

Abstract
Little is known about key pathological events preceding overt neuronal degeneration in Parkinson's disease (PD) and α-synucleinopathy. Recombinant adeno-associated virus 2-mediated delivery of mutant (A53T) human α-synuclein into the substantia nigra (SN) under a neuron-specific synapsin promoter resulted in protracted neurodegeneration with significant dopaminergic (DA) neuron loss by 17 weeks. As early as 4 weeks, there was an increase in a dopamine metabolite, DOPAC and histologically, DA axons in the striatum were dystrophic with degenerative bulbs. Before neuronal loss, significant changes were identified in levels of proteins relevant to synaptic transmission and axonal transport in the striatum and the SN. For example, striatal levels of rabphilin 3A and syntaxin were reduced. Levels of anterograde transport motor proteins (KIF1A, KIF1B, KIF2A, and KIF3A) were decreased in the striatum, whereas retrograde motor proteins (dynein, dynamitin, and dynactin1) were increased. In contrast to reduced levels in the striatum, KIF1A and KIF2A levels were elevated in the SN. There were dramatic changes in cytoskeletal protein levels, with actin levels increased and α-/γ-tubulin levels reduced. In addition to these alterations, a neuroinflammatory response was observed at 8 weeks in the striatum, but not in the SN, demonstrated by increased levels of Iba-1, activated microglia and increased levels of proinflammatory cytokines, including IL-1β, IFN-γ and TNF-α. These results demonstrate that changes in proteins relevant to synaptic transmission and axonal transport coupled with neuroinflammation, precede α-synuclein-mediated neuronal death. These findings can provide ideas for antecedent biomarkers and presymptomatic interventions in PD.
Introduction
α-Synuclein has been extensively studied in the pathogenesis of Parkinson's disease (PD). Aggregation of α-synuclein in intracytoplasmic Lewy bodies is a key pathological feature of both sporadic and familial PD. In addition, duplication, triplication or mutations in the α-synuclein gene cause some forms of familial PD (Singleton et al., 2003). Several groups have reported that viral delivery of human wild type or mutant (A53T or A30P) α-synuclein in rat causes progressive loss of dopaminergic (DA) neurons over 3 to 8 weeks (Kirik et al., 2002; Lo Bianco et al., 2002; Gorbatyuk et al., 2008). It is, however, not yet clear how pathological forms or levels of α-synuclein modify the function of neurons affected.
Recent studies suggest that axonal transport disruption and axonal degeneration precede neuronal death, and may be causal to disease progression in neurodegenerative diseases including Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD) (Li et al., 2001; Fischer et al., 2004; Stokin et al., 2005). In PD, axonal pathology has been observed in postmortem patient brain samples (Galvin et al., 1999) and a few in vitro studies have suggested that α-synuclein inhibits anterograde transport (Saha et al., 2004) and MPP+ toxicity inhibits anterograde and increases retrograde axonal transport (Morfini et al., 2007). The role of neuroinflammation in disease progression in PD (Chen et al., 2005) and animal models has been highlighted by recent studies. For example, in the presence of a pre-existing PD-relevant insult, including 6-OHDA, loss of parkin or α-synuclein overexpression, induction of neuroinflammation synergistically worsens the disease process (Sánchez-Pernaute et al., 2004; Frank-Cannon et al., 2008; Gao et al., 2008; Godoy et al., 2008; Koprich et al., 2008). However, whether α-synuclein overexpression itself can cause neuroinflammation before neuronal degeneration in vivo, has not been clearly demonstrated.
In this study, we have used a model of slow α-synucleinopathy in rat using the recombinant adeno-associated virus 2 (AAV2)-mediated nigral delivery of A53T α-synuclein under a neuron-specific synapsin promoter. This model exhibits a protracted cell death pattern where DA neuronal death was observed at 17 weeks with loss of striatal dopamine by 24 weeks, much slower degeneration than other viral α-synucleinopathy models. Moreover, use of the synapsin promoter limited the effect of α-synuclein to neurons, enabling us to assess the effect of α-synuclein overexpression in neurons by excluding the potential action of α-synuclein in surrounding glial cells. Taking advantage of the protracted neurodegenerative process, we investigated predegenerative changes (at 4 weeks and 8 weeks) long before cell death and striatal dopamine loss, aiming to identify pathogenic changes that are most likely causal to the disease, and not a by-product of the cell death process. Our findings reveal that before neuronal loss, A53T α-synuclein overexpression causes (1) dystrophic and bulging axons; (2) an increase in the striatal dopamine metabolite DOPAC and the ratio of DOPAC/dopamine; (3) dynamic changes in levels of proteins involved in synaptic transmission and axonal transport, implicating disruption in these cellular processes, and (4) neuroinflammation in the striatum marked by activated microglia and an increase in cytokine levels.
Materials and Methods
Recombinant adeno-associated virus 2 preparation.
The AAV2-A53T-α-synuclein vector contains the coding sequence for the human α-synuclein gene under the control of a synapsin promoter. The virus was produced by Harvard Gene Therapy Initiative. The final titer for the vector encoding A53T-α-syn was 1.8 × 1012 genome copies/ml, and green fluorescent protein (GFP), 1.5 × 1012 as determined by dot blot.
AAV2 nigral injection.
Female Sprague-Dawley rats weighing ∼280 g (Charles River Laboratories) were used in the animal experiments. These experiments had previously been approved by the McLean Hospital Institutional Animal Care and Use Committee. Stereotaxic coordinates for the surgeries were taken from the “Rats Atlas” by Paxinos and Watson (1986). Before surgery, the animals were anesthetized with xylazine and ketamine (3 mg/kg and 60 mg/kg, respectively). The animals were placed in a stereotaxic frame (Stoelting), where a 10 μl Hamilton syringe and 31 gauge needles were used as a delivery system. All injections were made into the substantia nigra using the following anteroposterior (AP), mediolateral (ML), and dorsoventral (DV) coordinates: first site, AP = −4.8 mm; ML = −2.0 mm; DV = −7.2 mm relative to dura, second site, AP = −5.5 mm; ML = −1.9 mm, DV = 7.0 mm relative to dura; toothbar (TB) set at −3.3 mm. Two microliters of AAV GFP or A53T α-synuclein was injected per site at a rate of 0.5 μl/min using microinfusion pumps (Stoelting), with a wait time of 10 min between injection.
Tissue preparation for immunostaining.
Animals were terminally anesthetized with an overdose of sodium pentobarbital (150 mg/kg, i.p.), and perfused intracardially with heparinized saline (0.1% heparin in 0.9% saline) followed by paraformaldehyde (4% in PBS). The brains were removed and postfixed for 8 h in 4% paraformaldehyde solution. Following postfixation, the brains were equilibrated in 20% sucrose in PBS, sectioned at 40 μm on a freezing microtome, and serially collected in PBS.
Immunohistochemistry.
All immunohistochemistry was performed on randomly selected series of sections. Sections were treated for 5 min in 3% hydrogen peroxide (Humco), washed three times in PBS, and incubated in 10% normal goat serum (NGS) and 0.2% Triton X-100 in PBS (PBS-T) for 1 h before overnight incubation at 4°C with the primary antibody diluted in 10% NGS and PBS-T. The primary antibodies used were rabbit anti-tyrosine hydroxylase (TH) (Pel Freez; 1:1000) and mouse anti-human α-synuclein (LB509, Zymed, 1:500). For light microscopy, biotinylated secondary antibodies (Vector Laboratories; 1:300) were used, followed by incubation in streptavidin-biotin complex (Vectastain ABC Kit Elite, Vector Laboratories) for 1 h at room temperature and visualized by incubation in 3,3′-diaminobenzidine (DAB) solution (Vector Laboratories). For immunofluorescence, actin (1:500, Sigma) or phalloidin (1:100, Molecular Probe) was used with Alexa 488, 564, and 647 as secondary antibodies (1:500, Invitrogen) for 1 h at room temperature. Confocal analysis was performed using a Zeiss LSM510/Meta station.
Tissue preparation for Western blot analysis.
Animals were terminally anesthetized the same way described above and perfused intracardially with heparinized saline (0.1% heparin in 0.9% saline). Striatum and substantia nigra (SN) were hand-dissected on ice-cold surface, assisted by a tissue chopper. Striatal tissue samples were homogenized using hand-held Polytron homogenizer in ice-cold TEVP buffer, pH 7.4 [10 mm Tris–HCl, 5 mm NaF, 1 mm Na3VO4, 1 mm EDTA, 1 mm EGTA, 320 mm sucrose and protease inhibitors (Sigma P8340, 1:100)]. The homogenate was centrifuged at 800 × g to separate P1 (nuclear and debris) and supernatant (S1) fraction. S1 fraction was centrifuged at 9200 × g to separate P2 (membranous fraction) and S2 (cytosolic fraction). Protein concentrations were determined by BCA assay (Pierce).
Western blot analysis.
10∼20 μg of protein from the tissue preparations were used to run a gel. The respective volumes were mixed 1:1 with sample buffer and then boiled for 5 min. After boiling, the samples were loaded into the Criterion precast 4∼15%, 10%, or 12.5% SDS polyacrylamide gel system (Bio-Rad). After the electrophoresis was completed, the proteins were transferred from the gel to a PVDF membrane electrically at 80 V for 1 h. Then, the membranes were washed in Tris-buffered saline with 0.1% Tween 20 (TBS-T). After at least 1 h of blocking in 5% nonfat dry milk, the membranes were incubated overnight at 4°C in various primary antibodies [α-synuclein (clone 42, BD Transduction Laboratories; 1:2000), GFP (Invitrogen; 1:5000) TH (Pel Freeze; 1:3000), dopamine transporter (DAT, Millipore Bioscience Research Reagents; 1:2000), Vesicular Monoamine Transporter 2 (VMAT2, Pel Freeze; 1:1000), SNAP-25 (Millipore Bioscience Research Reagents; 1:4000), Rabphilin3A (BD Transduction Laboratories; 1:2000), RAB3A (Affinity Bioreagent; 1:3000), syntaxin (Millipore Bioscience Research Reagents; 1:2000), synaptophysin (Santa Cruz; 1:500), synaptotagmin (BD Transduction Laboratories; 1:5000), synapsin (Millipore Bioscience Research Reagents; 1:5000), Munc-18 (Affinity Bioreagents; 1:3000), KIF1A (clone 16, BD Transduction Laboratories; 1:2000), KIF1B (Bethyl Laboratory; 1:2000), KIF2A (Abcam; 1:10,000), KIF3A (Abcam; 1:2000), KIF5 (Abcam; 1:1000), KIF17 (Abcam; 1:500), myosin Va (Sigma; 1:500), dynein (clone 74.1, Millipore Bioscience Research Reagents; 1:2000), dynamitin (Millipore Bioscience Research Reagents; 1:2000), dynactin1 (Novus,1:1000), α-tubulin (clone DM1A, Upstate; 1:5000), β-tubulin (Upstate; 1:5000), γ-tubulin (clone GTU-88, Sigma; 1:5000), Actin (clone C-2, Santa Cruz; 1:500), neurofilament 160 (clone NN18, Sigma; 1:1000), neurofilament 200 (clone N52, Sigma; 1:1000), and GAPDH (Millipore Bioscience Research Reagents; 1:5000)]. Then, the membranes were incubated in HRP-conjugated secondary antibodies for 1 h at room temperature. After washing with TBS-T, ECL or ECL plus (Amersham Biosciences) was used to probe for immunoreactive bands and exposed to film using the Kodak Biomax film system. Optical density analysis (NIH ImageJ) was used to determine the relative abundance of protein in each sample.
Cell counting.
Quantification of TH-positive neuronal number within the SN was performed using Stereo Investigator software (MBF Bioscience) and stereologic principles as previously described (Chung et al., 2007). Briefly, the anterior and posterior boundaries of the SN included in the analysis were defined according to the area transduced by the rAAV-eGFP in preliminary experiments [∼−4.80 mm through −6.00 mm from bregma (according to the rat brain atlas of Paxinos and Watson, 1986)]. Stereology was performed using a Zeiss Axiovert microscope coupled to an Optronics Microfire digital camera for visualization of tissue sections. The coefficients of error were calculated according to the procedure of West (1993), and values <0.10 were accepted.
HPLC analysis.
Animals were terminally anesthetized the same way as described above and perfused intracardially with heparinized saline (0.1% heparin in 0.9% saline). Small pieces of dorsolateral striatum were hand-dissected on ice-cold surface, assisted by a tissue chopper. Tissue pieces were sent to CMN/KC Neurochemistry Core Laboratory in Vanderbilt University for HPLC analysis. The brain sections were homogenized in 200–750 μl of 0.1 m TCA, which contains 10−2 m sodium acetate, 10−4 m EDTA and 10.5% methanol, pH 3.8, using a tissue dismembrator (Fisher Scientific). Samples were spun in a microcentrifuge at 10,000 × g for 20 min. The supernatant was removed and stored at –80 degrees. The pellet was saved for protein analysis. Supernatant was then thawed and spun for 20 min. Samples of the supernatant were then analyzed for biogenic monoamines and/or amino acids. Biogenic amines were determined by a specific HPLC assay using an Antec Decade II (oxidation: 0.5) electrochemical detector operated at 33°C. Twenty microliter samples of the supernatant were injected using a Water 717+ autosampler onto a Phenomenex Nucleosil (5 μ, 100A) C18 HPLC column (150 × 4.60 mm). Biogenic amines were eluted with a mobile phase consisting of 89.5% 0.1 m TCA, 10−2 m sodium acetate, 10−4 m EDTA and 10.5% methanol, pH 3.8. Solvent was delivered at 0.8 ml/min using a Waters 515 HPLC pump. Using this HPLC solvent the following biogenic amines eluted in the following order: noradrenaline, Adrenaline, DOPAC, dopamine, 5-HIAA, HVA, 5-HT, and 3-MT. HPLC control and data acquisition were managed by Waters Empower software.
Multiplex ELISA.
Tissue samples were collected and suspended in lysis buffer (TPER). In addition, phosphatase inhibitors I-II (1:100) and protease inhibitors (1:100) were added fresh before cell lysis (Sigma-Aldrich, P2850, P5276, and P8340 respectively). Following cell lysis, the homogenate was centrifuged, a portion of the supernatant was reserved for protein determination (BCA Assay, Pierce) and the remaining stored at −20°C. Samples were analyzed for the simultaneous detection of cytokines, chemokines and receptors using a multiplex ELISA based format and performed in duplicate. Testing was performed independently through the Searchlight Testing Service (Pierce, ThermoFisher Scientific).
Results
A rat model of α-synucleinopathy using AAV2 mediated A53T α-synuclein overexpression under a neuron-specific synapsin promoter
Using AAV2 mediated A53T α-synuclein delivery under a neuron-specific synapsin promoter, we established a model of α-synucleinopathy in rat. In this model, the majority of DA neurons in the SN were transduced with human A53T α-synuclein, demonstrated by human α-synuclein-specific antibody staining (Fig. 1A–E). This resulted in robust human α-synuclein-positive staining of the entire dorsolateral striatum by 4 weeks post AAV injection (Fig. 1F). Intensity of the human α-synuclein-positive staining in the striatum was maintained till 17 weeks but reduced by 24 weeks due to degeneration of DA terminals (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). The intensity of the AAV GFP staining in the striatum, however, was maintained until 24 weeks (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material), demonstrating that the transgene expression by AAV2 is robust and maintained until 24 weeks. While there was no reduction of TH-positive neurons and no apparent loss of presynaptic terminals in 4 and 8 weeks as determined by immunohistochemistry and Western blot analysis (Fig. 1J; supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), there was extensive fiber pathology including dystrophic and bulging neurites in the striatum of A53T α-synuclein injected rats (Fig. 1G,H). AAV A53T α-synuclein transduction produced a 32 ± 8% and a 56.3 ± 3% loss of DA neurons at 17 and 24 weeks, respectively, compared with AAV GFP injection (Fig. 1I,J). Reduction of TH-positive neurons at 24 weeks was accompanied by the loss of NeuN-positive cells, demonstrating that neuronal death has occurred by this time (supplemental Fig. 1E,F, available at www.jneurosci.org as supplemental material). Loss of striatal DA tissue content was only accompanied at 24 weeks (Fig. 1K). Striatal tissue DOPAC content was increased by A53T α-synuclein overexpression at 4, 8, and 17 weeks (Fig. 1L). The DOPAC content, however, was reduced at 24 weeks (Fig. 1L) probably due to significant loss of DA terminals (Fig. 1K). The ratios of DOPAC/dopamine were increased at 8, 17, and 24 weeks (Fig. 1M), suggesting an increase in dopamine turnover.
Figure 1.

AAV mediated A53T α-synuclein overexpression. A–E, At 4 weeks after AAV injection, human α-synuclein expression was detected in the SN area and was colocalized with TH-positive neurons. F, Human α-synuclein was highly expressed in the dorsolateral striatum where transduced DA neurons project their synaptic terminals. G, H, Many α-synuclein overexpressing axons showed swollen and bulging morphology compared with GFP expressing axons in the control striatum starting at 4 weeks and maintained till later stages (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). I, J, A53T α-synuclein overexpression resulted in a loss of TH-positive cells at 17 (32 ± 8%) and 24 (56.3 ± 3%) weeks compared with GFP overexpression. K, L, HPLC analysis in the striatal tissue demonstrated DOPAC content was significantly increased in the A53T α-synuclein overexpressing striatum at 4, 8, and 17 weeks (L) without changing dopamine (DA) content at these times (K). Striatal DA content was reduced only at 24 weeks after A53T α-synuclein overexpression (K). M, DOPAC/DA ratios were increased in A53T α-synuclein overexpressing striatum at 8, 17, and 24 weeks. Data are shown as means ± SEM (AAV GFP, n = 10∼12; AAV α-synuclein, n = 12∼15; *p < 0.05; **p < 0.01 two-tail t test).
Four weeks post AAV2 A53T α-synuclein injections
Since α-synuclein is highly expressed in presynaptic terminals and its overexpression causes axon pathology, we investigated the levels of various presynaptic proteins in the striatum by Western blot analysis. At 4 weeks post AAV A53T α-synuclein injection, a majority of synaptic proteins measured including synaptophysin, synapsin, synaptotagmin, syntaxin1a, munc-18, and RAB3A remained unchanged (Fig. 2A,B). However, levels of Rabphilin 3A, were mildly but significantly increased by 4 weeks (Fig. 2A,B). Since axonal transport defects have previously been shown to be causal to the type of axonal abnormalities observed here (Stokin et al., 2005), we investigated the expression levels of transport motor and cytoskeletal proteins in the striatum overexpressing GFP or A53T α-synuclein. Among anterograde transport microtubule motor proteins, KIF3A levels were mildly but significantly increased whereas KIF17 levels were significantly reduced (Fig. 2C,D). Levels of the retrograde transport microtubule motor protein, dynein were significantly decreased by A53T α-synuclein overexpression (Fig. 2C,D) while levels of dynamitin, a part of the dynactin complex which activates dynein function, was not altered (Fig. 2C,D). Levels of myosin Va, an actin based motor protein, were slightly increased (Fig. 2C,D). Among cytoskeletal proteins, actin levels were significantly increased in the α-synuclein overexpressing striata (Fig. 2C,D).
Figure 2.

Striatal changes in the levels of proteins involved in synaptic transmission and axonal transport at 4 weeks after AAV α-synuclein injection. Western blot was performed using membrane fraction (P2) of striatal lysate to measure levels of proteins involved in synaptic transmission. Among these proteins, rabphilin 3A levels were mildly increased (A, B). Western blot was performed using total lysate (T) of the striatum to measure levels of protein involved in axonal transport. KIF3A, myosin Va and actin levels were increased, whereas KIF17 and dynein levels were reduced (C, D). Optical densities of the individual bands were quantified using NIH ImageJ. Optical densities of α-synuclein overexpressing conditions were normalized by the averaged value of GFP expressing condition. Data are shown as mean ± SEM (AAV GFP, n = 4∼6; AAV A53T α-synuclein, n = 4∼6; *p < 0.05 two tail t test).
Eight weeks post AAV2 A53T α-synuclein injections
The presynaptic protein, rabphilin3A, which was increased at 4 weeks, was reduced by 8 weeks (Fig. 3A,B). Syntaxin levels were also reduced by A53T α-synuclein overexpression at this time (Fig. 3A,B). Overall, most of the synaptic proteins examined were not changed at 8 weeks. In contrast to presynaptic proteins, overexpression of A53T α-synuclein produced marked and extensive changes in the levels of transport motor and cytoskeletal proteins (Fig. 3C,D). Microtubule based motor proteins including KIF1A, KIF1B, KIF2A, and KIF3A showed dramatic reductions in expression levels, whereas levels of KIF5 remained unchanged. KIF17 levels, which were reduced by 4 weeks, were no longer different from the striatal levels of GFP expressing controls. Myosin Va, which was slightly increased at 4 weeks, was reduced by 8 weeks. In contrast to the anterograde transport motor proteins, levels of retrograde transport proteins including dynein, dynactin1 (p150Glued) and dynamitin (p50) were increased by 8 weeks.
Figure 3.

Striatal changes in the levels of proteins involved in synaptic transmission and axonal transport at 8 weeks post AAV α-synuclein injection. Western blot was performed using membrane fraction (P2) of striatal lysate to measure levels of proteins involved in synaptic transmission. Among these proteins, rabphilin 3A and syntaxin levels were reduced (A, B). Western blot was performed using total lysate (T) of the striatum to measure levels of protein involved in axonal transport. Levels of anterograde transport motor proteins including KIF1A, KIF1B, KIF2A, KIF3A and myosin Va were markedly decreased, whereas levels of retrograde transport motor proteins including dynein, dynamitin and dynactin1 were dramatically increased (C, D). Levels of cytoskeletal proteins such as actin, neurofilaments, α-tubulin and γ-tubulin, were altered (C, D). Optical densities of the individual bands were quantified using NIH ImageJ. Optical densities of α-synuclein overexpressing conditions were normalized by the averaged value of GFP expressing condition. Data are shown as mean ± SEM (AAV GFP, n = 4∼6; AAV A53T α-synuclein, n = 4∼6; *p < 0.05 two tail t test).
Cytoskeletal proteins were dramatically altered by A53T α-synuclein overexpression at 8 weeks. α-tubulin and γ-tubulin levels were strikingly reduced, whereas β-tubulin levels were not altered by A53T α-synuclein overexpression. One of the intermediate filaments, neurofilament 160 exhibited increased expression in the A53T α-synuclein overexpressing group. The increase in actin levels seen at 4 weeks was augmented by 8 weeks post AAV injection. Triple (human α-synuclein/TH/actin) immunostaining results revealed corresponding increased staining of actin in α-synuclein positive DA terminals (Fig. 4A). The actin increase was most prominent in dystrophic and swollen axons (Fig. 4A). These bulging axons were also positive for phalloidin, suggesting an accumulation of F-actin (Fig. 4B).
Figure 4.
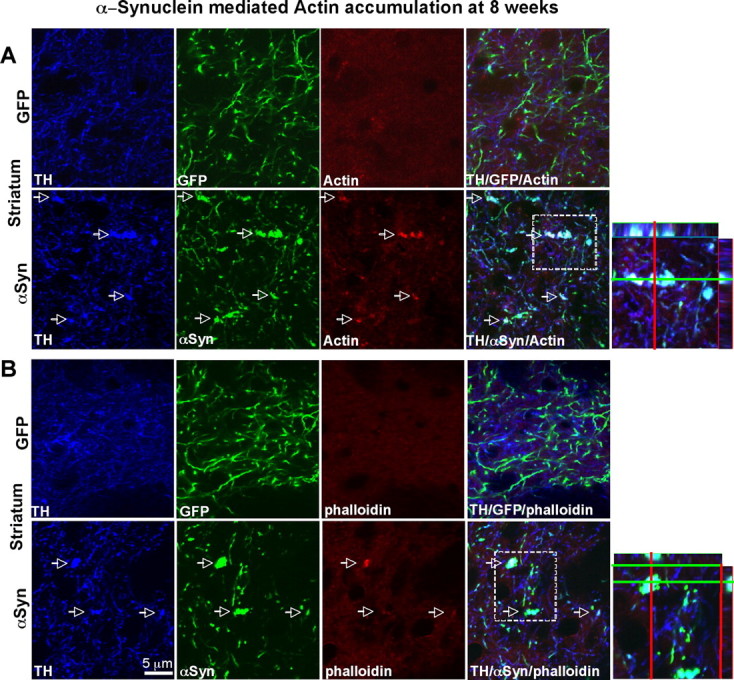
An increase in actin levels in α-synuclein overexpressing axons in the striatum. A, An increase in actin immunoreactivity colocalized with α-synuclein staining. B, Swollen and bulging parts of axons visualized with α-synuclein staining showed stronger staining of actin and phalloidin, suggesting the increase in F-actin levels.
In contrast to the results in the striatum, the expression levels of most of these proteins were not altered in the SN (Fig. 5A,B), suggesting that the decreased protein levels seen in the striatum was not a result of general reduction of protein production in the cell body, but most likely due to trafficking disruption. In addition, levels of KIF1A and KIF2A were significantly increased in the A53T α-synuclein overexpressing SN, indicating that transport of KIF1A and KIF2A to the presynaptic terminals was disrupted, causing accumulation of these proteins in the SN (Fig. 5A,B).
Figure 5.

Nigral changes in proteins involved in axonal transport at 8 weeks post AAV α-synuclein injection. Western blot was performed using total lysate (T) of the SN to measure levels of protein involved in axonal transport. Levels of KIF1A and KIF1B were increased in the α-synuclein overexpressing SN. Optical densities of the individual bands were quantified using NIH ImageJ. Optical densities of α-synuclein overexpressing conditions were normalized by the averaged value of GFP expressing condition. Data are shown as mean ± SEM (AAV GFP, n = 4∼6; AAV A53T α-synuclein, n = 4∼6; *p < 0.05 two tail t test).
Levels of Iba-I, a marker for microglia, were also increased in the striatum but not in the SN overexpressing A53T α-synuclein at 8 weeks, as determined by Western blot analysis (Fig. 6A,B). These results were confirmed by immunohistochemistry, showing that the intensity of Iba-I staining was augmented in the α-synuclein overexpressing striatum (Fig. 6C). Moreover, the Iba-I positive microglia were activated as evident by their larger soma with short swollen processes compared with the elongated and needle-like shape in the GFP expressing control striatum (Fig. 6C). However, microglial morphology in SN was not different between GFP and A53T α-synuclein overexpressing rats (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). In accordance with these results, levels of proinflammatory cytokines, IL-1β, interferon (IFN)-γ and TNF-α were significantly elevated in α-synuclein overexpressing striatum at 8 weeks whereas cytokine levels were not altered in the SN (Fig. 6D). Activated morphology of microglia was maintained in the A53T α-synuclein overexpressing striatum at 24 weeks (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material).
Figure 6.
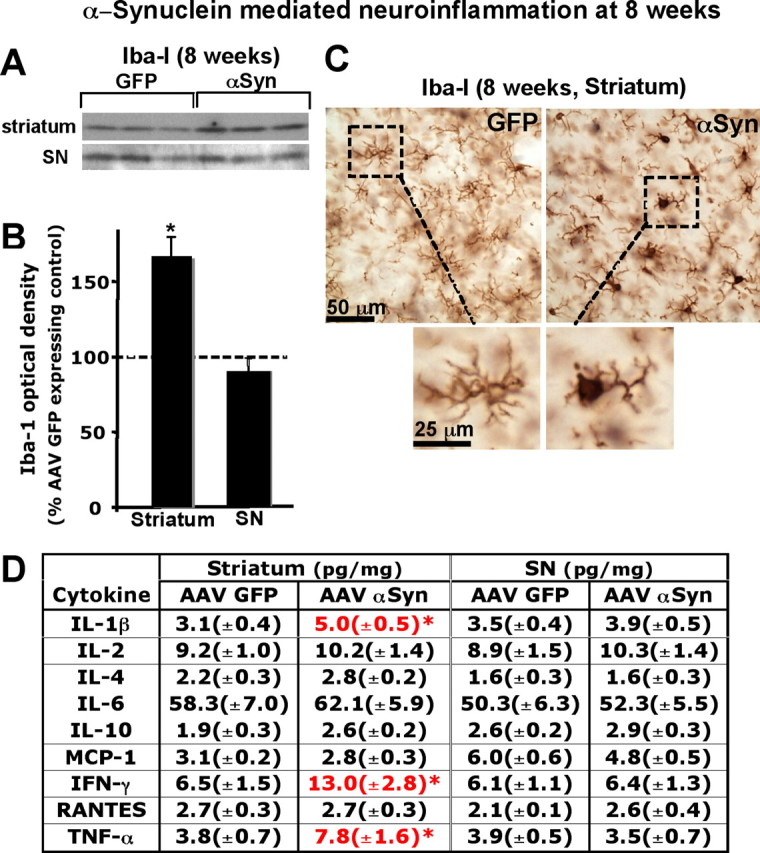
α-Synuclein overexpression causes neuroinflammatory responses in the striatum but not SN at 8 weeks post AAV injection. A, Western blot was performed using total lysate (T) of the striatum or SN. In the striatum, Iba-I levels were increased by α-synuclein overexpression whereas they remained unchanged in SN. B, Optical densities of the individual bands were quantified using NIH ImageJ. Optical densities of α-synuclein overexpressing conditions were normalized by the averaged value of GFP expressing condition. Data are shown as mean ± SEM (AAV GFP, n = 4∼6; AAV A53T α-synuclein, n = 4∼6; *p < 0.05 two tail t test). C, An increase in Iba-I levels by Western blot was confirmed by immunohistochemistry against Iba-I. Morphology of microglia in the α-synuclein overexpressing striatum suggests presence of activated microglia (C). Cytokine and chemokine levels were determined in the striatum and the SN at 8 weeks postinjection using ELISA. D, A53T α-synuclein overexpression caused elevated levels of IL-1β, IFN-γ and TNF-α in the striatum but not in SN. Data are shown as mean ± SEM (AAV GFP, n = 8; AAV A53T α-synuclein, n = 10; *p < 0.05 two tail t test).
Discussion
We demonstrated that levels of proteins involved in synaptic transmission, axonal transport, and neuroinflammation, are altered by overexpression of A53Tα-synuclein at early stages of disease progression before neuronal loss predominates. By studying the predegenerative process, we believe that the critical pathophysiological changes potentially causal to the disease progression can be identified without being confounded by cell death related events.
A53T α-Synuclein-mediated changes in levels of proteins involved in synaptic transmission
Over the course of the rat α-synucleinopathy, we detected a decrease in levels of proteins involved in synaptic vesicle exocytosis, including rabphilin 3A and syntaxin, by 2 months. Interestingly, rabphilin 3A levels were also reduced in an early degenerative state of HD and a mouse model of HD (Smith et al., 2005, 2007), suggesting an involvement of this protein in common neurodegenerative disease processes. Syntaxin is a part of the SNARE complex, and involved in exocytosis of synaptic vesicles. In accordance with our findings, levels of syntaxin were also dramatically reduced in brains of DLBD compared with control human cases (Kramer and Schulz-Schaeffer, 2007). Thus, these results provide potential components of synaptic vesicle exocytosis that may be affected by α-synucleinopathy. Given the role of rabphilin 3A and syntaxin in synaptic transmission and exocytosis, our findings indicates that an impairment of synaptic transmission and exocytosis occurs early on in the α-synuclein mediated degenerative process, causing disruption in neuronal circuitry.
Characterization of striatal tissue at different times after α-synuclein overexpression revealed dynamic changes in presynaptic and axonal transport related proteins. For example, levels of rabphilin3A, KIF3A and myosin Va were elevated at 4 weeks followed by a reduction at 8 weeks. Dynein, in contrast, was reduced at 4 weeks but increased at 8 weeks. Thus, at 4 weeks, the direction of the changes in these protein levels opposed the changes observed at 8 weeks, raising the possibility that compensatory changes occurred at 4 weeks to counteract the effects of A53T α-synuclein overexpression. It is plausible that such compensatory mechanisms were overcome by 8 weeks with progression of the disease process.
α-Synuclein-mediated neurochemical changes included increases in levels of DOPAC, a metabolite of dopamine, and altered DOPAC/dopamine ratios long before loss of dopamine in the striatum. Unlike the situation where DA terminals are acutely lost by toxins, which precipitously lower DA levels and cause DA transmission and turnover to increase in remaining terminals (Apicella et al., 1990), our data show that in predegenerative stages, A53T α-synuclein overexpression caused DOPAC levels to increase without loss of DA levels. Total DA tissue content measured by HPLC primarily reflects vesicular DA content in presynaptic terminals. We reason that based on our own observation, at presymptomatic stage, dystrophic DA terminals in the A53T α-synuclein overexpressing striatum are likely dysfunctional in DA transmission (Peschanski and Besson, 1987; Peschanski and Isacson, 1988) but still contain DA in synaptic vesicles since terminals are not lost. At the same time, remaining functional terminals will compensate for suboptimal DA release in synapses by increasing DA turnover, which will in turn increase DOPAC levels. This will result in increased DOPAC content without loss of DA content. These results suggest that, as reported in other models of PD, DOPAC levels and DOPAC/DA ratios may be important indicators of DA terminal dysfunction and predegenerative changes in α-synucleinopathy.
A53T α-Synuclein-mediated changes in levels of proteins involved axonal transport
Several anterograde transport motor proteins that carry synaptic vesicles (KIF1A, KIF1B, and myosin Va) and other cargo containing vesicles (KIF2A and KIF3A) to synaptic terminals were markedly reduced by A53T α-synuclein overexpression in the striatum, whereas retrograde transport proteins were upregulated at synaptic terminals. Moreover, some of the reduced motor proteins in the striatum such as KIF1A and KIF2A showed increased levels in the SN, suggesting that reduction of axonal transport for these motor proteins produced an accumulation of such proteins in the cell body region. These findings can be explained by reduction in anterograde axonal transport of certain cargoes in parallel with an increase in retrograde axonal transport, producing accumulation of cargoes in the cell body region. In effect, this would increase the total protein degradation burden in the cell body (Petrucelli et al., 2002; McNaught et al., 2003). Similar axonal transport defects to those mentioned have been reported in an MPP+ toxin model of PD (Morfini et al., 2007) and a mouse model of AD (Stokin et al., 2005). Stokin et al. also demonstrated axonopathy and transport defects early in the pathogenesis of AD cases and that axonal transport deficits are potentially causal to disease progression (Stokin et al., 2005). In vitro evidence showed that reduction in the anterograde motor protein, kinesin-1 produced a decrease in anterograde transport, and an increase in retrograde transport of amyloid precursor protein (Stokin et al., 2005). In this context, the dramatic reduction of several anterograde motor proteins along with increases in levels of retrograde transport motor proteins in the striatum suggest that elevated levels of α-synuclein may shift the axonal transport balance to the retrograde direction, causing abnormal accumulation and aggregation of proteins in the cell body, potentially causing formation of Lewy bodies in the cell body regions. The fact that axonopathy and axonal transport deficit have been observed in other models of PD and neurodegenerative diseases (Szebenyi et al., 2003; Stokin et al., 2005; Morfini et al., 2007), indicates that disrupted axonal transport is an important part of several neurodegenerative processes (Szebenyi et al., 2003; Stokin et al., 2005; Morfini et al., 2007). Although observed changes levels of transport motor proteins support axonal transport disruption in this model, we cannot exclude the possibility that reduction and accumulation of the certain protein levels observed in the striatum and the SN, may be due to the imbalance in protein degradation and synthesis rather than axonal transport deficit.
A53T α-Synuclein-mediated changes in levels of cytoskeletal proteins
In addition to the changes in the transport motor proteins, another striking outcome of A53T α-synuclein overexpression was changes seen in all categories of cytoskeletal proteins; microtubule, intermediate filaments such as neurofilaments, and actin. Alteration of α-tubulin has been shown to diminish the transport of a microtubule based motor protein, KIF1A and its cargo, synaptic vesicles to synaptic terminals, causing an impaired synaptic transmission (Ikegami et al., 2007). Therefore, the reduction in α-tubulin levels seen in the striatum following α-synuclein overexpression could contribute to the transport deficit of KIF1A and other microtubule based motor proteins, producing reduction of their protein levels observed in the striatal synaptic terminals. The transport defect on KIF1A caused by α-synuclein overexpression would diminish synaptic vesicle transport to synaptic terminals, and in turn disrupt dopaminergic synaptic transmission to medium spiny GABAergic neurons in the striatum. Recent studies also suggest a link between α-synuclein and tubulin: oligomeric α-synuclein inhibits tubulin polymerization (Chen et al., 2007). Interestingly, tubulin changes observed in our model were limited to α- and γ- tubulin, leaving β-tubulin unaffected, suggesting the presence of specificity on α-synuclein-mediated tubulin changes rather than generalized disruption of tubulin. This may reflect why some tubulin based motor proteins were affected by α-synuclein overexpression while others were not.
An increase in actin levels in the striatum was observed as early as 1 month post AAV α-synuclein injection when axon pathology emerges. The increase in levels was far more prominent at later stages of the α-synucleinopathy, and immunostaining results demonstrated that dystrophic axons were sites of actin accumulation, including the stabilized forms of actin, F-actin. Intriguingly, studies in yeast have demonstrated that increases in actin stabilization causes depolarization of the mitochondrial membrane and an increase in reactive oxygen species (ROS) production, resulting in cell death (Gourlay et al., 2004). Decreasing actin stabilization, in contrast, lowered levels of ROS, increasing the lifespan of yeast by 65% (Gourlay et al., 2004). Moreover, accumulation of F-actin was observed in both a drosophila and mouse model of tauopathy, and reduction of F-actin levels decreased tau-induced neurodegeneration in the drosophila model of tauopathy (Fulga et al., 2007). These results suggest that the early and persistent increase in actin levels in our model may be causally related to α-synuclein-mediated neurodegeneration.
A53T α-Synuclein-mediated neuroinflammation
Although many recent studies demonstrate that neuroinflammation aggravate degenerative processes in models of PD, including α-synuclein transgenic mice (Gao et al., 2008), it is unclear that expression of α-synuclein in DA neurons itself causes neuroinflammation in vivo. Our model demonstrates that an increase in activated microglia by α-synuclein overexpression at an early degenerative stage, suggesting that neuroinflammation is involved early in the disease progression of α-synucleinopathy rather than a result triggered by cell death. These data provide evidence that exclusive expression of A53T α-synuclein only in DA terminals in the striatum by the synapsin promoter-driven neuronal expression in the SN, is sufficient to create microglial activation over time, which will in turn aggravate on-going neurodegeneration. Cytokines that were elevated in α-synuclein overexpressing striatum (IL-1β, IFN-γ and TNF-α), are also known to be increased in PD patients, supporting that this α-synucleinopathy model closely recapitulates certain aspects of PD (Roodveldt et al., 2008). Importantly, although AAV α-synuclein was injected into the SN, the activation of microglia appears to start at DA terminal in the striatum rather than cell bodies in the SN. This observation as well as early signs of axonopathy raises an intriguing possibility that α-synuclein-mediated pathology may start at synaptic terminals and move onto cell bodies, creating a “dying-back” phenomenon.
Together, our data demonstrate that levels of proteins involved in synaptic vesicle exocytosis, axonal transport and neuroinflammation, are altered in a rat model of α-synucleinopathy long before cell death occurs. We believe that such early pathophysiological changes may provide valuable information about mechanisms that initiate cellular dysfunction leading to degeneration. Therefore, this approach may also yield clues to biomarkers for early degenerative stages, extending a window for potential early and disease-modifying treatment of neurodegenerative diseases.
Footnotes
This work was conducted at McLean Hospital and was supported by funds to O.I. from the National Institutes of Health/National Institute of Neurological Disorders and Stroke P50 (Grant NS39793), Parkinson's Disease Udall Research Centers of Excellence to McLean/Harvard Medical School, the Michael Stern Foundation for Parkinson's Disease Research, the Consolidated Anti-Aging Foundation, and Harold and Ronna Cooper Family. We thank Dr. Vikram Khurana for discussion and critical reading of this manuscript and Casper Reske-Nielsen, Kari Ording, Alyssa Yow, and Raymond Johnson for their excellent technical assistance. We also thank Dr. Pavel Oston for kindly providing AAV2 synapsin GFP construct.
The authors declare no competing financial interests.
References
- Apicella P, Trouche E, Nieoullon A, Legallet E, Dusticier N. Motor impairments and neurochemical changes after unilateral 6-hydroxydopamine lesion of the nigrostriatal dopaminergic system in monkeys. Neuroscience. 1990;38:655–666. doi: 10.1016/0306-4522(90)90059-d. [DOI] [PubMed] [Google Scholar]
- Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- Chen L, Jin J, Davis J, Zhou Y, Wang Y, Liu J, Lockhart PJ, Zhang J. Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem Biophys Res Commun. 2007;356:548–553. doi: 10.1016/j.bbrc.2007.02.163. [DOI] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Endo S, Isacson O. An endogenous serine/threonine protein phosphatase inhibitor, G-substrate, reduces vulnerability in models of Parkinson's disease. J Neurosci. 2007;27:8314–8323. doi: 10.1523/JNEUROSCI.1972-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Treviño I, O'Brien DE, Casey B, Goldberg MS, Tansey MG. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28:10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain. 2008;131:1880–1894. doi: 10.1093/brain/awn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay CW, Carpp LN, Timpson P, Winder SJ, Ayscough KR. A role for the actin cytoskeleton in cell death and aging in yeast. J Cell Biol. 2004;164:803–809. doi: 10.1083/jcb.200310148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K, Heier RL, Taruishi M, Takagi H, Mukai M, Shimma S, Taira S, Hatanaka K, Morone N, Yao I, Campbell PK, Yuasa S, Janke C, Macgregor GR, Setou M. Loss of alpha-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function. Proc Natl Acad Sci U S A. 2007;104:3213–3218. doi: 10.1073/pnas.0611547104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson's disease. J Neuroinflammation. 2008;5:8. doi: 10.1186/1742-2094-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Natl Acad Sci U S A. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson's disease. Exp Neurol. 2003;179:38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Opalach K, Serulle Y, Moreira JE, Sugimori M, Llinás RR, Brady ST. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci U S A. 2007;104:2442–2447. doi: 10.1073/pnas.0611231104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Peschanski M, Besson JM. Structural alteration and possible growth of afferents after kainate lesion in the adult rat thalamus. J Comp Neurol. 1987;258:185–203. doi: 10.1002/cne.902580203. [DOI] [PubMed] [Google Scholar]
- Peschanski M, Isacson O. Fetal homotypic transplant in the excitotoxically neuron-depleted thalamus: light microscopy. J Comp Neurol. 1988;274:449–463. doi: 10.1002/cne.902740311. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Roodveldt C, Christodoulou J, Dobson CM. Immunological features of alpha-synuclein in Parkinson's disease. J Cell Mol Med. 2008;12:1820–1829. doi: 10.1111/j.1582-4934.2008.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha AR, Hill J, Utton MA, Asuni AA, Ackerley S, Grierson AJ, Miller CC, Davies AM, Buchman VL, Anderton BH, Hanger DP. Parkinson's disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J Cell Sci. 2004;117:1017–1024. doi: 10.1242/jcs.00967. [DOI] [PubMed] [Google Scholar]
- Sánchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell AL, Isacson O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson's disease. J Neuroinflammation. 2004;1:6. doi: 10.1186/1742-2094-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smith R, Petersén A, Bates GP, Brundin P, Li JY. Depletion of rabphilin 3A in a transgenic mouse model (R6/1) of Huntington's disease, a possible culprit in synaptic dysfunction. Neurobiol Dis. 2005;20:673–684. doi: 10.1016/j.nbd.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Smith R, Klein P, Koc-Schmitz Y, Waldvogel HJ, Faull RL, Brundin P, Plomann M, Li JY. Loss of SNAP-25 and rabphilin 3a in sensory-motor cortex in Huntington's disease. J Neurochem. 2007;103:115–123. doi: 10.1111/j.1471-4159.2007.04703.x. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]