

Abstract
Classical dopaminergic signaling paradigms and emerging studies on direct physical interactions between the D1 dopamine (DA) receptor and the NMDA glutamate receptor predict a reciprocally facilitating, positive feedback loop. This loop, if not controlled, may cause concomitant overactivation of both D1 and NMDA receptors, triggering neurotoxicity. Endogenous protective mechanisms must exist. Here, we report that PSD-95, a prototypical structural and signaling scaffold in the postsynaptic density, inhibits D1–NMDA receptor subunit 1 (NR1) NMDA receptor association and uncouples NMDA receptor-dependent enhancement of D1 signaling. This uncoupling is achieved, at least in part, via a disinhibition mechanism by which PSD-95 abolishes NMDA receptor-dependent inhibition of D1 internalization. Knockdown of PSD-95 immobilizes D1 receptors on the cell surface and escalates NMDA receptor-dependent D1 cAMP signaling in neurons. Thus, in addition to its role in receptor stabilization and synaptic plasticity, PSD-95 acts as a brake on the D1–NMDA receptor complex and dampens the interaction between them.
Introduction
Dopaminergic and glutamatergic terminals converge onto the same dendritic spines of dopamineceptive neurons in dopamine (DA) target regions, forming “synaptic triads” (Freund et al., 1984; Goldman-Rakic et al., 1989; Carr and Sesack, 1996). This triadic heterosynaptic architecture provides a structural basis for a close interplay between DA and glutamate systems, which is essential for many cognitive and motivational processes (Berke and Hyman, 2000; Schultz, 2002). A balanced DA–glutamate interaction is, to a large degree, mediated by the functional cross talk between D1, the predominant subtype of the D1-class receptors, and the NMDA glutamate receptor in postsynaptic neurons. These receptors colocalize extensively at synaptic, parasynaptic, and nonsynaptic sites in dendritic spines and shafts (Hara and Pickel, 2005; Pickel et al., 2006). Classically, D1 receptor activation enhances NMDA receptor activity through the cAMP/protein kinase A (PKA)/dopamine and cAMP-regulated phosphoprotein-32 (DARPP-32)/protein phosphatase 1 (PP1) pathways (Lachowicz and Sibley, 1997; Missale et al., 1998; Greengard et al., 1999). D1 activation also induces rapid trafficking of intracellular NMDA receptors to the postsynaptic membrane, thus enhancing NMDA receptor function, via a tyrosine kinase signaling mechanism (Dunah and Standaert, 2001).
Recent studies reveal that NMDA receptors also reciprocally regulate D1 activity via direct physical coupling. D1 interacts with the NMDA receptor subunits 1 (NR1) through carboxyl tails of these receptors (Lee et al., 2002). Association with NMDA receptors facilitates D1 trafficking to the cell surface and inhibits D1 internalization (Scott et al., 2002; Fiorentini et al., 2003; Pei et al., 2004). Ligand-occupied NMDA receptors also constrain the mobility of laterally diffusing dendritic D1 receptors and recruit them to spines through a diffusion trap mechanism (Scott et al., 2006). Assuming that activation of NMDA receptors recruits D1 receptors to the plasma membrane, which in turn facilitates the activity and surface targeting of NMDA receptors, a positive feedback loop is created (Cepeda and Levine, 2006). This loop, if not controlled, might result in concomitant overactivation of both D1 and NMDA receptors, jeopardizing neuronal integrity and triggering neurotoxicity (Choi, 1988; Bozzi and Borrelli, 2006).
The postsynaptic scaffold PSD-95 interacts with NMDA receptor NR2 subunits through its first two PSD-95/Dlg/ZO-1 homology (PDZ) domains (Kornau et al., 1995; Niethammer et al., 1996), which may play a role in “functionally” localizing NMDA receptors in the synapse (Kennedy, 2000; Kim and Sheng, 2004) and in regulating synaptic efficacy (Migaud et al., 1998; Stein et al., 2003; Ehrlich and Malinow, 2004; Béïque et al., 2006; Xu et al., 2008). PSD-95 also interacts with D1 via the C terminus (CT) of the receptor and the N terminus (NT) of PSD-95, an interaction that regulates D1 trafficking (Zhang et al., 2007). Together with the demonstrated D1–NR1 association and overlapping subcellular distributions of these proteins (Valtschanoff et al., 1999; Aoki et al., 2001; Hara and Pickel, 2005), a tertiary protein complex containing these proteins may exist in the brain, in which the interplay between D1 and NMDA receptors is fine-tuned. Here, we provide evidence that PSD-95 associates with D1 and the NMDA receptor complex and negatively regulates the physical and functional interactions between these receptors.
Materials and Methods
Mice.
All experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of animals and with an approved animal protocol from the Harvard Medical Area Standing Committee on Animals. PSD-95 wild-type (WT) and knock-out (KO) mice (Yao et al., 2004) were housed under standard laboratory conditions (12 h light/dark cycle) with food and water provided ad libitum.
Plasmid constructs.
Plasmids encoding HA-D1 (hemagglutinin-tagged D1 receptors), PSD-95-GFP (green fluorescent protein-tagged PSD-95), NR1, NR2B, and PSD-95-GFP mutants ΔNT-GFP, Δ1&2-GFP, ΔNT, 1&2-GFP, and NT-GFP have been described previously (Tezuka et al., 1999; Zhang et al., 2007). PSD-95 small-hairpin (sh)RNA targeting sequence (Elias et al., 2006) in pLLox3.7 vector was a kind gift from Dr. Roger A. Nicoll (University of California, San Francisco, San Francisco, CA). The control shRNA (TCACAGTCGGATCCATCACTCAGTATA) was inserted into pLLox3.7. All constructs were generated by PCR and verified by automated sequencing.
Cell culture and transfection.
Human embryonic kidney (HEK) 293, HEK293T, and HEK293 cells stably expressing the rhesus monkey D1 receptor (D1-stable cells) were cultured and transfected as previously described (Zhang et al., 2007). Embryonic day 18–19 (E18–19) rat and mouse primary neurons were grown on poly-d-lysine-coated plates in neurobasal medium supplemented with B27 and 1% GlutaMax (Invitrogen). Hippocampal neurons were transfected with sh-PSD-95 or sh-control and HA-D1 by Lipofectamine 2000 (Invitrogen) or nucleofection (Amaxa).
Lentivirus production and neuronal infection.
HEK293T cells (2 × 106) were cotransfected with pLLox3.7 and helper vectors, pDelta8.9 and pVSV-G, using TransFectin (Bio-Rad). The supernatant was collected after 72 h and titer was determined by infection of HEK293T cells. Striatal neurons were infected by lentivirus particles expressing sh-PSD-95 or sh-control with a titer of MOI 2 at 11 days in vitro, and grown for 6 additional days before Western blot analyses and cAMP assays. The infection efficiency routinely reached >70%.
Immunocytochemistry, confocal microscopy, and immunofluorescence.
Hippocampal neurons were fixed in 4% paraforaldehyde/4% sucrose at room temperature for 15 min, permeabilized, and blocked, as previously described (Zhang et al., 2007). Cells were incubated with the following primary antibodies overnight at 4°C: anti-D1 (1:50; Sigma), anti-PSD-95 (1:200), and anti-NR1 (1:50; BD PharMingen), followed by incubation with secondary antibodies conjugated with appropriate Alexa dyes (1:500; Invitrogen) at room temperature for 1 h. Cells were mounted on glass slides.
D1 internalization was measured as described (Zhang et al., 2007). Briefly, HA-D1-transfected HEK293 cells or hippocampal neurons were incubated with a HA antibody (1:100, Covance) at 4°C or 15°C, respectively. After wash, cells were stimulated with SKF 81297 ((±)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide) (30 min, 37°C) in the presence or absence of NMDA receptor agonists and/or antagonists, as specified. Surface HA-D1 receptors were labeled with an Alexa Fluor 647-conjugated secondary antibody. The internalized HA-D1 receptor was recognized by an Alexa Fluor 568-conjugated secondary antibody after permeabilization. Confocal images were acquired using a Leica confocal microscope at the following excitation/emission wavelengths: 488/519 nm, 568/604 nm, or 647/669 nm. Image stacks were acquired under the same confocal settings along the z-axis, and were flattened into a single image using a maximum projection and analyzed with MetaMorph (Universal Imaging). Surface and internal D1 fluorescence intensities were measured as integrated pixel intensities, and the internalization index for each cell was defined as the ratio of the internalized fluorescence intensity to the total fluorescence intensity. HA-D1 internalization in neurons was performed at the soma.
For quantification of colocalization, dendritic segments of a neuron containing >50 fluorescence clusters were selected and traced. NR1 clusters were selected automatically in the pseudo-colored “blue” channel as discrete puncta of intensity >1.5-fold brighter than the background fluorescence. Selected clusters were transferred to the red channel to measure the D1 fluorescence. Colocalization of D1 over NR1 was measured as the percentage of integrated D1 pixel intensities that overlapped with the NR1 fluorescence in individual clusters and averaged for each neuron. All groups to be compared were run simultaneously using cells from the same culture preparations and transfection condition.
Immunoprecipitation and Western blotting.
HEK293(T) cells or cultured neurons were briefly sonicated in 25 mm Tris-HCl containing 150 mm NaCl, 3 mm KCl, 1 mm EDTA, and protease inhibitors, and the supernatants were extracted by centrifuge at 13,000 × g for 30 min. Mouse forebrain structures (i.e., striata, hippocampi, and cortices) were dissected, homogenized, and extracted in a deoxycholate (DOC) buffer as previously described (Zhang et al., 2007). Protein extracts were incubated with anti-D1 (10 μl; Sigma), anti-NR1 (4 μg; Upstate Biotechnology), or anti-HA (5 μl; Covance) antibodies at 4°C overnight with gentle rotation. Precipitated protein complexes were captured by Protein A/G agarose beads (Santa Cruz Biotechnology), immobilized to polyvinylidene difluoride (PDVF) membranes, incubated with anti-D1 (1:200), anti-PSD-95 (1:500; BD Transduction Laboratories), anti-NR1 (1:200), anti-NR2B (1:200; Upstate Biotechnology), anti-GST (glutathione S-transferase) (1:1000; Santa Cruz Biotechnology), anti-actin (1:1000; Millipore), or anti-GFP (green fluorescent protein) (1:1000; Santa Cruz Biotechnology) antibodies as specified. Horseradish peroxidase-conjugated secondary antibodies and signals were detected by an ECL-based LAS-3000 image system (Fujifilm). Densitometric analysis was performed within linear range using ImageGauge (Fujifilm).
GST fusion proteins and pull-down assay.
GST fusion proteins encoding D1 receptor C-terminal fragments CT1 (aa 361–389), CT2 (aa 385–415), or CT3 (aa 414–446) were generated by PCR and subcloned into pGEX6P-1 vector (GE Healthcare) in-frame. GST fusion protein production was induced by 0.5 mm isopropyl-β-d-thiogalactopyranoside (Promega) for 2 h in BL21 bacterium and immobilized on glutathione-Sepharose 4B agarose (GE Healthcare). Equal amounts of GST fusion proteins were incubated at 4°C overnight with lysates of HEK293 cells overexpressing PSD-95-GFP or cultured rat striatal neurons, followed by washes with ice-cold PBS containing 0.1% Triton X-100. The pulled-down proteins were resolved by SDS-PAGE and analyzed by Western blotting.
cAMP enzyme immunoassay.
D1-stable HEK293 cells transiently transfected with NR1 and NR2B cDNAs in the presence or absence of PSD-95-GFP coexpression, or striatal neurons infected with lentiviral particles expressing sh-PSD-95 or sh-control, were stimulated by SKF 81297 for 30 min in the presence or absence of NMDA receptor agonist/antagonist combinations, as specified. Whole-cell cAMP accumulation was measured using the Direct cAMP Enzyme Immunoassay Kit (Sigma or GE Healthcare) following manufacturers' instructions. cAMP concentrations were measured as optical density at 405 or 450 nm by a microplate reader (PerkinElmer).
Radioligand competition binding.
Cells were homogenized in 5 mm Tris-HCI containing 2 mm EDTA and protease inhibitors, and centrifuged at 3400 × g for 30 min at 4°C. The pellet was resuspended in binding buffer [5 mm Tris containing (in mm): 8.5 HEPES, 120 NaCl, 5.4 KCl, 1.2 CaCl2, 1.2 MgSO4, and 5 glucose, pH 7.4]. Sample (200 μg) was incubated with 0.4 nm [3H]SCH23390 (R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine) (86 Ci/mmol, GE Healthcare) in the presence of increasing concentrations of SKF 81297 (10−12–10−4 m) at 4°C for 2 h. Nonspecific binding was determined in the presence of 1 μm SCH39166 ((−)-trans-6,7,7α,8,9,13β-hexahydro-3-chloro-2-hydroxy-N-methyl-5H-benzo[d]naphtho[2,1-b]azepine). All experiments were performed in triplicates. Binding data were analyzed by fitting the data with a sigmoidal dose–response curve to derive Bmax and IC50 using GraphPad Prism software.
Results
The D1/PSD-95/NMDA receptor complex
We first tested the hypothesis that D1, PSD-95, and NMDA receptors reside in the same protein complex. HEK293T cells were transfected with various combinations of PSD-95-GFP, HA-D1 (Zhang et al., 2007), and NMDA receptors, which, in this study, represented coexpression of the NR1 and the NR2B subunits. Immunoblot analysis of whole-cell lysates showed that, when coexpressed, PSD-95, NR1, and D1 coprecipitated with antibodies against either HA or NR1 (Fig. 1A), suggesting that these proteins formed a multiprotein complex in these cells. NR1 or PSD-95 coprecipitated with the anti-HA antibody when each was coexpressed with HA-D1. Similarly, D1 or PSD-95 coprecipitated with the anti-NR1 antibody when each was coexpressed with NMDA receptors. These data suggest that the tertiary complex is assembled by protein–protein interactions at multiple sites (Fig. 1G).
Figure 1.

The D1, PSD-95, and NMDA receptor complex. A, Formation of D1/PSD-95/NMDA receptor complex in HEK293T cells. Cells were transfected with cDNA constructs encoding HA-D1, PSD-95-GFP, and/or NR1/NR2B. Coimmunoprecipitation was performed by incubation of cell lysates with the indicated antibodies followed by immunoblotting. B, Formation of D1, PSD-95, and NMDA receptor complex in the mouse brain. DOC extracts of mouse forebrain tissues were immunoprecipitated with an anti-D1 antibody and the blots were revealed by antibodies against PSD-95, NR1, NR2A, and NR2B, respectively. C, Colocalization of D1, PSD-95, and NR1 in subsets of spines or puncta in a dendritic process of a cultured mouse hippocampal neuron. Arrowheads indicate representative puncta in which D1, PSD-95, and NR1 colocalize. Scale bar, 2 μm. D, E, Schematics showing D1 domain structures (D) and GST fusion protein constructions (E). F, PSD-95 binds distal D1 CT domains. GST-D1 CT2 or GST-D1 CT3, but not GST-D1 CT1 or GST alone precipitated PSD-95 from HEK293 cells overexpressing PSD-95-GFP or mouse striatal cultures. Equal amounts of GST fusion proteins were used in the pull-down assays. IgG was used as a control in all immunoprecipitation experiments. Data in A, B, C, and F are representatives of three or more independent experiments. G, A model depicting molecular interactions (thick bars connecting various proteins) that assemble the D1/PSD-95/NMDA receptor complex. PSD-95 and NR1 interact with a partially overlapping region on the D1 CT.
To establish whether or not the D1/PSD-95/NMDA receptor complex exists in vivo, we performed coimmunoprecipitation experiments on mouse forebrain lysates (Fig. 1B). An anti-D1 antibody precipitated a protein complex that included PSD-95 and several NMDA receptor subunits, NR1, NR2A, and NR2B. To confirm the immunoprecipitation data, we also examined the subcellular distributions of D1, PSD-95, and NR1 in cultured hippocampal neurons using immunofluorescence confocal microscopy. D1, PSD-95, and NR1colocalized in a substantial portion of dendritic spines/clusters along dendritic processes (Fig. 1C). Together, these data provide support for the view that D1, PSD-95, and NMDA receptors coexist in the same protein complex in the brain.
PSD-95 and NR1 bind to an overlapping region on the D1 carboxyl tail
Our previous studies demonstrate that PSD-95, via its NT, directly interacts with the CT of D1 (D1 CT) (Zhang et al., 2007). A peptide fragment (L385–L415; D1 CT2) in the middle of D1 CT has been shown to interact with NR1 (Lee et al., 2002). We investigated whether PSD-95 also binds this domain of D1 CT using recombinant GST affinity purification assays (Fig. 1D–F). GST fusion proteins encoding the various fragments of D1 CT were constructed and used as baits to precipitate associated proteins (Fig. 1E). Incubation of GST, GST-D1 CT1, GST-D1 CT2, or GST-D1 CT3 fusion proteins with lysates prepared from HEK293 cells expressing PSD-95 revealed a copurification of PSD-95 and GST-D1 CT2 or GST-D1 CT3, but not GST-D1 CT1 or GST alone (Fig. 1F). Similarly, GST-D1 CT2 or GST-D1 CT3, but not GST-D1 CT1 or GST, was able to pull down PSD-95 from protein extracts prepared from mouse striatal cultures. These data suggest that although they bind to distinct sequences, PSD-95 and NR1 recognize an overlapping region on the D1 CT.
PSD-95 interferes with D1–NR1 interaction
By associating with a region on the D1 CT that also mediates D1–NR1 interaction, PSD-95 could interfere with the physical coupling between the two receptors. To test this hypothesis, HEK293T cells were transfected with cDNAs encoding D1 and NMDA receptors in the presence or absence of PSD-95 cotransfection (Fig. 2A–C). The strength of D1–NR1 association was measured by coimmunoprecipitation using an antibody against NR1. PSD-95 coexpression affected neither the total D1 nor NR1 expression (Fig. 2A,B). In the presence of PSD-95, however, the anti-NR1 antibody precipitated a significantly lower amount of D1 receptors, whereas the amount of coprecipitated NR1 protein remained the same (Fig. 2A,C). This result suggests that the presence of PSD-95 inhibited D1–NR1 association.
Figure 2.

PSD-95 inhibits D1–NR1 interaction. A, Coimmunoprecipitation of HA-D1 and NR1 in the presence or absence of PSD-95 overexpression in HEK293T cells. B, C, Densitometric analyses of total (B) and coprecipitated (C) NR1 and D1 receptors. n = 3–4; data represent mean ± SEM. *p < 0.05; two-tailed Student's t test. D, Schematic drawing of PSD-95 truncation constructs. E, Effects of PSD-95 mutants on D1–NR1 interaction, as measured by coimmunoprecipitation in the presence or absence of overexpression of different PSD-95 constructs. F, Densitometric analysis of coprecipitated D1 receptors from cell lysates prepared from cotransfected HEK293T cells. n = 6; data represent mean ± SEM. *p < 0.05, **p < 0.01 vs No PSD-95 control (D1+NR+mock); ##, p < 0.01 vs D1+NR+PSD-95; two-tailed Student's t tests. HEK293T cells were transfected with HA-D1, NR1/NR2B, and different PSD-95-GFP constructs or a mock vector. Coimmunoprecipitation was performed by incubating HEK293T cell lysates with an anti-NR1 antibody followed by immunoblotting with an anti-HA or an anti-NR1 antibody. Total levels of D1 and NR1 were measured by Western blots, and were used to assure that similar amounts of samples were used. For all densitometric analyses, results are presented in arbitrary units normalized to respective protein levels in D1–NR-transfected cells (used as controls; open bars in B, C, and first bar in F). In all cases, normalization involves two steps, a within-blot standardization across all samples on the blot followed by a between-blot normalization to the controls across all experiments.
To investigate whether the PSD-95 inhibition of D1–NR1 interaction is mediated by PSD-95 NT, we generated several PSD-95 truncation mutants (Fig. 2D) and analyzed their effects on D1–NR1 coprecipitation in cotransfected HEK293T cells (Fig. 2E,F). PSD-95 NT, a 72-amino acid peptide alone in the absence of the three PDZ, SH3, and GK domains, diminished the D1–NR1 association indistinguishable from that induced by the full-length PSD-95. Unexpectedly, a PSD-95 mutant lacking the NT (PSD-95 ΔNT) still inhibited, albeit with less effectiveness (Fig. 2F), the D1–NR1 association. However, a mutant lacking both NT and PDZ1 and -2 domains (PSD-95 ΔNT, 1&2) completely abolished the inhibition (Fig. 2E,F). These data suggest that the PSD-95 NT is sufficient but not necessary for inhibiting D1–NR1 interaction, and the first two PDZ domains that mediate PSD-95–NMDA NR2 receptor interaction also participate in the negative regulation of D1–NR1 interaction.
Removal of PSD-95 enhances D1–NR1 association
To confirm the inhibition of PSD-95 on D1–NR1 interaction in vivo, we performed coimmunoprecipitation and Western blots on forebrain protein extracts prepared from wild-type mice (PSD-95 WT) and their littermates that lacked PSD-95 (PSD-95 KO) (Fig. 3A–D). The total D1 and NR1 levels were unaltered in PSD-95 KO mice. An anti-D1 antibody precipitated a similar amount of D1 but a significantly higher amount of NR1 in PSD-95 KO mice, compared with the WT control. Thus, more NR1 was associated with a similar amount of D1 receptors in the absence of PSD-95.
Figure 3.

Removal of PSD-95 enhances D1–NR1 association in vivo. A, Protein levels of NR1, D1, PSD-95, and actin (as a loading control) in the forebrain of PSD-95 WT and KO littermates as measured by Western blots. B, Coimmunoprecipitation of D1 and NR1 in PSD-95 WT and KO mice. Coimmunoprecipitation was performed on forebrain extracts using an anti-D1 antibody and blotted with an anti-NR1 antibody. Total NR1 levels from the same samples are shown at the bottom. C, Densitometric analyses of total D1 and NR1 levels in PSD-95 WT and KO mice. n = 7. D, Densitometric analysis of NR1 levels (background-subtracted) coprecipitated with an anti-D1 antibody. n = 8. An area with equivalent size of the NR1 band at the top of each lane was used to estimate the background signal for the lane. Results for densitometric analyses are presented in arbitrary units normalized to corresponding WT controls using the two-step normalization procedures described in Figure 2. E, shRNA silencing of PSD-95 in primary hippocampal cultures. Cultured hippocampal neurons were transfected by electroporation with sh-PSD-95 or sh-control. Protein levels of PSD-95 and actin were analyzed by Western blots. Data are representative of three independent experiments. F, Confocal images of hippocampal neurons transfected with sh-control or sh-PSD-95 shRNAs (left; scale bar, 20 μm). Merged GFP, D1 (red), and NR1 (blue) florescence is shown. Representative endogenous D1 and NR1 clusters on dendritic processes of these neurons are shown on the right (scale bar, 2 μm). G, Quantification of D1 colocalization with NR1. n = 13–30 neurons. All data are expressed as mean ± SEM. **p < 0.01, ***p < 0.001; two-tailed Student's t tests.
To directly “visualize” the role of PSD-95 in D1–NR1 association, we examined the effect of shRNA-mediated PSD-95 knockdown on the colocalization of D1 and NMDA receptors in cultured hippocampal neurons (Fig. 3E–G). A shRNA carrying point mutations was used as a control (sh-control). Neurons were transfected with shRNA for PSD-95 (sh-PSD-95) or sh-control. Western blot analysis showed that sh-PSD-95 selectively silenced the expression of PSD-95 compared with sh-control (Fig. 3E) (Elias et al., 2006). Neurons expressing shRNAs were identified by their expression of GFP (Fig. 3F). D1 receptor colocalization with NR1 in dendritic puncta/clusters was significantly higher in neurons expressing sh-PSD-95 than in neurons expressing sh-control (Fig. 3F,G). This increase occurred without changes in the densities of D1 or NR1 puncta (data not shown). Collectively, these results suggest that PSD-95 fine-tunes the D1–NMDA receptor association within the same complex.
PSD-95 removes NMDA receptor inhibition of D1 internalization
The responsiveness of the D1 receptor, like most G-protein-coupled receptors (GPCRs), is controlled primarily by the classical β-arrestin- and GPCR kinase (GRK)-regulated desensitization process (Gainetdinov et al., 2004). Association with NMDA receptors immobilizes D1 receptors in the plasma membrane and abolishes agonist-induced D1 desensitization (Fiorentini et al., 2003). We thus investigated the functional significance of the PSD-95 interference of D1–NR1 association by determining the effect of PSD-95 on this D1 trafficking process using an immunocytochemistry-based internalization assay (Zhang et al., 2007). HEK293 cells were transfected with HA-D1 and NMDA receptors in the presence or absence of PSD-95-GFP. Surface HA-D1 receptors were labeled with an anti-HA antibody, and the internalization of these receptor-antibody complexes was monitored in live cells (Fig. 4). Consistent with previous studies (Fiorentini et al., 2003; Zhang et al., 2007), HA-D1, when expressed alone (Fig. 4A) or coexpressed with NMDA receptors (Fig. 4C), displayed little constitutive endocytosis but showed substantial spontaneous internalization when coexpressed with PSD-95 (Fig. 4B). Stimulation with SKF81297 (10 μm, 30 min), a full D1 agonist, induced robust internalization of the HA-D1 receptors, regardless of the presence of NMDA receptor and/or PSD-95 overexpression (Fig. 4).
Figure 4.
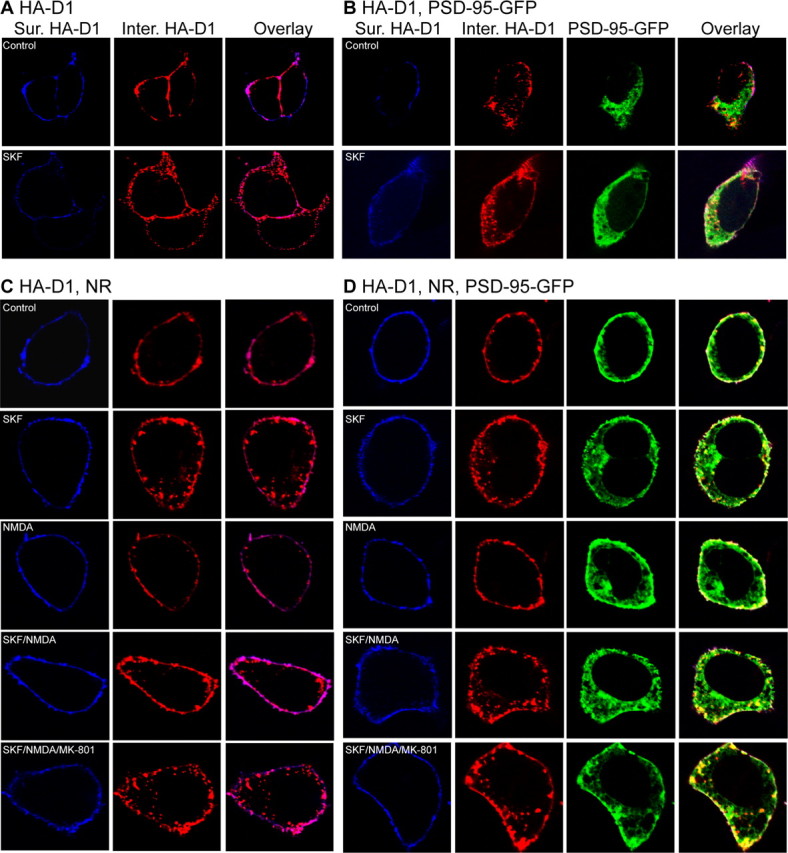
A–D, PSD-95 blocks NMDA receptor-dependent inhibition of D1 endocytosis. HEK293 cells were transfected with HA-D1 alone (A), HA-D1 and PSD-95-GFP (B), HA-D1 and NR1/NR2B (C), or HA-D1, PSD-95-GFP, and NR1/NR2B (D). Surface receptors were live-conjugated with an anti-HA antibody, and were allowed to undergo endocytosis (37°C, 30 min) under the indicated stimulation conditions. After internalization, cells were processed for differential staining of remaining surface (Sur., before permeabilization) and internalized (Inter., after permeabilization) receptors.
We then examined how PSD-95 might regulate D1 internalization as a consequence of NMDA receptor coactivation. In cells cotransfected with HA-D1 and NMDA receptors, simultaneous stimulation of NMDA receptors with NMDA (50 μm)/glycine (10 μm) inhibited the SKF 81297-induced D1 internalization (Fig. 4C, 5E). This inhibition was blocked by the NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) (10 μm), indicating the requirement for NMDA receptor activation (Fig. 4C). In contrast, this NMDA receptor-dependent inhibition of D1 internalization was abolished in cells cotransfected with D1, NMDA receptor, and PSD-95 (Fig. 4D, 5E). Notably, the SKF 81297-induced D1 internalization occurred regardless of NMDA receptor activation and persisted in the presence of MK-801 in these cells. These data suggest that PSD-95 uncoupled modulation of D1 trafficking by the NMDA receptor. Western blot analysis of total and surface-labeled (with Sulfo-NHS-SS-biotin) receptors indicated that overexpression of PSD-95 did not alter the total or the surface expression of either the D1 or the NMDA receptor in D1–NMDA receptor-expressing cells (data not shown). Thus, coexpression of PSD-95 with D1–NMDA receptors alters the rules that govern NMDA receptor modulation of D1 endocytosis without affecting the level or distribution of these receptors.
Figure 5.

PSD-95 blockade of NMDA receptor inhibition of D1 endocytosis requires PSD-95 NT and PDZ1 and -2 domains. A–D, SKF 81297-induced D1 internalization in HEK293 cells expressing D1, PSD-95-WT, and NR1/NR2B (A), D1, PSD-95-ΔNT, and NR1/NR2B (B), D1, PSD-95-Δ1&2, and NR1/NR2B (C), or D1, PSD-95-ΔNT, 1&2, and NR1/NR2B (D). HA-D1 and PSD-95-GFP were used. Note that only the PSD-95 mutant lacking both NT and PDZ1 and -2 domains failed to block the NMDA receptor-dependent inhibition of D1 internalization (D). Receptor internalization assay was performed as in Figure 4. E, Summary graph showing the effects of overexpressing the various PSD-95 constructs on NMDA receptor inhibition of D1 internalization. Internalization index was defined as the ratio of internalized to total fluorescence intensities. n = 15–17 cells for each group. Data are expressed as mean ± SEM. *p < 0.05; ***p < 0.0001; n.s., not significant; two-tailed Student's t tests.
Domain mapping of PSD-95 disinhibition of D1 internalization
We next investigated the domain mechanism by which PSD-95 removes, or disinhibits, the NMDA receptor-mediated inhibition of D1 internalization by analyzing the effect of PSD-95 truncation mutants on D1 internalization in cotransfected HEK293 cells (Fig. 5). Consistent with the involvement of both PSD-95 NT and PDZ1 and -2 domains in inhibiting D1–NR1 interaction, deletion of either domain, but not both, still mimicked full-length PSD-95 in disinhibiting the NMDA receptor-dependent inhibition of D1 internalization. In particular, either PSD-95 ΔNT (Fig. 5B,E) or PSD-95 Δ1&2 (Fig. 5C,E) blocked the NMDA receptor-dependent inhibition of D1 internalization in a manner indistinguishable from that induced by the full-length PSD-95 (Fig. 5A,E). In contrast, PSD-95 ΔNT, 1&2 (Fig. 5D) failed to block the NMDA receptor-mediated inhibition of D1 internalization and instead induced D1 internalization patterns similar to those of cells expressing HA-D1 and NMDA receptors in the absence of PSD-95 coexpression (Fig. 5E). These data suggest that both the NT and PDZ1 and -2 domains of PSD-95 are involved in disinhibiting the NMDA receptor-dependent inhibition of D1 internalization.
PSD-95 knockdown enables inhibition of D1 internalization by NMDA receptors
We further investigated the role of PSD-95 in the NMDA receptor-dependent modulation of D1 trafficking in neurons using a receptor internalization assay (Fig. 6). Cultured hippocampal neurons were cotransfected with HA-D1 and sh-PSD-95 or sh-control, live labeled with an anti-HA antibody, stimulated, and processed for differential labeling and measurements of surface and internalized HA-D1 receptors, respectively (Fig. 6A,B). In both control (expressing sh-control) and PSD-95 knockdown (expressing sh-PSD-95) neurons, SKF 81297 (10 μm) alone induced robust HA-D1 internalization, whereas NMDA (50 μm)/glycine (10 μm) alone had no effect (Fig. 6C). The SKF 81297-induced HA-D1 internalization was abolished by the D1 antagonist SCH23390 (10 μm), suggesting that this process was specifically mediated by D1 (data not shown). However, simultaneous stimulation of NMDA receptors during D1 stimulation significantly inhibited the SKF81297-induced D1 internalization in PSD-95 knockdown neurons, but had no significant effect in control neurons (Fig. 6C). These data are consistent with the idea that under normal conditions, a physiological role for PSD-95 is to uncouple modulation of D1 trafficking by NMDA receptors.
Figure 6.

PSD-95 knockdown unmasks NMDA receptor-dependent inhibition of D1 endocytosis in hippocampal neurons. A, B, Agonist-induced D1 internalization in hippocampal neurons cotransfected with HA-D1 and sh-control (A) or sh-PSD-95 (B). HA-D1 internalization was measured by an immunofluorescence-based antibody uptake assay. Representative images of the surface (left column) and internalized (middle column) HA-D1 receptors in sh-control- or sh-PSD-95-transfected neurons (identified by GFP expression, right column) under different stimulation conditions are shown. Surface HA-D1 receptors were live-conjugated with an anti-HA antibody and were allowed to undergo endocytosis (37°C, 30 min) in normal culture medium or medium containing SKF 81297 (10 μm), NMDA (50 μm)/glycine (10 μm), or both. C, Quantification of HA-D1 internalization in neurons. n = 10–15 neurons per group. Data are expressed as mean ± SEM. *p < 0.05; n.s., not significant; two-tailed Student's t tests.
PSD-95 blocks NMDA receptor modulation of D1 signaling
The strength of D1 signaling is typically measured by the level of cAMP produced as a consequence of receptor activation. Because receptor trafficking is an integral regulatory mechanism of GPCR signaling, we hypothesized that PSD-95 may also uncouple D1 cAMP signaling from NMDA receptor modulation. A stable HEK293 cell line constitutively expressing the rhesus macaque D1 receptor (Zhang et al., 2007) was transiently transfected with NMDA receptors in the presence or absence of PSD-95 overexpression. D1 signaling was assessed by dose–response curves of D1-mediated cAMP production in response to various concentrations of SKF 81297 stimulation (Fig. 7). Consistent with a previous study (Pei et al., 2004), activation of NMDA receptors overexpressed in D1-stable cells significantly enhanced the SKF 81297-stimulated, D1-mediated cAMP accumulation (Fig. 7A,C). NMDA receptor activation increased the maximal cAMP levels (Bmax) induced by saturating doses of SKF 81297, but did not significantly alter the EC50 of SKF 81297 (no stimulation: 4.4 ± 2.4 × 10−8 m; stimulation: 6.4 ± 3.2 × 10−8 m; p > 0.05). The potentiation was blocked by the NMDA receptor antagonist MK-801, confirming that NMDA receptor activation was required for this potentiation (Fig. 7A). In contrast, NMDA receptor activation-dependent potentiation of D1 function was absent (or slightly reversed) in D1-stable cells coexpressing both NMDA receptors and PSD-95 (Fig. 7B,C). These results suggest that PSD-95 association with the D1–NMDA heteroreceptor complex abolished the ability of NMDA receptor activation to potentiate D1 cAMP signaling.
Figure 7.

PSD-95 blocks NMDA receptor modulation of D1 cAMP signaling in HEK293 cells. A, Dose–response curves of cAMP production in NR1/NR2B-transfected D1-stable cells. Cells were stimulated with increasing concentrations of SKF 81297 for 30 min. Stimulation of NMDA receptors by NMDA (50 μm)/glycine (10 μm) increased the maximum cAMP accumulation. B, Dose–response curves of cAMP production in D1-stable cells transfected with NR1/NR2B and PSD-95. Stimulation of NMDA receptors failed to increase the maximum cAMP accumulation. C, Summary of D1-mediated cAMP production induced by 10 μm SKF 81297 from independent experiments (n = 3–5). Results are presented in arbitrary units normalized to cAMP levels in cells not stimulated by NMDA/glycine. *p < 0.05; n.s., not significant; two-tailed Student's t tests. D, Dose–response curves of cAMP production in cells transfected with NMDA receptor, or both NMDA receptors and PSD-95, in the absence of NMDA/glycine stimulation. Inset, summary of D1-mediated cAMP production induced by 10 μm SKF81297 from 4 to 7 independent experiments. *p < 0.05 vs D1 stable; #p < 0.05 vs D1/NR; two-tailed Student's t tests. E, Unaltered competition binding curves for D1-stable cells transfected with NR1/NR2B in the presence or absence of PSD-95 cotransfection. F, PSD-95 coexpression did not affect IC50 or the total binding determined at saturating doses. The data represent the mean ± SEM of three independent experiments. Competition binding experiments were performed using 400 pm [3H]SCH23390 and nine concentrations of competing SKF 81297 ranging from 1 pm to 100 μm. Each data point in dose–response curves in A, B, D, and E represents the mean ± SEM of three replications.
To determine whether the PSD-95 abolishment of NMDA receptor modulation of D1 cAMP signaling requires activation of the NMDA receptor per se, we repeated the above experiments in the absence of NMDA receptor stimulation (Fig. 7D). We found that, when transfected into the D1-stable cells, unstimulated NMDA receptors significantly suppressed the SKF 81297-induced, D1-mediated cAMP accumulation (Fig. 7D). This finding is consistent with the current evidence that most physical heteroreceptor interactions lead to mutual inhibitory effects (Ginés et al., 2000; Liu et al., 2000; Hillion et al., 2002; Lee et al., 2002). PSD-95 removed, at least partially, this NMDA receptor-mediated inhibition of D1 signaling in D1-stable cells cotransfeccted with both NMDA receptor and PSD-95. These data indicate that activation of these receptors or their Ca2+-coupled downstream signaling is not necessary for the PSD-95 interference of D1–NMDA receptor interaction.
Finally, we investigated whether PSD-95 might modulate the agonist binding efficacy of the NMDA receptor-associated D1 receptor. Radioligand competition binding experiments were performed to quantify the number as well as agonist binding parameters of D1 in D1-stable cells transfected with NMDA receptors in the presence or absence of PSD-95 coexpression, using the ligand [3H]SCH23390 and increasing concentrations of SKF 81297 (Fig. 7E). PSD-95 overexpression had no effect on either the total number of D1 receptors expressed or the IC50 of SKF 81297 (Fig. 7F), suggesting that the ligand-binding properties of the receptor were not modified by PSD-95. Together, the effects of PSD-95 on NMDA receptor-dependent modulation of D1 signaling are independent of a modification of D1 pharmacological profiles.
PSD-95 knockdown escalates NMDA receptor-dependent D1 cAMP signaling in striatal neurons
The role of endogenous PSD-95 in NMDA receptor-dependent modulation of D1 signaling was examined in cultured striatal neurons infected with lentivirus (Lois et al., 2002) expressing sh-PSD-95. To rule out potential viral-associated side effects, lentivirus expressing sh-control was used as a control. sh-PSD-95 eliminated the vast majority of endogenous PSD-95 in striatal neurons compared with sh-control (Fig. 8A). This represents an underestimate of the knockdown efficiency in infected cells, because our infection efficiency routinely reached 70%. In control neurons, stimulation of the D1-class receptors with SKF 81297 (10 μm, 30 min) increased cAMP accumulation (Fig. 8B). Simultaneous activation of NMDA receptors by NMDA (50 μm)/glycine (10 μm) during D1 stimulation did not further increase, and in fact inhibited, the SKF 81297-induced, D1-mediated cAMP production (Fig. 8B,C), an effect that was blocked by MK-801 (10 μm) (Fig. 8B). This result suggests that an endogenous protective mechanism may exist under normal conditions. Acute PSD-95 knockdown in sh-PSD-95 expressing cultures retained SKF 81297-induced cAMP production activity (Fig. 8B). However, concomitant stimulation of NMDA receptors elicited a significant increase of SKF81297-induced cAMP levels (Fig. 8B,C), which was partially reversed by MK-801 (10 μm) (Fig. 8B). These data support the hypothesis that a normal role for PSD-95 is to prevent excessive potentiation of D1 signaling by NMDA receptors.
Figure 8.
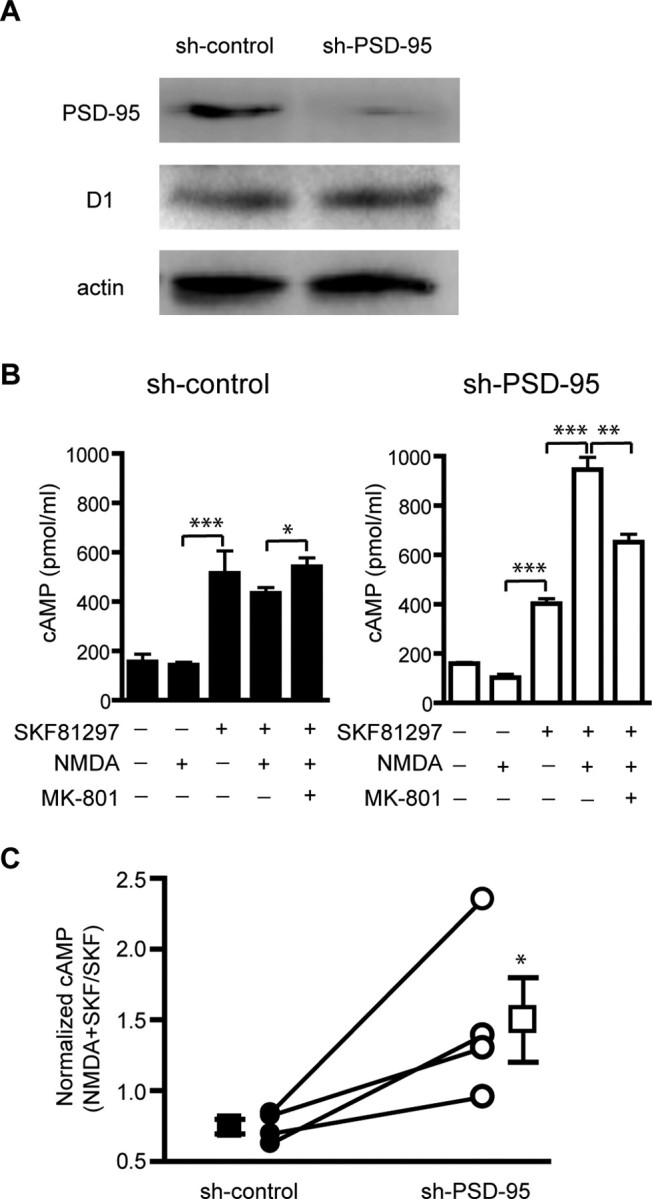
PSD-95 knockdown enhances NMDA receptor modulation of D1 signaling in striatal neurons. A, shRNA silencing of PSD-95 in striatal cultures. Dissociated striatal neurons were infected with sh-PSD-95 or sh-control lentivirus. Protein levels of PSD-95, D1, and actin were analyzed by Western blots. B, D1-mediated cAMP production in striatal neurons infected with sh-PSD-95 or sh-control lentiviruses. Neurons were stimulated (30 min) with combinations of SKF 81297 (10 μm), NMDA (50 μm)/glycine (10 μm), and MK-801 (10 μm) as indicated. Data represent mean ± SEM of three replications. C, Summary of effect of PSD-95 knockdown on NMDA receptor-mediated modulation of D1 cAMP signaling from four independent experiments. The y-axis represents the ratio of SKF 81297/NMDA-induced cAMP to that induced by SKF 81297 alone. Data collected under different infection conditions from the same batch of neuron cultures are connected by a line. *p < 0.05; **p < 0.01; ***p < 0.001; two-tailed Student's t test.
Discussion
In this study, we identified a novel gain of function for the prototypical synaptic scaffold PSD-95 and illustrated a mechanism by which the physical and functional interplay between DA and glutamate systems can be fine-tuned by this glutamatergic scaffold. We show that PSD-95, D1, and NMDA receptors are components of a multiprotein complex. Within the complex, PSD-95 inhibits the physical association between D1 and NMDA receptors and functionally uncouples D1 receptor trafficking and signaling from modulation by NMDA receptors. This PSD-95 interference may represent an effective means to weaken the constitutive D1–NMDA receptor interaction and to prevent this interaction from being abnormally strengthened by DA and/or glutamate during neural activity. Because concomitant overactivation of both D1 and NMDA receptors can be detrimental to functional and structural integrity of neurons, our study illustrates a mechanism by which the D1–NMDA receptor coupling can be dampened to afford neuroprotection (Fig. 9).
Figure 9.

Proposed mechanism by which PSD-95 interferes with D1–NMDA receptor interaction and uncouples modulation of D1 function by the NMDA receptor. A, When activated by DA, stand-alone D1 receptor stimulates adenylate cyclase, increases the production of cAMP, and signals through the PKA/DARPP-32/PP1 cascade to modulate various downstream effectors and substrates, including NMDA receptors. D1 receptors also undergo agonist-induced internalization via the classical dynamin-dependent, vesicle-mediated endocytic pathway (not shown) to regulate the surface availability and responsiveness of the receptor. B, Association with activated NMDA receptors can enhance D1 surface availability and signaling through multiple mechanisms, e.g., inhibition of D1 endocytosis, facilitation of D1 exocytosis (not shown), or allosteric trapping by NMDA receptors (not shown), leading to a reciprocal facilitation of the NMDA receptor through the PKA/DARPP-32/PP1 cascade. C, Presence of PSD-95 in the D1/NMDA receptor complex places a physical as well as a functional (dotted line) barrier between these receptors, weakening their interaction. This barrier could in theory abolish NMDA receptor-dependent D1 surface recruitment, regardless of the mechanisms involved. As a result, D1-mediated cAMP signaling is dampened, NMDA receptor potentiation is suppressed, and the positive D1–NMDA receptor feedback is ultimately antagonized. PSD-95 also promotes constitutive D1 endocytosis, adding another level of inhibition on D1 signaling. Line thickness and arrowhead size indicate signaling strength. Red and blue colors indicate stimulation and inhibition, respectively.
At least three distinct interactions may contribute to the assembly of the D1/PSD-95/NMDA receptor complex: the D1–NR1 interaction mediated by the CTs of these receptors, the D1–PSD-95 interaction mediated by the NT of PSD-95 and CT of D1, and the PDZ-mediated interactions between PSD-95 and NR2 subunits. Our data demonstrate that these protein–protein interactions are not always cooperative to stabilize a multiprotein complex, and, to the contrary, some may even be antagonizing to destabilize formation of the complex. In particular, by associating with both D1 and NMDA receptors, PSD-95 interferes with the interaction between these receptors. Our deletion analyses suggest that the PSD-95 interference is mediated by both the NT and PDZ1 and -2 domains of PSD-95. Although PSD-95 NT may conceivably inhibit NR1 association with the D1 receptor because the two proteins bind an overlapping region on the D1 CT, the mechanism by which the PDZ1 and -2 domains also contribute to the disruption of D1–NMDA receptor complex formation is less clear.
Two mechanisms, perhaps acting in a synergistic manner, may mediate the PSD-95 interference of NMDA receptor modulation of D1 signaling. First, the interference may be achieved through a direct, physical obstruction of D1–NR1 coupling by the presence of PSD-95, independent of intracellular signaling (Fig. 9C). Previous work indicates that the enhancement of D1 cAMP signaling by NMDA receptor activation depends on the physical interaction between the two receptors, because it is abolished by overexpression of mini genes encoding either D1 or NR1 CT fragments that disrupt D1–NR1 interaction (Pei et al., 2004). The ability of PSD-95 to mimic these peptide fragments in inhibiting D1–NR1 association and blocking the suppression of D1 cAMP signaling by unstimulated NMDA receptors suggests that PSD-95 can serve as a simple physical barrier.
A second mechanism by which PSD-95 may interfere with NMDA receptor modulation of D1 signaling is through downstream signaling complexes that may be recruited by PSD-95. These signaling modalities may respond to NMDA receptor-mediated Ca2+ influx and participate in regulation of D1-mediated signaling (Fig. 9C). Indeed, both our in vitro (Fig. 7) and in vivo (Fig. 8) experiments reveal a slight, yet consistent suppression of D1-mediated cAMP signaling in cells expressing D1, PSD-95, and NMDA receptors. This suppression is most likely related to activation of NMDA receptors that subsequently engages the PSD-95-recruited intracellular signaling. More specifically, four different configurations of the D1 receptor, in terms of its association with NMDA receptors and PSD-95, are likely to be differentially distributed across the subcellular compartments of a neuron. These include stand-alone D1 receptors, D1/PSD-95 complexes, D1–NMDA heteroreceptors, and D1/PSD-95/NMDA receptor tertiary complexes. Among these, stand-alone D1 receptors and PSD-95-associated D1 receptors are not sensitive to NMDA receptor activation. In contrast, stimulation of NMDA receptors in D1–NMDA heteroreceptors results in enhanced D1 cAMP signaling, whereas stimulation of D1/PSD-95/NMDA receptor tertiary complexes may not only abolish the enhancement of D1 signaling by NMDA receptor stimulation, but also activate downstream signaling that could be inhibitory to D1-mediated cAMP production. The overall D1-mediated DA signaling of a neuron is thus determined by spatial and temporal summations of signaling mediated by these distinct D1 receptor configurations.
NMDA receptor-mediated D1 signaling enhancement can be explained, at least in part, by surface recruitment/retention of D1 receptors after NMDA receptor stimulation. In both heterologous cells and cultured neurons, NMDA receptor activation recruits D1 receptors to the plasma membrane (Scott et al., 2002; Pei et al., 2004), perhaps via a SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor)-dependent mechanism (Pei et al., 2004). In cultured striatal slices, ligand-occupied NMDA receptors have been shown to recruit laterally diffusing D1 receptors to dendritic spines through a diffusion-trap mechanism (Scott et al., 2006). Here, we found that NMDA receptors also immobilize D1 receptors on the cell surface. In all three cases, interestingly, stimulation of NMDA receptors is necessary for their action (but see Fiorentini et al., 2003). In this context, it is of interest to note that stimulated and nonstimulated NMDA receptors exert opposite modulations on D1 signaling in cells coexpressing D1 and NMDA receptors. That is, stimulated NMDA receptors potentiate, whereas nonstimulated receptors inhibit D1-mediated cAMP signaling. The mechanism(s) underlying this inhibition is currently unknown, but could involve downregulation of total D1 receptor expression, suppression of surface D1 receptor level, alteration of D1 pharmacological profiles, and/or clamp of D1 at a less effective state (e.g., via conformational changes). Regardless of the mechanisms, it appears that ligand occupancy of NMDA receptors can release this “clamp” through, for example, an allosteric conformational change of the receptor that leads to increased D1–NMDA receptor association (Pei et al., 2004; Scott et al., 2006), which can be further fine-tuned by PSD-95. This may represent a delicate switch by which glutamate can gate D1 signaling through opening D1-associated NMDA receptors.
Activation of D1 receptors is long recognized to enhance NMDA receptor-mediated responses in the cortex (Cepeda et al., 1992; Seamans et al., 2001; Gonzalez-Islas and Hablitz, 2003; Chen et al., 2004) and striatum (Cepeda et al., 1993; Blank et al., 1997; Snyder et al., 1998; Flores-Hernández et al., 2002), involving primarily the cAMP/PKA/DARPP-32/PP1 cascade. Together with the reciprocal facilitation of D1 receptor function as a consequence of NMDA receptor activation, a positive feedback would be created that, if left uncontrolled, could result in overactivation of both D1 and NMDA receptors. Excessive activation of the NMDA receptor mediates excitotoxicity associated with neurodegenerative diseases and traumatic brain injuries (Choi, 1988; Lipton and Rosenberg, 1994). Elevated DA tone is also neurotoxic, contributing to the degeneration of postsynaptic DA-receiving neurons, via receptor-independent mechanisms involving oxidative stress-induced apoptosis and largely undefined receptor-dependent mechanisms (Cyr et al., 2003; Bozzi and Borrelli, 2006). Among all DA targets, the striatum is the most densely innervated and a particularly susceptible region for degeneration. We showed that NMDA receptor activation failed to enhance, and in fact suppressed, the D1 agonist-induced cAMP signaling in normal striatal neurons but increased this response in PSD-95 knockdown striatal neurons. Interestingly, activation of NMDA receptors enhances D1 function in hippocampal cultures (Pei et al., 2004). These studies suggest that the positive coupling between D1 and NMDA receptor is normally under control, at least in the striatum, and that the PSD-95 interference may represent such a negative control mechanism in this region.
The PSD-95-mediated interference reported here provides an efficient and novel means to constitutively dampen excessive D1–NMDA receptor stimulation. Another pharmacologically distinct family of receptors that mediates DA actions is the D2-class receptors (D2, D3, and D4). These receptors couple to Gi/Goα and inhibit the cAMP/PKA/DARPP-32/PP1 cascade (Missale et al., 1998; Greengard et al., 1999). Some of these receptors, e.g., the predominant D2 subtype, are localized in dendritic spines (for review, see Yao et al., 2008). Activation of D2-class receptors could therefore counteract the D1–NMDA receptor coupling by antagonizing the D1-mediated cAMP cascade and reducing NMDA receptor responses, providing a classical case of neuroprotection against DA-related neurological diseases (Bozzi and Borrelli, 2006). However, despite evidence of overlapping expressions of D1- and D2-class receptors in individual neurons (Gaspar et al., 1995; Vincent et al., 1995; Surmeier et al., 1996), colocalization of receptors from both classes in the same spines remains to be seen. Moreover, D1 and D2 receptors are segregated into different populations of neurons in the striatum, with D1 present in the striatonigral neurons of the direct pathway and D2 in the striatopallidal neurons of the indirect pathway of the basal ganglion circuitry (Gerfen, 1992). In this context, the PSD-95-mediated interference identified in our study could protect synapses to which D2 receptors are not targeted and in neurons in which D2 receptors are not expressed, thus substituting the classical D2-mediated neuroprotection. This interference mechanism could operate in essentially every synapse in which D1 and NMDA receptors are colocalized, provided that PSD-95 is sufficiently abundant to occupy these synapses, and can be more dominant in cells that express few D2 receptors, such as the striatonigral neurons in the striatum.
Footnotes
This work was supported by National Institutes of Health Grants RR00168 [New England Primate Research Center (NEPRC)], NS39793 (O.I.), DA021420, and NS057311, a National Alliance for Research on Schizophrenia and Depression Young Investigator Award, and a Williams F. Milton Fund of Harvard University (W.-D.Y.). We thank Drs. Roger Nicoll for the PSD-95 shRNA construct, Fang Liu for GST D1 CT plasmids, and Tadashi Yamamoto for the PSD-95 mutant cDNA lacking PDZ1 and -2. We thank Dr. Roger Spealman and members of the Division of Neuroscience at NEPRC for helpful discussions.
References
- Aoki C, Miko I, Oviedo H, Mikeladze-Dvali T, Alexandre L, Sweeney N, Bredt DS. Electron microscopic immunocytochemical detection of PSD-95, PSD-93, SAP-102, and SAP-97 at postsynaptic, presynaptic, and nonsynaptic sites of adult and neonatal rat visual cortex. Synapse. 2001;40:239–257. doi: 10.1002/syn.1047. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A. 2006;103:19535–19540. doi: 10.1073/pnas.0608492103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Teichert U, Kügler H, Behrsing H, Fienberg A, Greengard P, Spiess J. The phosphoprotein DARPP-32 mediates cAMP-dependent potentiation of striatal N-methyl-D-aspartate responses. Proc Natl Acad Sci U S A. 1997;94:14859–14864. doi: 10.1073/pnas.94.26.14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? Trends Neurosci. 2006;29:167–174. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Hippocampal afferents to the rat prefrontal cortex: synaptic targets and relation to dopamine terminals. J Comp Neurol. 1996;369:1–15. doi: 10.1002/(SICI)1096-9861(19960520)369:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE. 2006:pe20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Radisavljevic Z, Peacock W, Levine MS, Buchwald NA. Differential modulation by dopamine of responses evoked by excitatory amino acids in human cortex. Synapse. 1992;11:330–341. doi: 10.1002/syn.890110408. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Cyr M, Beaulieu JM, Laakso A, Sotnikova TD, Yao WD, Bohn LM, Gainetdinov RR, Caron MG. Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc Natl Acad Sci U S A. 2003;100:11035–11040. doi: 10.1073/pnas.1831768100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci. 2001;21:5546–5558. doi: 10.1523/JNEUROSCI.21-15-05546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Fiorentini C, Gardoni F, Spano P, Di Luca M, Missale C. Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-D-aspartate receptors. J Biol Chem. 2003;278:20196–20202. doi: 10.1074/jbc.M213140200. [DOI] [PubMed] [Google Scholar]
- Flores-Hernández J, Cepeda C, Hernández-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Freund TF, Powell JF, Smith AD. Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience. 1984;13:1189–1215. doi: 10.1016/0306-4522(84)90294-x. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Ginés S, Hillion J, Torvinen M, Le Crom S, Casadó V, Canela EI, Rondin S, Lew JY, Watson S, Zoli M, Agnati LF, Verniera P, Lluis C, Ferré S, Fuxe K, Franco R. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci U S A. 2000;97:8606–8611. doi: 10.1073/pnas.150241097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Leranth C, Williams SM, Mons N, Geffard M. Dopamine synaptic complex with pyramidal neurons in primate cerebral cortex. Proc Natl Acad Sci U S A. 1989;86:9015–9019. doi: 10.1073/pnas.86.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Hablitz JJ. Dopamine enhances EPSCs in layer II-III pyramidal neurons in rat prefrontal cortex. J Neurosci. 2003;23:867–875. doi: 10.1523/JNEUROSCI.23-03-00867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Hara Y, Pickel VM. Overlapping intracellular and differential synaptic distributions of dopamine D1 and glutamate N-methyl-D-aspartate receptors in rat nucleus accumbens. J Comp Neurol. 2005;492:442–455. doi: 10.1002/cne.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, Canela EI, Zoli M, Agnati LF, Ibanez CF, Lluis C, Franco R, Ferre S, Fuxe K. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem. 2002;277:18091–18097. doi: 10.1074/jbc.M107731200. [DOI] [PubMed] [Google Scholar]
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Lachowicz JE, Sibley DR. Molecular characteristics of mammalian dopamine receptors. Pharmacol Toxicol. 1997;81:105–113. doi: 10.1111/j.1600-0773.1997.tb00039.x. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chéry N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;111:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Liu F, Wan Q, Pristupa ZB, Yu XM, Wang YT, Niznik HB. Direct protein-protein coupling enables cross-talk between dopamine D5 and gamma-aminobutyric acid A receptors. Nature. 2000;403:274–280. doi: 10.1038/35002014. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Lee FJ, Moszczynska A, Vukusic B, Liu F. Regulation of dopamine D1 receptor function by physical interaction with the NMDA receptors. J Neurosci. 2004;24:1149–1158. doi: 10.1523/JNEUROSCI.3922-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickel VM, Colago EE, Mania I, Molosh AI, Rainnie DG. Dopamine D1 receptors co-distribute with N-methyl-D-aspartic acid type-1 subunits and modulate synaptically-evoked N-methyl-D-aspartic acid currents in rat basolateral amygdala. Neuroscience. 2006;142:671–690. doi: 10.1016/j.neuroscience.2006.06.059. [DOI] [PubMed] [Google Scholar]
- Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–263. doi: 10.1016/s0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- Scott L, Kruse MS, Forssberg H, Brismar H, Greengard P, Aperia A. Selective up-regulation of dopamine D1 receptors in dendritic spines by NMDA receptor activation. Proc Natl Acad Sci U S A. 2002;99:1661–1664. doi: 10.1073/pnas.032654599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L, Zelenin S, Malmersjö S, Kowalewski JM, Markus EZ, Nairn AC, Greengard P, Brismar H, Aperia A. Allosteric changes of the NMDA receptor trap diffusible dopamine 1 receptors in spines. Proc Natl Acad Sci U S A. 2006;103:762–767. doi: 10.1073/pnas.0505557103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezuka T, Umemori H, Akiyama T, Nakanishi S, Yamamoto T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc Natl Acad Sci U S A. 1999;96:435–440. doi: 10.1073/pnas.96.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtschanoff JG, Burette A, Wenthold RJ, Weinberg RJ. Expression of NR2 receptor subunit in rat somatic sensory cortex: synaptic distribution and colocalization with NR1 and PSD-95. J Comp Neurol. 1999;410:599–611. [PubMed] [Google Scholar]
- Vincent SL, Khan Y, Benes FM. Cellular colocalization of dopamine D1 and D2 receptors in rat medial prefrontal cortex. Synapse. 1995;19:112–120. doi: 10.1002/syn.890190207. [DOI] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41:625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- Yao WD, Spealman RD, Zhang J. Dopaminergic signaling in dendritic spines. Biochem Pharmacol. 2008;75:2055–2069. doi: 10.1016/j.bcp.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Vinuela A, Neely MH, Hallett PJ, Grant SG, Miller GM, Isacson O, Caron MG, Yao WD. Inhibition of the dopamine D1 receptor signaling by PSD-95. J Biol Chem. 2007;282:15778–15789. doi: 10.1074/jbc.M611485200. [DOI] [PMC free article] [PubMed] [Google Scholar]