

Abstract
Both the large arteries and microvascular beds of the central nervous system respond to injury by initiating processes compatible with Virchow’s triad: alterations in the microvascular permeability barrier, reduction in flow with the target bed, and/or thrombosis of brain-supplying arteries and of the microvasculature. This is particularly true during focal cerebral ischemia. The temporal and topographical coincidence of neuron injury and microvessel response during focal ischemia has suggested that neuron-microvessel interactions could be bidirectional. The neurovascular unit offers a conceptual and structural framework with which to examine events within the microvasculature and their impact on neuron integrity, with the participation of the intervening astrocytes, matrix, and other supportive cells (eg, pericytes and oligoden-droglia). Activation of the endothelium and of coagulation, capture of leukocytes, and increased microvessel permeability lead to the focal “no-reflow” phenomenon. Decreased shear stress is a component of the evolving ischemia. Strategies that inhibit the interactions within the microvasculature have been shown to prevent no-reflow and improve neurological outcome. It is, therefore, possible that addressing the processes of Virchow’s triad in the setting of focal ischemia could promote neurovascular function.
Keywords: Focal “no-reflow”, Microvessels, Neurons, Neurovascular unit, Virchow’s triad
Most ischemic strokes result from thromboemboli that originate from atheroma-containing lesions of the proximal brain-supplying arteries from structures within the heart. The resulting focal cerebral ischemia significantly perturbs the integrity of microvessels within the “at-risk” arterial territories, and simultaneously affects neuron and microvessel components of the neurovascular units supplied by these arteries. These events emphasize the notion that ischemic stroke is a vascular disorder, and that the neurological consequences depend upon vascular flow. This article explores the recent observations that the plasma-microvessel interface participates in changes in the integrity and function of the neurovascular unit during focal ischemia.
Virchow’s Triad
For the cerebral vasculature the 3 elements of Virchow’s triad are a very suitable starting point for considering the large artery-microvessel interrelationships that typify the responses to ischemic injury in the central nervous system (CNS). The triad consists of alterations in or injury to the vascular endothelium, activation of the coagulation system, and/or reduction in flow within the target vascular bed. In clinical medicine the appearance of 2 of 3 of these elements increases the risk of vascular thrombosis (eg, deep venous thrombosis).
The principles suggested by Virchow apply well to the CNS, although the 3 elements have not been formally considered in the context of focal brain ischemia before. They can be simplified as alterations in the microvascular permeability barrier, reduction in total blood flow within the target vascular bed, and/or thrombosis of brain-supplying arteries and of the microvasculature. The targets, then, are the endothelium, flow within individual vessels and vascular regions, and blood elements (ie, leukocytes, platelets, and hemostasis/ coagulation) and their activation (Table 1).
Table 1.
Elements of Virchow’s Triad and Their Targets
| Triad Elements | Targets |
|---|---|
| Injury to vascular endothelium | Endothelium |
| Activation of coagulation | Blood elements |
| Reduction in flow within vascular bed | Blood flow |
Thrombosis and endothelial responses to injury occur at 2 levels of the cerebral vasculature. For large artery atheroma formation (eg, the internal carotid artery), alterations of the endothelium and the progressing architectural changes in the vascular wall lead to local flow disturbances, with activation of platelets and coagulation. These further complicate the vascular lesion. At the other end of the vascular tree, reductions in flow through microvascular beds and loss in the microvascular permeability barrier during focal ischemia expose the plasma to the perivascular tissue (astrocytes and the neuropil) that contains the procoagulant tissue factor (TF). TF interacts with circulating factor VII to generate the TF:VIIa complex that initiates (extrinsic) coagulation system activation, and thrombosis. Platelet-fibrin thrombi can occlude capillaries within the microvascular bed early following the onset of focal ischemia. The endothelium can react by expressing receptors or adhesive proteins that capture leukocytes (eg, P-selectin, intercellular adhesion molecule-1 [ICAM-1], E-selectin) and platelets (eg, von Willebrand factor [vWF]). This article examines the interrelated events that alter flow within the microvasculature during ischemia and increase the risk of thrombosis of the ischemic cerebral vascular beds.
Vascular Arrays Within the Cerebral Hemispheres
The architectural arrangement of the cerebral vasculature is determined during brain development and by the regional location of the microvasculature within the anterior cerebral circulation that accommodates these flow dynamics. During CNS development the relative positions of microvessels and neurons to each other evolve along lines of extracellular matrix (ECM) (eg, laminin).1-5 For capillaries, astrocytes and endothelial cells interact to form the intervening basal lamina barrier and the inter-endothelial tight junctions that form a part of the permeability barrier (Figure 1).6-8 Elegant xenograft experiments have shown that the permeability barrier phenotype can be transplanted, and that its integrity requires the close interaction of endothelial cells with astrocyte end-feet.9 Within the corpus striatum (of primates), capillaries are located within a mean of 30 μm from the nearest neighboring neuron.10 The microvessel-neuron distance distribution is highly ordered and predictable.10 This capillary arrangement also has branch points at approximately 30 μm intervals.10 The intersections of the capillaries allow diversion of flow to patent capillaries/microvessels and thereby around occluded microvessels. For the cortex, Bär11,12 described a hierarchical organization of the arterial supply of the cerebral gray matter as a descending series of stacked hexagonal arrays from the pial supply to the white matter border. The branching within these microvessel arrays allows diversion of flow around impediments or occlusions. Within the white matter, capillaries are arranged in line with axons, and comprise approximately 10% of the density of capillaries found within the gray matter.13 Therefore, there are region-specific arrangements of the microvasculature. These accord with differences in regional cerebral blood flow (rCBF) in which flow is lowest in the striatum and highest in the gray matter.14 Importantly, the hierarchy of vessels within the CNS implies that although similar processes are involved in the injury of larger arteries and microvessels, the consequences are likely to be different.
Figure 1.
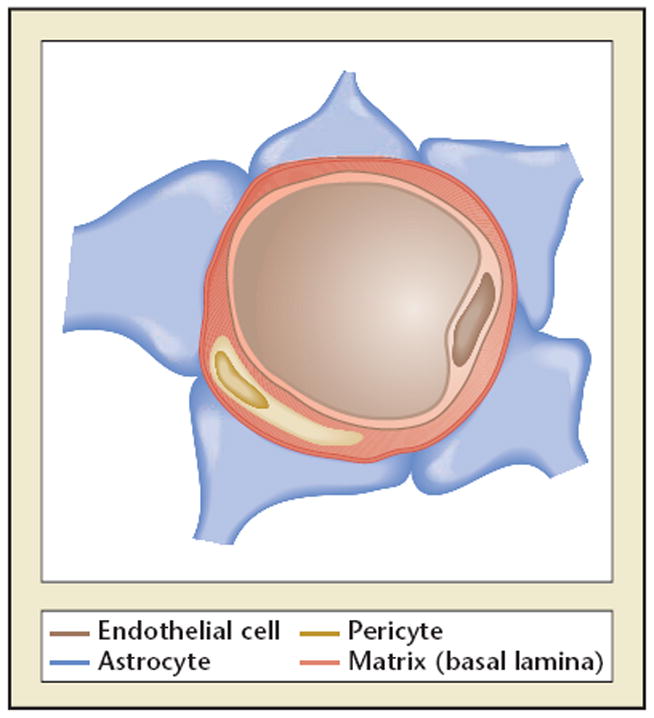
Cerebral capillary. Astrocytes and endothelial cells comprise capillaries throughout the brain. These cells are separated by the matrix proteins of the basal lamina. Embedded in the matrix are pericytes.
The Neurovascular Unit
The control of regional and local flow in the absence of ischemic injury depends upon neurovascular coupling.15 To some degree neurovascular coupling reflects the presence and form of neuronal activation, and reflects the functional efforts of intact neurons. The proximity of endothelial cells to astrocyte end-feet within microvessels and the support of astrocytes for neurons suggest that communication could also be directed from microvessels to the neurons they supply.13 This hypothesizes a conceptual “neurovascular unit” that consists of microvessels (endothelial cell-basal lamina matrix-astrocyte end-feet and pericytes), astrocytes, neurons, and their axons, in addition to other supporting cells (eg, microglia and oligodendroglia) (Figure 2). The resilience of the unit to reductions in flow or to flow cessation is unclear, but is likely to be complex as adjacent units are connected through their common microvessels and through dendritic connections. This provides for intercommunication and protected perfusion at the same time. Alterations in microvessel integrity could have other follow-on effects within the neurovascular unit that impact on neuronal function. For instance, it is possible that sustained hypertension, hyperglycemia, amyloid deposition, and other processes that can injure the endothelium also affect microvessel integrity, and hence neuron function.
Figure 2.
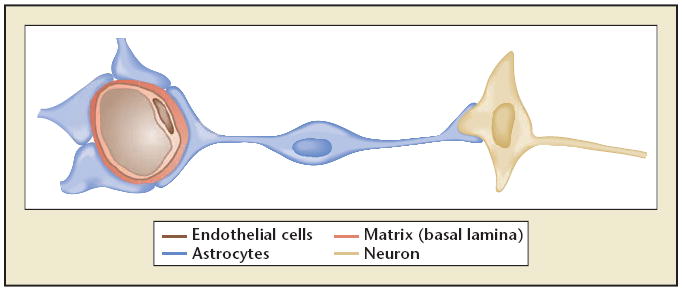
The neurovascular unit. A simplified depiction of the relationship between neuron and axon location and a supply microvessel. The astrocyte end-feet form the abluminal portion of capillaries and small microvessels via their attachment to the basal lamina matrix, and astrocyte arborizations contact neurons.
Vascular Events in the CNS
Two elements of Virchow’s triad—alteration in endothelial cell integrity and activation of coagulation—are interrelated, and contribute to disturbance of blood flow and complete vascular occlusion. Rosenberg16 hypothesized that organ-specific hemostasis can respond to specific types of injury. The forms of hemostasis that this might take in the CNS are not known, although the evidence for a CNS-specific form of hemostasis rests so far in the unique structure of the microvasculature.
Atherogenesis is primarily found in brain-supplying arteries at sites of branching and turbulence of flow. This is an example of a sustained procoagulant state. The complex atherogenic and ischemic events share processes common to coagulation system activation. Endothelial injury, platelet activation and fibrin formation, and altered flow contribute to the vascular causes of stroke and its microvascular consequences during brain ischemia.
For the microvasculature, a little-considered aspect of the integrity of the unit is reflected by the known distribution of components of the extrinsic coagulation system: TF, tissue factor pathway inhibitor (TFPI), and protease nexin-1 (PN-1).17-19 These are localized around microvessels. TF is expressed by astrocytes at their end-feet and is localized around non-capillary microvessels.17 The molecule is found in approximately 2-fold higher (protein) content, or approximately 6-fold higher activity, in the gray matter than in white matter of the cerebral hemispheres of nonhuman primates.17 The diffuse distribution of the antigen in gray matter suggests that TF expression is broadly transcribed. Teleologically, the distribution of TF around non-capillary microvessels may reflect the need to limit thrombus formation in capillaries where leakage occurs, as this could reduce blood flow and hence oxygen exchange in capillaries and the need to limit hemorrhage originating with leakage from large vessels. The TF distribution in brain tissue also reflects the higher microvessel density in gray matter and the potentially higher risk of hemorrhage that would need to be contained. In addition, TF is generated by activated cells of the monocyte/macrophage lineage in the circulation, and can “finger print” onto PMN leukocytes, a leukocyte subset that does not express TF. The principal inhibitor of TF—TFPI—is expressed in gray matter, but at low levels, as suggested by immunohisto-chemistry. The impact of injury on TFPI levels in cerebral tissue is not known. During CNS ischemia when plasma is exposed to perivascular TF, with leakage of the microvasculature, thrombin is generated. In the CNS the antithrombin equivalent to the primary inhibitor of thrombin in the circulation is PN-1.18,19 Its perivascular distribution affords a layer of protection for the cerebral tissue from thrombin, a molecule that is neuron toxic. The arrangement of TF and its associated inhibitors within cerebral tissues is reminiscent of other control systems (eg, regulators of coagulation system activation) that have evolved to manage or limit potentially harmful effects of minor injury or inflammatory lesions. Here, the goal is the maintenance of local CBF and oxygen exchange at the capillary level.
Large Artery Thrombosis
The bifurcation of the carotid artery is a predilection site for atheroma formation.20 The hemodynamics at the flow divider lead to turbulence against the walls of the internal and external carotid arteries, local platelet activation, endothelial cell injury, accumulation of lipid-laden macrophages and inflammatory cells, TF expression, and distortion of the otherwise smooth arterial wall.21 With growth of the lesion, alterations in the arterial surface and loss of the endothelial cell protection promote fibrin deposition and platelet-fibrin formation. Embolization of thrombus-atheroma material leads to downstream occlusion and ischemia. Similar processes occur at the origins of the vertebral arteries. Within the basilar artery, atheroma formation can be more diffuse, leading to in situ thrombotic obstruction of penetrating arteries that supply the brain stem and its structures.
The relevance of Virchow’s triad in the central nervous system is best observed in the microvasculature, and the differences in their reactivity under conditions of normoxia and ischemia.
The Cerebral Microvessel Bed
The relevance of Virchow’s triad in the CNS is best observed in the microvasculature, and the differences in their reactivity under conditions of normoxia and ischemia.
Normoxia
Under conditions of normoxia cerebral microvessels and the hemostatic system together maintain local blood flow. Cerebral microvessel endothelial cells can generate tissue plasminogen activator (t-PA), reserve leukocyte adhesion receptors (eg, P-selectin), and vWF in storage granules (ie, Weibel-Palade bodies), and maintain a permeability barrier with the aid of tight junctions and matrix adhesion. Notable have been observations of plasma flow in some microvessels during normoxia and focal ischemia (the so-called “plasmatic capillaries”). These can be explained under normoxia by rather slow cellular flow through capillaries temporarily obstructed by PMN leukocytes that must pass through the smaller capillaries, allowing only plasma to flow transiently within obstructed segments. This condition is common in other noncerebral vascular beds. The microvascular flow conditions are altered considerably during focal ischemia. It should be remembered that CBF does not fall to nil following obstruction of a major supply artery in the anterior circulation; rather it is maintained at 10% to 20% of normal.22 Nonetheless, activation of the microvasculature, coagulation, platelet, and protease systems occurs rapidly, with the accumulation of microvessel obstructions.
Regional flow is managed by precapillary arteriolar constriction due to neural firing or by autocrine activities.15,23,24 These promote shunting of blood and protection of the tissue from ischemia. Neurovascular coupling manages CBF both locally and at a distance.15 In experimental preparations, when the pial arterial anastomoses and superficial gray matter microvessels are observed directly, flow through capillaries (4.0-7.5 μm diameter) is transiently interrupted by trafficking PMN leukocytes (10-11 μm diameter).25 However, this is a normal occurrence observed in other vascular beds and does not lead to ischemic injury, as the endothelium is not activated.
Microvessel integrity depends upon the proximity of astrocyte end-feet to the endothelium: both are required for the formation of the basal lamina matrix, and for the formation of the endothelial permeability barrier (blood-brain barrier).26 Interendothelial tight junctions are purported to constitute the vascular portion of the blood-brain barrier. The matrix also provides a barrier to the transmigration or leakage of blood cells: erythrocytes during hemorrhage and leukocytes in response to inflammatory stimuli (eg, the inflammatory phase of ischemia). The integrity of the microvasculature is also affected by the presence of pericytes within the matrix or vessel wall histiocytes within larger vessels.27 The ultrastructure and integrity of cerebral capillaries depends upon their location and regional tissue composition. Cellular matrix receptors are central to the integrity of the microvasculature. Integrin and dystroglycan receptors are positioned to bind endothelial cells and astrocyte end-feet to the intervening matrix components of the basal lamina (eg, fibronectin, laminins, collage type IV, perlecan). αβ-dystroglycan is expressed by astrocyte end-feet on all cerebral vessels, whereas integrin α6β4 is expressed on astrocytes around all microvessels in the white matter and only large penetrating vessels of the gray matter. β1-integrins are expressed by the endothelium of all cerebral microvessels in gray and white matter.28 Disruption of the permeability barrier has been associated with changes in tight junction protein expression, although those changes are not rapid.
Focal ischemia
Occlusion of the proximal middle cerebral artery (MCA) produces an abrupt decrease in regional CBF. Flow does not reach nil, but usually persists at a low level. So, residual flow through the ischemic region exists even despite no flow through the brain-supplying artery. Nonetheless, significant alterations in both microvessel integrity and in the blood occur within the first minutes of flow cessation. These lead to obstruction of microvessels within the territory-at-risk, focal loss of permeability barriers, and changes in endothelium-astrocyte relationships.13
Within hours of proximal MCA occlusion in the nonhuman primate, obstruction within the microvasculature occurs in the striatum (Figure 3).25,29 This focal “no-reflow” results when the endothelium is activated leading to expression of the leukocyte adhesion receptors P-selectin and ICAM-1, the activation of PMN leukocytes, and their lodgment in the microvessel bed. These microvessel defects of focal no-reflow can be prevented by inhibition of PMN leukocyte β2-integrin-ICAM-1 interactions.29 The microvessel obstructions also variously contain activated platelets and fibrin, caused by generation of thrombin.30,31 The deposition of fibrin within the microvasculature originates from exposure of perivascular TF to the plasma column and activation of prothrombin (factor II) to thrombin. Fibrin formation is decreased significantly by blocking TF-factor VIIa interactions and preventing thrombin generation.30 Platelet activation entails interaction of the platelet surface integrin receptor αIIbβ3 with fibrin(ogen). Inhibition of these interactions via organic inhibitors or arginine-glycine-aspartic acid (RGD)-containing peptides can inhibit focal no-reflow in a dose-dependent manner in rodent or nonhuman primate focal ischemia models.31 With increased concentrations of the inhibitor, significant symptomatic hemorrhage results. Those findings indicate that normal platelet function is required to suppress the risk of hemorrhage during focal ischemia, that the risk of hemorrhage can be manipulated by altering platelet function, and that the range of the risk to benefit is relatively narrow when platelet-fibrinogen interactions are disrupted.
Figure 3.
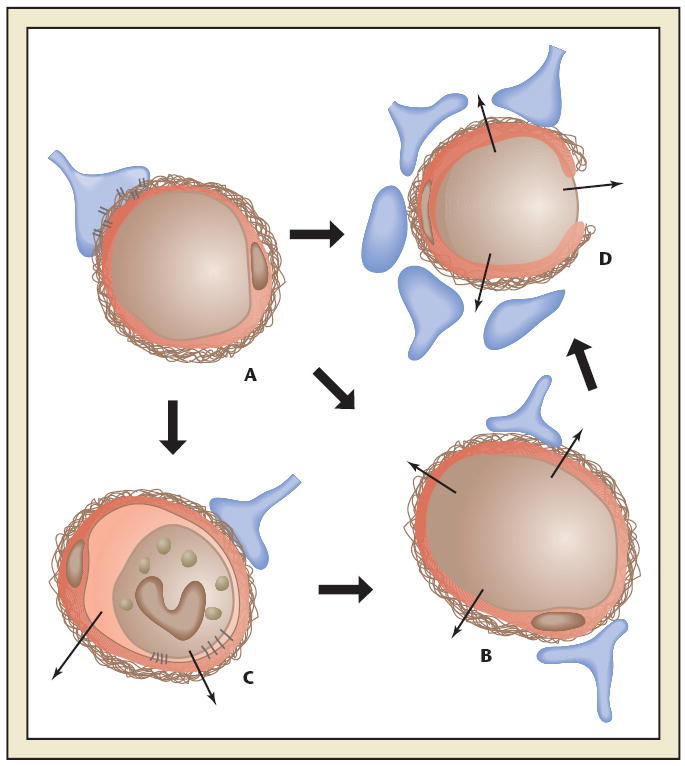
Schematic diagram of the effect of ischemia on microvessel permeability and integrity. (A) Normal cerebral microvessel. Endothelial cells and astrocyte end-feet bound to basal lamina by integrin and dystroglycan adhesion receptors. Blood-brain barrier intact. (B) Breakdown of the blood-brain barrier. (C) Leukocyte adhesion by receptors on endothelium and granulocytes. Increased permeability caused by granule release. (D) Breakdown of basal lamina with loss of astrocyte and endothelial cell contacts. Permeability to erythrocytes and other blood borne cells. Reprinted with permission from del Zoppo et al.44
Whether inhibiting PMN leukocyte adherence within the microvasculature or preventing fibrin deposition can lead to a reduction in neuron injury has not yet been studied. However, separate experiments that have examined the impact of the acute use of low molecular weight heparins (LMWHs; eg, enoxaparin) in rodent models of MCA occlusion have shown significantly decreased residual volumes of infarction and improvement in behavioral outcome.32 Early studies in nonhuman primates had already shown that heparin (and ticlopidine in combination) could significantly reduce the density of microvessel occlusions in the ischemic territory. Those studies suggest that by limiting coagulation system activation, and blocking the effects of endothelial cell activation, the effects of Virchow’s triad can be reduced.
The microvessel wall also displays dynamic changes. Significant changes in the matrix integrity of the basal lamina and in matrix receptors occur simultaneously with neuron injury. The expression of the matrix constituents of the basal lamina, including laminin-1 and -5, collagen IV, cellular fibronectin, and perlecan, are decreased substantially.33 The endothelial cell β1-integrin receptor and integrin α6β4 on astrocyte end-feet decrease substantially in the first 60 minutes following MCA occlusion.28,34 The alternate receptor dystroglycan is lost from the end-feet in the same timeframe.29 Separation of the end-feet from the matrix of select microvessels occurs in this period.35
Control of flow depends upon constriction at the precapillary arterioles, in response to local neural control, autocrine secretion, nitric oxide (NO) generation, and as-yet undefined local factors.
Those observations indicate that within the target regions of ischemic injury, the endothelium is activated and the coagulation system generates thrombin, fibrin, and activated platelets. These events contribute to intravascular occlusions that would be expected to reduce local flow. Although a small number of experimental studies have failed to show persistent decrease in residual flow, the level of resolution of these techniques is not sufficient to detect no-reflow. It is certain that ischemia initiates major alterations in the integrity of microvessels in the same territory-at-risk.
Flow and Shear Stress in the Cerebral Circulation
The cerebral circulation is a low pressure-high flow circuit. Fenstermacher and colleagues14 demonstrated that local CBF through the cortical gray matter exceeds that of the white matter and the deep brain (eg, the corpus striatum). The circle of Willis, pial anastomoses, and internal microvascular cross-communication provide protection of the cerebral vascular beds by allowing reversal of flow in this low pressure system. In particular, the capillary architecture in the gray matter is quite different from that of the corpus striatum.12,13 Bär12 defined the cortical microvasculature in the rodent as a hierarchical hexagonal array in conjunction with vertical neuronal arrays. Within the striatal gray matter the arrangements are more complex, with multiple bifurcations within the capillaries. Both systems allow rerouting of flow that is compatible with the ultrastructure of the tissue (ie, hierarchical within cortical gray matter columns and apparently radial within the striatum). Within the white matter the microvasculature follows the orientation of the axon bundles.
Control of flow depends upon constriction at the precapillary arterioles, in response to local neural control, autocrine secretion, nitric oxide (NO) generation, and as-yet undefined local factors. Neurovascular coupling connects neuron activation with either local or distant vascular flow control.15 The contribution of shear stress to the reactivity of the vascular wall endothelium in isolated systems appears to depend upon the diameter and construction of the vessel.
Analysis of shear and its effects in the cerebral vasculature in vivo is still quite limited. Using closed cranial window preparations the peak shear stress of pial surface vessels is 39 ± 14 dynes/cm−2.36 This reflects flow in penetrating arteries to capillaries only within the brain surface. Observations of pial capillaries are unlikely to reflect the status of microvessels within the white matter or the corpus striatum, however. Capillaries achieve and sustain high shear that is largely nonpulsatile, whereas large supply arteries have lower shear, but are subject to cyclic distortion during the systolic pulsation.
Studies of the impact of lower shear on the endothelial cell expression of vasoactive substances have largely involved systems of cultured endothelial cells from noncerebral sources. Endothelial NO synthase (eNOS) is expressed in the endothelium from medium to large arteries.37 Hence, in vitro systems are intended to mimic this setting. Pulsatile flow of 2 to 12 dynes/cm−2 over confluent endothelial cells stimulates significant increases in eNOS expression and NO generation over static flow.38 Turbulent flow inhibits this increase so that no expression of NO occurs. In separate studies pulsatile flow over human umbilical vein endothelial cells can reduce NO production, whereas asynchronous pulsatile flow, mimicking the transverse distortion of a large artery during pulse waves and relaxation, initially decreases eNOS and prostaglandin I2 expression within 5 hours of initiation of the pulsations, but is followed later by recovery and increases in their expression.39 Those observations suggest that pulsation in large arteries normally stimulates the endothelium to maintain an antithrombotic milieu.
The condition in capillaries is not so readily known, but the pulsation in large arteries is not uniformly transmitted through capillaries. Instead, capillary flow is generally constant. When shear decreases substantially (particularly in postcapillary venules), adhesion of platelets and PMN leukocytes to their respective receptors is more probable. The initial interaction of PMN leukocytes with P-selectin on activated endothelium is responsible for rolling of the cells, which precedes firm attachment via adherence of its β2-integrins to the endothelial cell counter-receptor ICAM-1.40 Secondary capture of PMN leukocytes via L-selectin appears to occur when shear is reduced to approximately 1 dyne/cm-2.41 Accumulation of platelet-leukocyte aggregates within a narrow vessel could obstruct flow, and the obstruction of a number of capillaries during ischemia could substantially reduce flow in the larger bed.
The general features of the contribution of shear to cell-vessel interactions are appreciated, but the exact events within the vasculature are presumed from static observations. Hence, the third element of Virchow’s triad is presumed to play a role in expansion/extension of the ischemic lesion.
It is now apparent that dissolution or remodeling of the basal lamina matrix can allow extravasation or transmigration of cells, including erythrocytes (hemorrhage) and leukocytes (inflammation).
Related and Characteristic Events
The 3 elements of Virchow’s triad are applicable to both the supply arteries and the microvasculature of the CNS under conditions of focal ischemia. In addition, cerebral microvessels can undergo ultrastructural changes that are unique: 1) changes in the matrix adhesion receptors of both endothelial cells and astrocytes in the microvessels, 2) increased permeability of the microvessel wall, and 3) hemorrhagic transformation.
Adhesion Receptors
In the early moments of focal ischemia the presentation of β1 integrins on endothelial cells and of integrin α6β4 and dystroglycan on astrocyte end-feet decreases substantially in the ischemic regions.34,42 Astrocyte end-feet appear to detach from the matrix, and matrix components are altered or lost.35
Permeability
Leakage of solute and fluid into the neuropil begins within hours of the onset of focal ischemia in the striatum. Although a large molecule (360 kDa), fibrinogen appears in the extravascular space within the first hour of MCA occlusion, indicating that leakage can be initiated rapidly as some microvessels are displaying evidence of occlusion (focal no-reflow). These events have been attributed to disruption of the interendothelial tight junctions. It is now apparent that dissolution or remodeling of the basal lamina matrix can allow extravasation or transmigration of cells, including erythrocytes (hemorrhage) and leukocytes (inflammation).43
Hemorrhagic Transformation
Hemorrhagic transformation in the areas of ischemic injury occurs in up to 65% of patients.44 These hemorrhagic events are also observed in select animal models of focal cerebral ischemia.45 Hemorrhagic infarction (HI; petechial to confluent petechial hemorrhages) appears to represent local leakage of erythrocytes from microvessels within the ischemic core regions when the basal lamina matrix is degraded, whereas parenchymal hemorrhage (PH; hematomas causing mass effect and most often clinical worsening) appears to result from degradation of the structure of large microvessels under arterial pressure. The disruption of the microvascular matrix barrier is hypothesized to be tied to matrix protease generation by proximity, timing, sources, and the up-regulation of the protease activation systems.46 Importantly, the risk of detectable hemorrhage appears to depend upon the presence of antithrombotic agents, and upon their dose and target. For instance, highest incidences of PH are associated with the use of plasminogen activators (eg, recombinant t-PA), that generate plasmin from circulating plasminogen: the substrates of plasmin include, in addition to fibrin(ogen), the matrix proteins laminin and collagen (as well as myelin basic protein). Hemorrhage would be accentuated when plasmin degrades extravascular fibrin, thereby preventing the sustained formation of a fibrin network.
Reversal of Occlusions
The conceptualization by Virchow that defined vascular factors promote intravascular thrombosis implies that strategies against these targets could substantially alter microvessel occlusions. Currently, no strategies applied during thromboembolic ischemic stroke have been shown to alter microvessel events acutely. However, as noted above, specific inhibitors of platelet activation, fibrin generation, and inflammatory cell adhesion (eg, PMN leukocyte-endothelial cell interactions) have been shown to prevent or abort the development of microvascular obstructions in the nonhuman primate model of focal ischemia, as well as in several rodent models. Inhibition of platelet-fibrin binding in a murine model confirmed the reduction in microvessel occlusions in the ischemic territory, and promoted a significant reduction in ischemic injury and in persistent neurological deficits.31,47 Similarly, inhibition of PMN leukocyte adhesion and/or leukocyte activation (and the respiratory burst) substantially decreases microvessel occlusions, and decreases cerebral injury volume during focal ischemia.48 Clinical studies of similar strategies, for a number of reasons to the contrary, have not yet properly tested the hypothesis that reduction in focal no-reflow could produce detectable reduction in cerebral injury.29 The experimental studies support the requirement for acute intervention, for proper dosing and dose delivery, and for a full understanding of the risks of the interventions.
Agents that might simultaneously affect more than 1 element of the triad could prove beneficial in reducing the consequences of ischemic injury.
Agents that might simultaneously affect more than 1 element of the triad could prove beneficial in reducing the consequences of ischemic injury. For instance, compounds that affect platelet function directly, and indirectly via action at the vascular wall, could prove useful in this setting. Dipyridamole can increase the platelet half-life during vascular thrombotic processes by direct action through a phosphodiesterase (PDE)-5 mechanism, and also indirectly by inhibition of endothelial cell-mediated metabolism of adenosine, thereby increasing endothelial cell-dependent adenosine concentrations.49 The fixed combination of aspirin and extended-release dipyridamole has proven beneficial in secondary prevention of ischemic stroke.50 Although untested in acute management of stroke, cilostazol acts as an antiplatelet agent and has other vascular effects based on PDE-3-dependent mechanisms.51 These serve as examples of multifunctional agents that affect more than 1 target of Virchow’s triad as applied to CNS ischemia.
Conclusions
Focal cerebral ischemia presents at least 2 conditions that satisfy Virchow’s triad in the CNS. First is the development of atheroma-based thrombosis within the brain-supplying arteries, where altered flow and coagulation system activation lead to fibrin deposition on the endothelium of the irregular atheroma surface. Second is the conversion of the usually high flow of the target microvasculature to a low flow prothrombotic state during ischemia. This entails the elements of the triad: 1) coagulation system activation caused in part by the exposure of the plasma to perivascular TF (thrombin generation and fibrin formation), 2) endothelial cell activation, and 3) resultant reduction in flow due to partial obstruction of the microvessel bed. Endothelial cell activation results in presentation of leukocyte and platelet receptors, adhesion of these cells, and generation of a prothrombotic milieu. The endogenous protective mechanisms that might limit these processes, or through their abatement could promote the elements of the triad, are still not fully understood. It is unknown whether there is a particular brain or brain-region specific form of hemostasis. So far this does not seem likely; however, special to the cerebral microvasculature are alterations in the permeability barriers at the interendothelial cell tight junctions and the endothelial cell-matrix interfaces, hemorrhage, and loss of microvessel integrity. These events accompany neuronal injury, and occur within the same time frame and in the same location. With increasing knowledge of the conditions at the microvessel wall more refined approaches to limiting focal no-reflow are quite possible.
Main Points.
An increasingly useful formulation links microvessel and neuron responses to ischemic injury: the neurovascular unit.
Focal ischemia initiates processes in the cerebral microvasculature that are consistent with Virchow’s triad: endothelial cell activation (with leukocyte adherence and transmigration), activation of coagulation (with fibrin deposition and thrombotic occlusion of the microvasculature), and alteration of flow (reduction in local cerebral blood flow).
Ischemic injury alters the integrity of the cerebral microvasculature by initiating processes that degrade matrix content, decrease endothelial cell and astrocyte matrix receptor expression, and increase permeability of the blood-brain barrier.
Matrix proteases can participate in some of the processes that lead to changes in microvascular integrity.
Footnotes
The author has no real or apparent conflicts of interest to report.
References
- 1.Liesi P. Do neurons in the vertebrate CNS migrate on laminin? EMBO J. 1985;4:1163–1170. doi: 10.1002/j.1460-2075.1985.tb03755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herken R, Gotz W, Thies M. Appearance of laminin, heparan sulphate proteoglycan and collagen type IV during initial stages of vascularisation of the neuroepithelium of the mouse embryo. J Anat. 1990;169:189–195. [PMC free article] [PubMed] [Google Scholar]
- 3.Grant DS, Kleinman HK. Regulation of capillary formation by laminin and other components of the extracellular matrix. EXS. 1997;79:317–333. doi: 10.1007/978-3-0348-9006-9_13. [DOI] [PubMed] [Google Scholar]
- 4.Engvall E, Davis GE, Dickerson K, et al. Mapping of domains in human laminin using monoclonal antibodies: localization of the neurite-promoting site. J Cell Biol. 1986;103:2457–2465. doi: 10.1083/jcb.103.6.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.David S, Braun PE, Jackson DL, et al. Laminin overrides the inhibitory effects of peripheral nervous system and central nervous system myelin-derived inhibitors of neurite growth. J Neurosci Res. 1995;42:594–602. doi: 10.1002/jnr.490420417. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein JJ, Getz R, Jefferson M, Kelemen M. Astrocytes secrete basal lamina after hemisection of rat spinal cord. Brain Res. 1985;327:135–141. doi: 10.1016/0006-8993(85)91507-0. [DOI] [PubMed] [Google Scholar]
- 7.Nagano N, Aoyagi M, Hirakawa K. Extracellular matrix modulates the proliferation of rat astrocytes in serum-free culture. GLIA. 1993;8:71–76. doi: 10.1002/glia.440080202. [DOI] [PubMed] [Google Scholar]
- 8.Webersinke G, Bauer H, Amberger A, et al. Comparison of gene expression of extracellular matrix molecules in brain microvascular endothelial cells and astrocytes. Biochem Biophys Res Commun. 1992;189:877–884. doi: 10.1016/0006-291x(92)92285-6. [DOI] [PubMed] [Google Scholar]
- 9.Hurwitz AA, Berman JW, Rashbaum WK, Lyman WD. Human fetal astrocytes induce the expression of blood-brain barrier specific proteins by autologous endothelial cells. Brain Res. 1993;625:238–243. doi: 10.1016/0006-8993(93)91064-y. [DOI] [PubMed] [Google Scholar]
- 10.Mabuchi T, Lucero J, Feng A, et al. Focal cerebral ischemia preferentially affects neurons distant from their neighboring microvessels. J Cereb Blood Flow Metab. 2005;25:257–266. doi: 10.1038/sj.jcbfm.9600027. [DOI] [PubMed] [Google Scholar]
- 11.Bär T. Morphometric evaluation of capillaries in different laminae of rat cerebral cortex by automatic image analysis: changes during development and aging. In: Cervos-Navarro J, editor. Advances in Neurology. 20. New York: Raven Press; 1978. pp. 1–9. [PubMed] [Google Scholar]
- 12.Bär T. The vascular system of the cerebral cortex. Adv Anat Embryol Cell Biol. 1980;59(IVI):1–62. doi: 10.1007/978-3-642-67432-7. [DOI] [PubMed] [Google Scholar]
- 13.del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–894. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- 14.Fenstermacher J, Nakata H, Tajima A, et al. Functional variations in parenchymal microvascular systems within the brain. Magn Reson Med. 1991;19:217–220. doi: 10.1002/mrm.1910190205. [DOI] [PubMed] [Google Scholar]
- 15.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 16.Rosenberg RD. Vascular-bed-specific hemostasis and hypercoagulable states. N Engl J Med. 1999;340:1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 17.del Zoppo GJ, Yu J-Q, Copeland BR, et al. Tissue factor localization in non-human primate cerebral tissue. Thromb Haemost. 1992;68:642–647. [PubMed] [Google Scholar]
- 18.Cunningham DD, Donovan FM. Regulation of neurons and astrocytes by thrombin and protease nexin-1. Relationship to brain injury. Adv Exp Med Biol. 1997;425:67–75. doi: 10.1007/978-1-4615-5391-5_7. [DOI] [PubMed] [Google Scholar]
- 19.Vaughan PJ, Cunningham DD. Regulation of protease nexin-1 synthesis and secretion in cultured brain cells by injury-related factors. J Biol Chem. 1993;268:3720–3727. [PubMed] [Google Scholar]
- 20.Croft RJ, Ellam LD, Harrison MJG. Accuracy of carotid angiography in the assessment of atheroma of the internal carotid artery. Lancet. 1980;1:997. doi: 10.1016/s0140-6736(80)91438-5. [DOI] [PubMed] [Google Scholar]
- 21.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 22.Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia - the ischemic penumbra. Stroke. 1981;12:723–725. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- 23.Horiuchi T, Dietrich HH, Hongo K, et al. Role of endothelial nitric oxide and smooth muscle potassium channels in cerebral arteriolar dilation in response to acidosis. Stroke. 2002;33:844–849. doi: 10.1161/hs0302.104112. [DOI] [PubMed] [Google Scholar]
- 24.Niwa K, Araki EMSG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000;20:763–770. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.del Zoppo GJ, Schmid-Schönbein GW, Mori E, et al. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–1284. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- 26.Risau W, Hallmann R, Albrecht U, Henke-Fahle S. Brain astrocytes induce the expression of an early cell surface marker for blood-brain barrier specific endothelium. EMBO J. 1986;5:3179–3183. doi: 10.1002/j.1460-2075.1986.tb04627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dore-Duffy P. Isolation and characterization of cerebral microvascular pericytes. Meth Mol Med. 2003;89:375–382. doi: 10.1385/1-59259-419-0:375. [DOI] [PubMed] [Google Scholar]
- 28.Tagaya M, Haring H-P, Stuiver I, et al. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. 2001;21:835–846. doi: 10.1097/00004647-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 29.Mori E, del Zoppo GJ, Chambers JD, et al. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–718. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- 30.Okada Y, Copeland BR, Fitridge R, et al. Fibrin contributes to microvascular obstructions and parenchymal changes during early focal cerebral ischemia and reperfusion. Stroke. 1994;25:1847–1853. doi: 10.1161/01.str.25.9.1847. [DOI] [PubMed] [Google Scholar]
- 31.Abumiya T, Fitridge R, Mazur C, et al. Integrin alpha(IIb)beta(3) inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke. 2000;31:1402–1410. doi: 10.1161/01.str.31.6.1402. [DOI] [PubMed] [Google Scholar]
- 32.Stutzmann JM, Mary V, Wahl F, et al. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Review. 2002;8:1–30. doi: 10.1111/j.1527-3458.2002.tb00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;26:2120–2126. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- 34.Wagner S, Tagaya M, Koziol JA, et al. Rapid disruption of an astrocyte interaction with the extracellular matrix mediated by integrin alpha 6 beta 4 during focal cerebral ischemia/reperfusion. Stroke. 1997;28:858–865. doi: 10.1161/01.str.28.4.858. [DOI] [PubMed] [Google Scholar]
- 35.Heo JH, Han SW, Lee SK. Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med. 2005;39:51–70. doi: 10.1016/j.freeradbiomed.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 36.Rovainen CM, Woolsey TA, Blocher NC, et al. Blood flow in single surface arterioles and venules on the mouse somatosensory cortex measured with videomicroscopy, fluorescent dextrans, nonoccluding fluorescent beads, and computer-assisted image analysis. J Cereb Blood Flow Metab. 1993;13:359–371. doi: 10.1038/jcbfm.1993.49. [DOI] [PubMed] [Google Scholar]
- 37.Fish JE, Matouk CC, Rachlis A, et al. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem. 2005;280:24824–24838. doi: 10.1074/jbc.M502115200. [DOI] [PubMed] [Google Scholar]
- 38.Noris M, Morigi M, Donadelli R, et al. Nitric oxide synthesis by cultured endothelial cells is modulated by flow conditions. Circ Res. 1995;76:536–543. doi: 10.1161/01.res.76.4.536. [DOI] [PubMed] [Google Scholar]
- 39.Dancu MB, Berardi DE, Vanden Heuvel JP, Tarbell JM. Asynchronous shear stress and circumferential strain reduces endothelial NO synthase and cyclooxygenase-2 but induces endothelin-1 gene expression in endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:2088–2094. doi: 10.1161/01.ATV.0000143855.85343.0e. [DOI] [PubMed] [Google Scholar]
- 40.Okada Y, Copeland BR, Mori E, et al. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke. 1994;25:202–211. doi: 10.1161/01.str.25.1.202. [DOI] [PubMed] [Google Scholar]
- 41.Paschall CD, Lawrence MB. L-selectin shear thresholding modulates leukocyte secondary capture. Ann Biomed Eng. 2008;36:622–631. doi: 10.1007/s10439-008-9468-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milner R, Hung S, Wang X, et al. The rapid decrease in astrocyte-associated dystroglycan expression by focal cerebral ischemia is protease-dependent. J Cereb Blood Flow Metab. 2008 doi: 10.1038/sj.jcbfm.9600585. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamann GF, Okada Y, del Zoppo GJ. Hemorrhagic transformation and microvascular integrity during focal cerebral ischemia/ reperfusion. J Cereb Blood Flow Metab. 1996;16:1373–1378. doi: 10.1097/00004647-199611000-00036. [DOI] [PubMed] [Google Scholar]
- 44.del Zoppo GJ, von Kummer R, Hamann GF. Ischemic damage of brain microvessels: inherent risks for thrombolytic treatment in stroke. J Neurol Neurosurg Psychiatry. 1998;65:1–9. doi: 10.1136/jnnp.65.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.del Zoppo GJ, Copeland BR, Harker LA, et al. Experimental acute thrombotic stroke in baboons. Stroke. 1986;17:1254–1265. doi: 10.1161/01.str.17.6.1254. [DOI] [PubMed] [Google Scholar]
- 46.Chang DI, Hosomi N, Lucero J, et al. Activation systems for matrix metalloproteinase-2 are up-regulated immediately following experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:1408–1419. doi: 10.1097/01.WCB.0000091765.61714.30. [DOI] [PubMed] [Google Scholar]
- 47.Choudhri TF, Hoh BL, Zerwes HG, et al. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor-mediated platelet aggregation. J Clin Invest. 1998;102:1301–1310. doi: 10.1172/JCI3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walder CE, Green SP, Darbonne WC, et al. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidate. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- 49.Fitzgerald GA. Dipyridamole. N Engl J Med. 1987;316:1247–1257. doi: 10.1056/NEJM198705143162005. [DOI] [PubMed] [Google Scholar]
- 50.Diener H, Cunha L, Forbes C, et al. European Stroke Prevention Study 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci. 1996;143:1–13. doi: 10.1016/s0022-510x(96)00308-5. [DOI] [PubMed] [Google Scholar]
- 51.Kimura Y, Tani T, Kanbe T, Watanabe K. Effect of cilostazol on platelet aggregation and experimental thrombosis. Arzneimittelforschung. 1985;35:1144–1149. [PubMed] [Google Scholar]