation of fatty acids differ in terms of the substrates on which they act. To date, the enzymes specifically involved in the biosynthesis of polyunsaturated fatty acids have not yet been identified. In an attempt to identify a gene(s) encoding an enzyme(s) specific for the elongation of γ-linolenic acid (GLA) (18:3n-6), a cDNA expression library was made from the fungus Mortierella alpina. The cDNA library constructed in a yeast expression vector was screened by measuring the expressed elongase activity [conversion of GLA to dihomo-GLA (20:3n-6)] from an individual yeast clone. In this report, we demonstrate the isolation of a cDNA (GLELO) whose encoded protein (GLELOp) was involved in the conversion of GLA to dihomo-GLA in an efficient manner (60% conversion). This cDNA contains a 957-nucleotide ORF that encodes a protein of 318 amino acids. Substrate specificity analysis revealed that this fungal enzyme acted also on stearidonic acid (18:4n-3). This report identifies and characterizes an elongase subunit that acts specifically on the two Δ6-desaturation products, 18:3n-6 and 18:4n-3. When this GLELO cDNA was coexpressed with M. alpina Δ5-desaturase cDNA in yeast, it resulted in the conversion of GLA to arachidonic acid (20:4n-6) as well as the conversion of stearidonic acid to eicosopentaenoic acid (20:5n-3). Thus, this GLELO gene may play an critical role in the bio-production of both n-6 and n-3 polyunsaturated fatty acids.
Polyunsaturated fatty acids (PUFAs) are important components of cellular structure and function and serve as precursors to eicosanoids, including prostaglandins and leukotrienes (1). In mammals, these eicosanoids are involved in inflammatory responses, regulation of blood pressure, and reproductive function (2). Many of these PUFAs are derived from essential fatty acids such as linoleic acid (LA) (18:2n-6) and α-linolenic acid (18:3n-3) by an alternating series of desaturation and elongation reactions. The major metabolite product of the n-6 pathway in mammals is arachidonic acid (AA) (20:4n-6), and the major end products of the n-3 pathway are eicosapentanoic acid (EPA) (20:5n-3) and docosahexanoic acid (22:6n-3). AA is produced from dihomo-γ-linolenic acid (DGLA) (20:3n-6) by a Δ5-desaturase. The step prior, in which DGLA is produced from γ-linolenic acid (GLA) (18:3n-6), is catalyzed by an elongase complex. Similarly, EPA is produced from eicosatetraenoic acid (20:4n-3) by the Δ5-desaturase. Eicosatetraenoic acid is synthesized from stearidonic acid (STA) (18:4n-3) by the action of an elongase complex.
The key elongation and desaturation steps involved in the production of AA and EPA are outlined below (1):
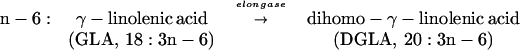 |
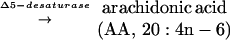 |
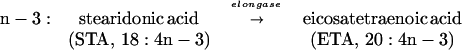 |
 |
Among these PUFAs, AA is known to be responsible for fetal cognitive development; diminished levels of AA could lead to impaired fetal growth, as well as impaired nerve transmission in the developing infant (3–5). EPA, on the other hand, has been shown to modulate some of the inflammatory responses in clinical trials (2). AA can be found in significant amounts in animal liver and adrenal gland and is also produced in filamentous fungus Mortierella alpina and red alga Porphyridium cruentum (6). EPA can be found in fish oil and other marine organisms (6). However, many of these sources are not easily available for human use. Because plant seed oils are currently the largest source of PUFAs in the human diet, modification of the fatty acid biosynthetic pathways by genetic manipulation to produce desired PUFAs in an oilseed crop would provide an economical source of these important fatty acids. Major advances in the cloning and manipulation of fatty acid desaturase genes from various organisms (7) have been made over the last several years, and these recombinant enzymes offer the prospect of producing a desired fatty acid in oilseed crops (7). Nevertheless, the production of a designer oil containing either AA or EPA remained hindered because of a lack of availability of a recombinant enzyme(s) involved in the elongation of PUFA.
In plants and mammals, it is believed that the fatty acid elongation is a four-step process. The first step involves condensation of malonyl-CoA with a long chain acyl-CoA to yield carbon dioxide and a β-ketoacyl-CoA in which the acyl moiety has been elongated by two carbon atoms. Subsequent reactions include reduction to β-hydroxyacyl-CoA, dehydration to an enoyl-CoA, and a second reduction to yield the elongated acyl-CoA. The initial condensation reaction catalyzed by β-ketoacyl-CoA synthase (KCS) is not only the substrate-specific step but also the rate limiting step (8). To date, several enzymes from plant and yeast have been identified and characterized by their involvement in the elongation of fatty acids with 18 carbon atoms or longer. The plant enzymes, Jojoba KCS, and Arabidopsis fatty acid elongation 1 show activity with monounsaturated and saturated fatty acid substrates having carbon chain lengths from 18 to 22 (8, 9). The yeast enzyme ELO2 appears to be involved in the elongation of saturated or monounsaturated fatty acids up to 24 carbons whereas ELO3 apparently has broader substrate specificity and is essential for the conversion of 24-carbon saturated fatty acids to 26-carbon species (10). None of them, however, is involved in the elongation of polyunsaturated fatty acids. Despite the demonstration of the PUFA elongase activities in both lower and higher eukaryotes (11, 12), the gene(s) for the PUFA elongase has not been yet identified and characterized.
In this study, we selected M. alpina as a source of the GLA-specific elongase activity based on the fact that this filamentous fungus is capable of producing significant amounts of AA (30–50% of total fatty acids) (11). In addition, several desaturase enzymes, specifically Δ6- and Δ5-desaturase, which are involved in the PUFA biosynthesis, had previously been isolated from this fungus (13–15). Hence, the polyA(+) RNA isolated from this organism was used for the construction of a cDNA expression library. Baker's yeast (Saccharomyces cerevisiae), which is incapable of producing PUFAs endogenously because of the absence of PUFA-specific desaturases and elongases, was used as the host for expressing M. alpina cDNA library. Here, we report the isolation of a gene from M. alpina and demonstrate the involvement of its encoded protein in the elongation of polyunsaturated fatty acids specifically with a chain length of 18 carbon atoms.
Materials and Methods
Strains and Growth Conditions.
M. alpina (American Type Culture Collection catalog no. 32221) was plated onto cornmeal agar and was grown at room temperature for 3–4 days. Once fungus growth was visible, it was inoculated into 50 ml of potato dextrose broth (Difco), and the culture was grown by shaking gently at room temperature for 72 h. The 50-ml inoculum was then inoculated into a 1-liter culture of the same medium and was grown again for 72 h. Cells were collected by filtering through sterile gauze and immediately were frozen into liquid nitrogen for RNA extraction. The S. cerevisiae strain used was SC334 (Matα pep4-3 prb1-1122 ura3-52 leu2-3, 112reg-501 gal1) (16). SC334 was grown either in yeast extract/peptone/dextrose (Difco) or selective medium [dropout broth (DOB)/uracil (URA) or leucine (LEU); Bio 101], depending on the experiment.
Construction of Mortierella alpina cDNA Expression Library.
Total RNA was isolated from M. alpina cell pellet by using a hot phenol/LiCl extraction method (17). Poly(A)(+) RNA was prepared from the total RNA according to the standard protocol by using oligo(dT) cellulose affinity purification (18). The poly(A)(+) RNA was reverse-transcribed with oligo(dT) primer containing a XhoI restriction site using avian myeloblastosis virus reverse transcriptase. After first strand cDNA synthesis, the second strand of cDNA was synthesized by adding E. coli DNA polymerase, E. coli DNA ligase, and RNase H (enzymes from Boehringer Mannheim). The EcoRI adapter oligos were ligated into the blunt-ended cDNA by T4 DNA ligase (Boehringer Mannheim). The sample was kinased by using T4 polynucleotide kinase (Boehringer Mannheim) and was digested with XhoI, was diluted with column buffer, and was passed through a Sephacryl S-400 column. The DNA samples were eluted by high salt buffer. Samples containing DNA from 400–5,000 bp were pooled and used for ligation into pYES2 vector (Invitrogen). The cDNA fragments were ligated into EcoRI/XhoI-digested pYES2 vector by using T4 DNA ligase. A large scale ligation reaction was carried out because a large amount of the ligated DNA (2–3 μg) was used directly in the transformation of yeast.
To transform yeast cells directly with the cDNA/pYES2 ligated mixture, competent S. cerevisiae SC334 cells were prepared by using lithium acetate/single-stranded carrier DNA/polyethylene glycol transformation method (19). An aliquot of 750 μl of competent SC334 cells was transferred into a 15-ml tube. Approximately 2 μg of cDNA/pYES2-ligated DNA was added to the cells along with carrier DNA and was mixed gently. To the cells, 3 ml of sterile 40% polyethylene glycol/lithium acetate was added and mixed gently but thoroughly. The cells were incubated with shaking at 30°C for 30 min and subsequently were given heat shock at 42°C for 15 min. The cells were cooled, pelleted, and resuspended in 5 ml of 1 × TE. A 100-μl aliquot of the above cells was plated onto fifty 150-mm minimal agar plates (DOB/URA) and was incubated at 30°C for 3 days. A total of 8 × 105 primary clones were obtained. Five colonies were pooled at a time and were grown in 1 ml of DOB/URA medium for 6–7 h, and a glycerol stock was prepared for storage at −80°C.
Library Screening.
Glycerol stocks were thawed, and approximately 0.5 ml of each pool was added to a 5-ml DOB/URA medium and was grown at 30°C for 24 h. The entire 5-ml culture was then transferred into 50 ml of DOB/URA containing 2% galactose and 25 μM GLA (substrate for the PUFA elongase). No detergent was added with the substrate. Each culture was induced by galactose for 24 h at 25°C with shaking. The induced culture was harvested by centrifugation, and each cell pellet was subjected to fatty acid analysis for determining the GLA elongation activity. A culture of SC334 containing pYES2 vector was used as control. The rate of conversion of the substrate GLA to the product DGLA [conversion rate = product/(substrate + product)] was calculated to reflect the GLA elongation activity present in an individual pool or individual clone.
Fatty Acid Analysis.
Fatty acid contents of yeast cells were analyzed as described (14). In brief, each cell pellet from a 50-ml culture was vortexed with 6 ml of methanol, followed by the addition of 12 ml of chloroform and 100 μg of tridecanoin (as internal standard). The mixture was incubated for at least 1 h at room temperature or at 4°C overnight. The chloroform layer was extracted and filtered through a Whatman filter with 1 g of anhydrous sodium sulfate to remove particulates and residual water. The organic solvents were evaporated to dryness at 40°C under a stream of nitrogen. The extracted lipids were derivatized to fatty acid methyl esters (FAME) for gas chromatography (GC) analysis according to the method described elsewhere (14). In brief, 2 ml of 0.5 N KOH in methanol was added to the extracted lipid, was heated to 95–100°C for 30 min, and was cooled to room temperature. Approximately 2 ml of 14% boron triflouride in methanol was added, and heating was repeated. After the mixture was cooled, 2 ml of water and 1 ml of hexane were added to extract FAME. The gas chromatography-mass spectrometry was carried out as described (14). The mass spectrum of a new peak obtained was compared with that of authentic standards and those in database NBS75K.L (National Bureau of Standards).
DNA Sequencing.
The recombinant plasmid was isolated from yeast clone P-708-2 that showed GLA-specific elongase activity by bead beating method (20). To eliminate RNAs and any protein contamination, it was further purified by using the Qiagen (Valencia, CA) plasmid miniprep kit according to the manufacturer's protocol. The recombinant plasmid DNA isolated was designated as pRPB2 and was reamplified in Escherichia coli for restriction analysis and DNA sequencing. E. coli TOP10 (Invitrogen) cells were transformed by the recombinant plasmid, and the plasmid DNA was isolated by using Qiagen miniplasmid kit according to the manufacturer's protocol. The size of the cDNA present in the recombinant plasmid was confirmed by digesting with EcoRI and XhoI. The cDNA present in pRPB2 was sequenced completely from both 5′ and 3′ ends. The DNA sequencing was performed by using ABI 373A stretch DNA sequencer according to the manufacturer's protocol. Sequence analysis was carried out by using the programs available in the Genetic Computer Group package (Madison, WI) with parameters used in default settings unless indicated otherwise. The cDNA in pRPB2 was designated as “GLELO” and its encoded protein as GLELOp.
Coexpression of GLELOp and Δ5-Desaturase.
Two recombinant plasmids pRPB2 and pRPE31 were used to cotransform into SC334 for the coexpression of GLELOp and Δ5-desaturase in yeast. pRPB2 contains the M. alpina GLELO cDNA in pYES2(URA+) vector under the control of GAL1 inducible promoter whereas pRPE31 contains M. alpina Δ5-desaturase cDNA (14) in the pYX242(LEU+) vector (Novagen) under the control of TP1 constitutive promoter. The cotransformation was done by using Alkali-Cation yeast transformation kit (Bio 101). The cotransformed yeast cells, designated as 334(pRPB2/pRPE31), were plated onto DOB/URA/LEU/agar and were grown for 72 h at 30°C. An individual colony was inoculated into 5 ml of DOB/URA/LEU medium and was grown at 30°C for 24 h. The culture was then transferred into 50 ml of the same medium containing 25 μM of either GLA or STA and 2% galactose. After growing for 24 h at 25°C, the culture was harvested, and the cell pellet was subjected to fatty acid analysis as described above. A cotransformation of pYES2 and pYX242 into S. cerevisiae 334 was done in parallel, was designated as 334(pYES2/pYX242), and was used as the control.
Results
Identification of a cDNA Encoding PUFA Elongase Subunit(s).
The findings that the filamentous fungus M. alpina produces significant amount of AA prompted us to believe that this organism might have high level of GLA elongating activity. Therefore, this fungus was selected as an abundant mRNA source of PUFA-specific elongation enzymes for the construction of a cDNA expression library. We decided to construct the library directly in yeast instead of making the library first in E. coli. The advantage of making a direct yeast library is to avoid amplification of primary clones, which happens when the library is made in E. coli as an intermediate. We optimized the efficiency of transformation in different S. cerevisiae strains by using a cDNA/vector-ligated mix because the transformation efficiency in yeast is lower than that in E. coli. The plasmid pYES2 was used as a yeast expression vector for the construction of the M. alpina cDNA library. In this plasmid, the expression of the cDNA in S. cerevisiae is driven by the GAL1 promoter. The best results were obtained with a yield of 4–5 × 105 transformants per microgram of ligated DNA in S. cerevisiae strain SC334. To maximize screening efficiency, five individual yeast colonies/clones were pooled. Each pool was used to screen for PUFA elongase activity by growing in a 50-ml culture in the presence of GLA as substrate. After induction with galactose for 24 h, the elongase activity was determined by measuring the amount of DGLA produced in cells.
After screening approximately 750 individual pools by using GC-FAME analysis, one pool, P-708, appeared to have significant elongase activity in converting GLA to DGLA. The ability to convert GLA to DGLA was found to be approximately 5-fold higher than the control (SC334 containing pYES2 vector only) in terms of DGLA/GLA ratio (data not shown). This sample was tested again under identical assay condition to confirm the initial findings. As expected, the repeat experiment showed around 10% conversion of GLA to DGLA, which was around 5-fold higher than the control (data not shown). These results strongly indicated that P-708 pool contained a M. alpina cDNA clone expressing an enzyme that is involved in the elongation of GLA to DGLA. Because this pool was a mixture of five individual clones, it was necessary to isolate the putative cDNA clone(s) from this pool. To do this, the P-708 pool was plated onto a DOB/URA agar plate. Thirty individual colonies were picked randomly and were grown in DOB/URA + 2% galactose medium in the presence of 25 μM GLA. The cell pellet obtained from each induced culture was then subjected to fatty acid analysis as described above. The GC-FAME analysis revealed that 5 of the 30 individual clones showed elongase activity by converting GLA to DGLA with a range of 50–60% substrate conversion. Among them, an individual clone, P-708-2, showed a maximum 60% conversion of GLA substrate to DGLA (Fig. 1). No significant amount of DGLA was found in yeast containing pYES2 vector alone (Fig. 1). The GC-MS analysis confirmed the identity of DGLA by its retention time (24.8 min), mass peak at m/z = 320 (the expected molecular mass for DGLA), and fragmentation pattern identical to that of the DGLA standard (data not shown). It is important to note that, in Fig. 1, a small peak very close to DGLA peak was found in pYES2 vector only control. Because of the insufficient amount of the fatty acid recovered from this peak, we could not determine the identity of this small peak by MS.
Figure 1.

Gas chromatogram of fatty acid methyl esters (FAME) from the lipid fraction in yeast containing pYES2 or in yeast clone P-708-2. All yeast strains were grown in selective medium supplemented with exogenous GLA at a final concentration of 25 μM. The arrows indicate the substrate as well as the newly synthesized DGLA in the culture of yeast clone P-708-2. The front end of the chromatogram is not shown.
Characterization of the cDNA-Encoding PUFA Elongase Subunit.
The recombinant plasmid pYES2 containing the cDNA corresponding to PUFA elongase subunit was isolated from P-708-2 clone and was designated as pRPB2. The size of the cDNA insert in the pRPB2 was confirmed by digesting pRPB2 with EcoRI and XhoI. The insert size appeared to be around 1.0 kb, as judged by agarose gel electrophoresis. The cDNA insert in pRPB2 was fully sequenced in both directions and was found to contain a 1,066-bp-long sequence that was deposited in GenBank under accession no. AF206662. The longest ORF deduced from the entire cDNA sequence in the pRPB2 clone appeared to be 957 bp in length, which encodes a protein of 318 amino acids (Fig. 2). The identified M. alpina gene has been designated as GLELO and its encoded protein as GLELOp because this fungal gene product is involved in the elongation of GLA. After searching the GenBank as well as Swissprot databases, no homologous sequence was found, indicating that the GLELO gene had not been identified previously. The only homology (25% identity in 295-amino acid overlap) was found with yeast ELO2 protein, which is involved in the elongation of saturated and monounsaturated fatty acids. The amino acid sequence of GLELOp showed several regions of identity, including a common histidine box motif with ELO2 (Fig. 2). The hydropathy plot (Kyte-Doolitle) of GLELOp indicates that this protein is hydrophobic in nature and contains several presumptive trans-membrane domains (data not shown).
Figure 2.

Comparison of the deduced amino acid sequence of the M. alpina GLELOp with yeast ELO2p. Identical residues are shaded. The conserved histidine box is underlined.
Characterization of GLELO-Encoded Enzyme.
To optimize the expression of recombinant GLELOp in yeast, the SC334(pRPB2) culture was grown at various temperatures in the presence of a specific substrate concentration (25 μM GLA). After induction with galactose for 24 h, the amount of DGLA produced in cells of each culture was measured. The results of GC-FAME analysis indicated that the maximum conversion of GLA to DGLA was achieved at 25°C. At this temperature, the conversion of GLA to DGLA was 55% for SC334(pRPB2) and 2.9% for SC334(pYES2) (data not shown). To analyze the optimal substrate concentration, we performed similar experiments as described above with three different concentrations of GLA: 25 μM, 50 μM, and 100 μM. The cultures were induced for 24 h at 25°C followed by fatty acid analysis. The results indicated that, at any substrate concentration, an almost identical amount of DGLA was synthesized with an average conversion of 55% GLA to DGLA (data not shown).
Next, we determined the substrate specificity of GLELOp. To do this, we tested the ability of SC334(pRPB2) culture to elongate various substrates including PUFAs from both n-6 and n-3 pathways (Fig. 3). The cultures were induced for 24 h at 25°C in the presence of different substrates at a final concentration of 25 μM, and the substrate elongation was measured by cellular fatty acid analysis. Under identical assay conditions, the only substrate other than GLA used by the GLELOp was STA (18:4n-3), a counterpart PUFA from the n-3 pathway. GLELOp was able to convert 73% of STA to eicosatetranoic acid (20:4n-3) (Fig. 3). From these results, it can be concluded that GLELOp has substrate specificity for both GLA and STA, indicating that it is involved in the elongation of PUFAs along both n-6 and n-3 pathways.
Figure 3.

Substrate specificity of the M. alpina GLELOp. The SC334(pRPB2) culture was grown in the presence of indicated substrates at a final concentration of 25 μM. Percent conversion was calculated as [product/(substrate + product)] × 100.
Previously, we have demonstrated the coexpression of the M. alpina Δ12- and Δ6- desaturase cDNAs in yeast that resulted in the production of GLA from endogenous oleic acid (18:1) (13). Based on these findings, we were interested in determining the feasibility of producing AA from GLA in yeast by coexpressing M. alpina GLELO and Δ5-desaturase cDNAs. It was assumed that, once DGLA (20:3n-6) is produced from GLA by the action of GLELOp, the Δ5-desaturase could then convert it to AA (20:4n-6) in a desired coexpression system. This hypothesis was tested by cotransforming SC334 with recombinant plasmids pRPB2 (containing GLELO gene under the control of the GAL1 promoter) and pRPE31 (pYX242 plasmid containing M. alpina Δ5-desaturase gene under the control of the TPI promoter). The cotransformed yeast culture was supplemented with 25 μM GLA and was analyzed for AA synthesis. The results in Table 1 indicate that the sequential action of GLELOp and Δ5-desaturase on GLA substrate resulted in an average conversion of 17.6% GLA to AA. To determine whether STA could also be converted to EPA in the n-3 pathway, similar coexpression experiments were carried out in the presence of 25 μM STA. The results indicate that around 21% of STA was converted to EPA (Table 1). Thus, the recombinant M. alpina GLELOp and Δ5-desaturase were active on both n-6 and n-3 fatty acid substrates.
Table 1.
Production of AA and EPA in yeast
| SC334 containing | Total lipid, μg* | GLA, μg† | DGLA, μg | AA, μg | Total lipid, μg* | STA, μg‡ | ETA, μg | EPA, μg |
|---|---|---|---|---|---|---|---|---|
| pRPB2/pRPE31 | 254 | 2.60 | 3.55 | 1.32 | 257 | 2.93 | 4.13 | 1.87 |
| pYES2/pYX242 | 258 | 5.60 | 0.17 | ND | 304 | 4.57 | 0.22 | 0.10 |
pRPB2/pRPE31 indicates SC334 strain containing pRPB2 (GLELO cDNA in pYES2) and pRPE31 (Δ5-desaturase cDNA in pYX242) recombinant plasmids whereas pYES2/pYX242 indicates SC334 containing pYES2 and pYX242 vectors only. ND, not detected.
The volume of culture used for lipid extraction was 50 ml.
Exogenous GLA concentration used was 25 μM.
Exogenous STA concentration used was 25 μM.
Discussion
Polyunsaturated fatty acids, particularly 18- to 22-carbon PUFAs, have become high profile in the biomedical and nutraceutical areas because of their specific therapeutic roles in certain clinical conditions. Besides pharmaceutical applications, public awareness on eating healthy has also brought these PUFAs to the attention of the consumer.
In the past few years, it has become clear that the synthesis of important PUFAs such as GLA or AA involves a subclass of microsomal fatty acid desaturase enzymes, the so called “front-end” desaturases (7). With the recent identification of several front-end desaturase genes, the scope for further investigation of the precise mechanisms of desaturation in PUFA biosynthesis and the enzymes involved has been increased (7). However, the lack of identity of an enzyme responsible for the elongation of PUFA has hindered the understanding of the elongation mechanism involved in the biosynthetic pathway.
In this study, we reported the identification of a gene (GLELO) whose encoded protein (GLELOp) is involved specifically in the elongation of GLA to DGLA and STA to eicosatetraenoic acid. The GLELO gene was identified and isolated from the fungus Mortierella alpina through extensive library screening in yeast by measuring the GLA elongation activity from pools of yeast clones. After screening approximately 3,750 individual cDNA clones in pools, one clone, P-708-2, showed significant activity in converting GLA to DGLA (Fig. 1). The PUFA elongase activity found in this particular clone showed high conversion (≈60%) of GLA to DGLA (Fig. 1).
The search for a PUFA-specific elongation enzyme in M. alpina initially began based on a review of the homologies shared between Jojoba KCS and Arabidopsis fatty acid elongation 1 or yeast ELO enzymes (ELO 1, 2, and 3). These enzymes are involved in the elongation of saturated or monounsaturated fatty acids with varying chain length (8, 10). Because Jojoba KCS or yeast ELO2 is involved in the elongation of fatty acids with 18 carbons or longer, it is conceivable that one component of the PUFA elongase complex might also share a common domain that is involved in the elongation reaction. We designed primers based on shared homologies either between plant enzymes (KCS, fatty acid elongation 1) or among yeast enzymes (ELO 1, 2, and 3) and attempted to amplify out elongase related gene(s) from M. alpina cDNA library by PCR technique. Using the primers based on yeast ELO enzymes, we were able to identify a yeast ELO2-like gene from M. alpina cDNA library, whose encoded protein showed an overall 40% amino acid sequence identity with yeast ELO2 peptide. The yeast culture expressing putative M. alpina ELO2-like protein mainly elongated saturated and monounsaturated fatty acids of 16–18 carbons (data not shown). This PCR-based screening, however, did not result in the identification of a gene that encodes PUFA-specific elongation enzyme. These findings led us to hypothesize that the PUFA-specific elongation enzyme might be unique in amino acid sequence and different from that of saturated or monounsaturated specific enzymes. The overall amino acid sequence identity (< 25%) between the isolated GLELOp and yeast ELO2 shown in Fig. 2 indeed support our contention that the enzyme responsible for the elongation of PUFA is unique. Nonetheless, both GLELOp and ELO2p contain a conserved histidine box motif HXXHH (Fig. 2). The histidine box motif has been implicated in the binding of iron atoms in the case of desaturases, which might be a characteristic of membrane-bound enzymes. The precise biological role or functional ramifications of this histidine box present in GLELOp remains to be determined.
The results of the substrate specificity analysis strongly indicate the GLELOp is highly specific for 18-carbon PUFAs: i.e., GLA and STA (Fig. 3). Interestingly, the rate of conversion of STA (73%) was higher than that of GLA (62%), indicating that GLELOp might work better along the n-3 pathway when expressed in yeast. Previously, we were able to convert 35–40% of endogenous oleic acid to GLA in yeast (13) as well as in canola seed oil by coexpressing M. alpina Δ12- and Δ6-desaturase genes (13). The present results prompted us to evaluate the feasibility of producing endogenous AA and EPA from GLA and STA, respectively, by coexpressing GLELOp and Δ5-desaturase of M. alpina in yeast. Indeed, results in this study clearly demonstrate that the coexpression of GLELOp and Δ5-desaturase could produce endogenous AA or EPA when the respective substrate (GLA or STA) was supplemented in the yeast culture (Table 1). It can be noted that the activity of each enzyme in the coexpression system appeared to be identical compared with when expressed individually (60–65% elongation activity and 17–20% Δ5-desaturation activity; Table 1). In light of these findings, it would be interesting to demonstrate whether the endogenous substrate oleic acid could be converted to AA by coexpressing all three desaturases (Δ12-, Δ6-, and Δ5-) and GLELOp in yeast and, eventually, in oilseed plants.
In summary, we have isolated a cDNA sequence that encodes a PUFA-specific elongation enzyme from M. alpina by using a cDNA library screening strategy. This enzyme demonstrates high activity and substrate specificity on both n-6 and n-3 PUFAs. The availability of a recombinant PUFA elongation enzyme as well as desaturases offers the exciting possibility of producing a wide range of PUFAs in oilseed crops. Moreover, the GLELO sequence can be used to identify homologues in other organisms. Finally, of biotechnological importance, the production of designer oil in transgenic plants should become a reality and should help satisfy the demands of the nutraceutical and pharmaceutical industries in the near future.
Abbreviations
- PUFA
polyunsaturated fatty acid
- LA
linoleic acid
- GLA
γ-linolenic acid
- STA
stearidonic acid
- DGLA
dihomo-γ-linolenic acid
- AA
arachidonic acid
- EPA
eicosapentaenoic acid
- DOB
dropout broth
- URA
uracil
- LEU
leucine
- MS
mass spectrometry
- KCS
β-ketoacyl-CoA synthase
- FAME
fatty acid methyl ester
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
The GLELO sequence reported in this paper has been submitted in the GenBank database (accession no. AF206662).
References
- 1.Smith W L, Borgeat P. In: Biochemistry of Lipids and Membranes. Vance D E, Vance J E, editors. Menlo Park, CA: Benjamin/Cummings; 1985. pp. 325–360. [Google Scholar]
- 2.Horrobin D F. Rev Contemp Pharmacother. 1990;1:1–45. [Google Scholar]
- 3.Horrobin D F. Diabetes. 1997;46, Suppl. 2:S90–S93. doi: 10.2337/diab.46.2.s90. [DOI] [PubMed] [Google Scholar]
- 4.Carlson S E, Werkman S H, Peeples J M, Cooke R J, Tolley E A. Proc Natl Acad Sci USA. 1993;90:1073–1077. doi: 10.1073/pnas.90.3.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birch E E, Hoffman D R, Uauy R, Birch D G, Prestidge C. Pediatr Res. 1998;44:201–209. doi: 10.1203/00006450-199808000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Gunstone F D, Harwood J L, Padley F B. The Lipid Handbook. London: Chapman & Hall; 1994. pp. 47–223. [Google Scholar]
- 7.Napier J A, Michaelson L V, Stobart A K. Curr Opin Plant Biol. 1999;2:123–127. doi: 10.1016/s1369-5266(99)80025-9. [DOI] [PubMed] [Google Scholar]
- 8.Lassner M W, Lardizabal K, Metz J G. Plant Cell. 1996;8:281–292. doi: 10.1105/tpc.8.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millar A A, Kunst L. Plant J. 1997;12:121–131. doi: 10.1046/j.1365-313x.1997.12010121.x. [DOI] [PubMed] [Google Scholar]
- 10.Oh C S, Toke D A, Mandela S, Martin C E. J Biol Chem. 1997;272:17376–17384. doi: 10.1074/jbc.272.28.17376. [DOI] [PubMed] [Google Scholar]
- 11.Bajpai P K, Bajpai P, Ward O P. J Ind Microbiol. 1991;8:179–185. doi: 10.1007/BF01575851. [DOI] [PubMed] [Google Scholar]
- 12.Chapkin R S, Coble K J. Biochim Biophys Acta. 1991;1085:365–370. doi: 10.1016/0005-2760(91)90141-4. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y S, Chaudhary S, Thurmond J M, Bobik E G, Jr, Yuan L, Chan G M, Kirchner S J, Mukerji P, Knutzon D S. Lipid Res. 1999;34:649–659. doi: 10.1007/s11745-999-0410-8. [DOI] [PubMed] [Google Scholar]
- 14.Knutzon D S, Thurmond J M, Huang Y S, Chaudhary S, Bobik E G, Jr, Chan G M, Kirchner S J, Mukerji P. J Biol Chem. 1998;273:29360–29366. doi: 10.1074/jbc.273.45.29360. [DOI] [PubMed] [Google Scholar]
- 15.Michaelson L V, Lazarus C M, Griffiths G, Napier J A, Stobart A K. J Biol Chem. 1998;273:19055–19059. doi: 10.1074/jbc.273.30.19055. [DOI] [PubMed] [Google Scholar]
- 16.Hoveland P, Flick J, Johnston M, Sclafani R A. Gene. 1989;83:57–64. doi: 10.1016/0378-1119(89)90403-4. [DOI] [PubMed] [Google Scholar]
- 17.Ausubul F, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Short Protocols in Molecular Biology. New York: Wiley; 1995. pp. 4.8–4.9. [Google Scholar]
- 18.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, New York: Cold Spring Harbor Lab. Press; 1989. pp. 8.3–8.32. [Google Scholar]
- 19.Gietz R D, Schiestl R H. Methods Mol Cell Biol. 1995;5:255–269. [Google Scholar]
- 20.Hoffman C, Winston F. Gene. 1987;57:267. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]