

Abstract
Prostaglandins (PGs) are potent autocrine and paracrine oxygenated lipid molecules that contribute appreciably to physiologic and pathophysiologic responses in almost all organs, including brain. Emerging data indicate that the PGs, and more specifically PGE2, play a central role in brain diseases including ischemic injury and several neurodegenerative diseases. Given concerns over the potential toxicity from protracted use of cyclooxygenase inhibitors in the elderly, attention is now focused on blocking PGE2 signaling that is mediated by interactions with four distinct G protein-coupled receptors, EP1-4, which are differentially expressed on neuronal and glial cells throughout the central nervous system. EP1 activation has been shown to mediate Ca2+-dependent neurotoxicity in ischemic injury. EP2 activation has been shown to mediate microglial-induced paracrine neurotoxicity as well as suppress microglia internalization of aggregated neurotoxic peptides. Animal models support the potential efficacy of targeting specific EP receptor subtypes in Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and ischemic stroke. However promising these preclinical studies are, they have yet to be followed by clinical trials targeting any EP receptor in neurologic diseases.
Keywords: Prostaglandins, PGE2, CNS, neurodegeneration, EP receptors
PROSTAGLANDIN SIGNALING
In response to varying physiologic and pathophysiologic stimuli, phospholipase (PL) A2 hydrolyzes the ester linkage that bonds arachidonic acid (AA) to glycerol in phospholipids [1]. Catalyzed oxygenation of AA produces the derivative class of molecules termed eicosanoids, which includes the subclasses of prostanoids and leukotrienes. The two predominant groups of enzymes that catalyze the oxygenation of hydrolyzed AA to yield initial eicosanoid products are the cyclooxygenase (COX) isozymes and lipoxygenases (LOXs), respectively. COX isozymes catalyze the formation of the prostanoid prostaglandin (PG) H2, which is the committed step in PG synthesis and involves formation of the intermediate PGG2 with the release of an oxidizing radical [2]. The bioactivity of PGH2 is a consequence of three independent mechanisms. First, PGH2 may be converted to the prostanoids PGD2, PGE2, PGF2α, PGI2, and thromboxane (Tx) A2 by cell-specific synthases or isomerases. These potent prostanoid signaling molecules exert their effects through autocrine and paracrine stimulation of eight specific G protein-coupled receptors (GPCRs) designated DP, EP1-4, FP, IP, and TP, respectively [3]. Differentially restricted expression of the prostanoid synthases, isomerases and receptors allow these prostanoids to achieve a wide variety of biological actions in different cell types and tissues. Second, PGH2 itself is an agonist for the TP receptor. Third, PGH2 may spontaneously rearrange to form levuglandins (LGs). LGs are highly chemically reactive compounds that form irreversible adducts with protein lysyl residues leading to protein-protein crosslinks [4]. An example of this potentially harmful activity is the ability of LGs to accelerate the oligomerization of Alzheimer's disease related Aβ peptides in vitro [5].
Compelling epidemiologic data have repeatedly shown that protracted use of non-steroidal anti-inflammatory drug (NSAID) inhibitors of COX isoforms prior to the onset of dementia can substantially (50% or more) lower the risk of subsequently developing dementia [6]. Unfortunately, a large clinical trial aimed at testing this associataion with both a non-selective and COX2-selective NSAID, the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT), was terminated early because of increased “thrombotic events” (myocardial infarct and ischemic stroke) in individuals randomized to drug [7]. While there is an ongoing debate over the risks associated with protracted use of NSAIDs in the elderly [8,9], many academic and industry laboratories have turned their attention to individual PG receptors, rather than suppression of the entire PG pathway by NSAIDs, as a more specific approach to retain therapeutic effect while limiting toxicity.
E PROSTANOID (EP) RECEPTORS
EP Receptor Expression
PGE2 is a unique prostanoid because it has four receptor subtypes, EP1-4, which are widely expressed and linked to functionally antagonistic second messenger systems. Multiple PGE2 receptor subtypes with differential ligand affinity, cellular and tissue receptor expression profile, and coupling to opposing second messengers systems allows PGE2 its signaling versatility and often opposing actions in tissues and cells (reviewed in [10]). For example, EP3 is the only EP receptor that has three transcriptional splice variants, EP3α, EP3β, and EP3γ. The EP3β receptor is unique because it does not desensitize and thus displays persistent signaling when exposed to ligand [10,11]. General signaling characteristics of the EP receptors are summarized in Table 1. PGE2 binds to EP1 and EP2 with much lower affinity than EP3 and EP4. In humans, PGE2 binding affinities are EP3 (kd=0.3nM) > EP4 (kd=0.8nM) > EP2 (kd=4.9nM) >EP1 (kd=9.1nM). In mice, PGE2 binding affinities are EP3 (kd=0.9nM) > EP4 (kd=1.9nM) > EP2 (kd=12nM) > EP1 (kd=20nM). Several relatively selective ligands with differential activities exist for all four EP receptors. However, it is difficult to directly compare the structure-activity relationship of each class based on the existing literature due to differing receptor expression systems and reported binding efficacies. EP1 agonists include ONO-DI-004, the prostacyclin analogue Iloprost, and 17-phenyl-trinor PGE2. EP1 antagonists include SC 51322, SC 51089, and ONO-8713. EP2 agonists include Butaprost, 11-deoxy PGE1, AH 13205, and ONO-AE1-259. There are currently no selective EP2 antagonists available. EP3 agonists include Sulprostone, Misoprostol, SC 46275, ONO-AE-248, and M&B 28767. EP3 antagonists include ONO-AE3-240 and L-826266. EP4 agonists include ONO-AE1-329 and PGE1-alcohol. EP4 antagonists include AH 23848, ONO-AE3-208, and CJ-023,423. Selected EP1-4 ligands and their structures are shown in Table 2. As mentioned previously, EP receptors are differentially expressed on almost all organs, including the central nervous (CNS). However, there are conflicting data concerning the expression of EP receptor subtypes in the CNS [12-17]. This appears to result from the specificity of commercially available antibodies and the purity of some primary cultures used for PCR. We are skeptical of conclusions resulting from Western blot and immunohistochemical studies because many currently available antibodies are not specific, presenting multiple bands from brain homogenates probed by Western blot and showing comparable immunoreactivity in wild type and homozygous EP receptor deficient mice (unpublished data).
Table 1.
Signaling of EP receptor subtypes
| EP1 | EP2 | EP3 | EP4 | |
|---|---|---|---|---|
| PGE2 Kd (nM)/Human (Mouse) | 9.1 (20) | 4.9 (12) | 0.3 (0.9) | 0.8 (1.9) |
| G-protein Coupling | Unknown (↑ Ca2+) | Gs | Gs, Gi, Gq | Gs |
Table 2.
Structures of PGE2 and relatively selective EP receptor ligands
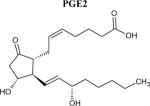 |
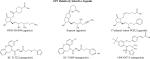 |
Other ligands not shown include, but are not limited to, ONO -AE3-240 (EP3 antagonist), L-826266 (EP3 antagonist), M&B 28767 (EP3 agonist), and CJ-023,423 (EP4 antagonist).
Every EP receptor is present in the rodent brain, each with its own regional and cell-specific difference in expression and activity (expression summarized in Table 3). Experiments have shown that EP1, EP2, and EP3 are expressed by neurons in multiple regions of brain while neuronal EP4 expression is restricted to only some hypothalamic nuclei [12,18-22]. Differential expression of EP receptors has been reported for both primary microglia and astocyte cell cultures. EP2 is functionally expressed on microglia in culture and on activated microglia in vivo. EP1 and EP3 (inducible) may be expressed on microglia, but those who have observed these EP receptors have yet to show any function in microglia. Two groups have not observed a change of microglial response to lipopolysaccharide (LPS) in EP1-/- microglia compared to wild type [12,23]. A confound of these expression data comes from the fact that culturing of microglia leads to some level of `activation' that is not present in vivo. This may lead to artificial differences in EP receptor expression in glia [5,10,13,24,25]. EP2, EP3 (inducible), and EP4 appear to be expressed by astrocytes
Table 3.
CNS cellular expression of EP receptor subtypes
| EP1 | EP2 | EP3 | EP4 | |
|---|---|---|---|---|
| Neuron | Parietal cerebral cortex, cerebellum, hypothalamus | Stria terminalis, lateral septum, subfornical organ, ventromedial hypothalamic nucleus, amygdala, locus coeruleus, the area postrema | Widespread throughout the brain | Some hypothalamic nuclei |
| Microglia | Yes | Yes | May be induced | No |
| Astrocytes | No | Yes | Yes | Yes |
EP Receptors in the CNS
The role of PGE2 and EP receptors in CNS pathophysiology of neuronal damage continues to be a focus of several academic and industrial laboratories. These mechanisms include both direct and indirect neurotoxicity due to excitotoxicity, neurotoxic protein aggregation, innate immune activation, and clearance of neurotoxic peptides.
Excitotoxicity
Glutamate is the major excitatory neurotransmitter in the CNS. Its effects are mediated mainly through ionotropic receptors that are named after their selective pharmacologic ligands, including the N-methyl-D-aspartic acid (NMDA) receptor, the kainate receptor, and the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor. Excitotoxicity results in selective neuron damage and ultimately death from high levels of synaptic glutamate. Relevant to our discussion, excitotoxicity has been proposed to contribute to many neurologic diseases including ischemic damage, traumatic brain injury, and neurodegenerative disease.
The NMDA receptor regulates COX2 neuronal expression, which provides a mechanistic link between PG signaling and excitatory neurotransmission [26]. Inhibition of COX2 activity early after gluatamate exposure suppresses NMDA mediated neuronal excitotoxicity [27-29]. The EP1 receptor antagonist SC51089 reduces NMDA mediated neurotoxicity in mice, indicating that EP1 is a downstream excitotoxic effector [12]. Furthermore, EP1 enhances neuronal Ca2+ dysregulation induced by NMDA receptor activation through impairment of Na+-Ca2+ exchange in vitro. Conversely, EP2 and EP4 demonstrate a neuroprotective effect during excitotoxic conditions. Organotypic culture preparations from rat hippocampus or spinal cord show neuroprotection in excitotoxic conditions when treated with the EP2 agonist butaprost or the EP4 agonist ONO-AE1-329 [30,31].
Neurotoxic protein aggregation
The amyloid hypothesis of Alzheimer's disease (AD) pathogenesis states that neurodegeneration is mediated, at least in part, by the direct neurotoxicity of accumulated Aβ peptide aggregates. There are several studies showing that aggregated Aβ peptides are directly neurotoxic to primary neuronal cell cultures (just two examples of many are [32,33]). Treating mouse primary cerebral cortical neurons with the EP2 agonist butaprost or the EP3/EP4 agonist 1-hydroxy-PGE1 suppresses aggregated Aβ peptide mediated neurotoxicity [33]. In one study, primary neuronal cell culture derived from EP2-/- mice show no difference in aggregated Aβ peptide mediated neurotoxicity when compared to wild type [25]. It is important to note here the pleiotropic effects of aggregated Aβ peptide and its ability to cause indirect neurotoxicity through microglial activation. This can make interpreting the relative contribution of direct and indirect neurotoxicity of aggregated Aβ peptide in vivo less clear.
Innate immune response
The innate immune response in the CNS is thought to be a major effector in several chronic neurodegenerative diseases, such as AD, Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS). The innate immune response has multiple arms of action including cytokine secretion, paracrine neurotoxicity, and clearance of toxic aggregated proteins. This provides a response that can be both beneficial and deleterious to the surrounding neurons. CNS glial cells, especially microglia, are the main effectors of the innate immune response. Lipopolysaccharide (LPS)-stimulated microglial activation occurs mainly through the activation of the co-receptors CD14 and TLR4 which in turn activate the NFκB- and p38-MAPK signaling pathways [34,35]. These co-receptors respond to additional endogenous stimuli including aggregated Aβ peptides and neoantigens expressed by apoptotic cells [36-38]. LPS provides a convenient and reliable means to selectively activate a pathologic process via receptors that are similarly activated by endogenous ligands found in neurologic disease states.
LPS-induced neuroinflammation can negatively impact several aspects of rodent behavior, including enhancing both age-dependent cognitive decline and reactive oxygen species (ROS) mediated impairments in age-dependent learning [39]. Intracerebroventricular (ICV) LPS injection in mice causes hippocampal CA1 pyramidal neurons to display a reversible decrease in dendrite length and spine density without cell death [40,41]. This phenomenon is completely blocked by either NSAID treatment or genetic ablation of EP2 [40,41]. Further evidence for the importance of PG signaling in CNS innate immunity is the ability of NSAIDs to suppress the effector molecules produced by LPS-activation of microglia primary cultures. In addition, primary microglia cell cultures prepared from EP2-/- mice suppress the secretion of over 20 cytokines and chemokines examined [42]. It then appears that EP2 may have opposite effects on neuron survival depending on its expression; on neurons EP2 is protective against excitotoxicity whereas activated microglia EP2 promotes paracrine neurotoxicity. In vivo experiments have yet to show a role for EP1 in innate immune response [12,23]. Beyond cytokines and chemokines, microglial activation includes an increased expression and activity of a number of enzymes that generate free radicals. Microglia themselves are relatively well protected from increased free radical stress that promotes neurotoxicity through their robust anti-oxidant defenses [43]. Again highlighting the role of PGs, NSAIDs suppress LPS-stimulated microglia mediated paracrine oxidative damage to neurons both in vivo and ex vivo [41,42].
Accumulation of aggregated Aβ peptides is deleterious to neurons through direct effects and microglial mediated indirect effects. Therefore, enhanced internalization and clearance of these neurotoxic peptides provides one way to provide neuroprotection. Mouse primary cultures of EP2-/- microglia show increased internalization of fluorescently labeled Aβ and are highly efficient at clearing Aβ species from human AD brain slices without prior opsonization [32]. This latter point is important because wild type mouse microglia have no detectable phagocytic clearance of the Aβ peptides from human AD tissue without prior opsonization [44].
PROSTANOIDS AND EP RECEPTORS IN HUMAN NEUROLOGIC DISEASE
Body fluids are typically used to assess changes in PG production in disease states because PGs in brain tissue are highly labile due to their rapid metabolism. Increased PGE2 levels have been demonstrated in cerebrospinal fluid (CSF) from several neurologic diseases, including AD, ALS, Creutzfeldt-Jakob Disease (CJD), ischemic stroke, and HIV associated dementia (HAD) (summarized studies in Table 4). Several studies have shown that the EP receptors play key roles in several neuropathophysiologic processes [28]. Combined, these data indicate an important role for PGE2 and its EP receptors in several diseases of the CNS. This rationale has lead to several laboratories investigating the role of EP receptors in rodent models of human CNS diseases.
Table 4.
PGE2 levels in CSF from patients with neurologic disease
| Disease | CSF PGE2 Levels | Reference |
|---|---|---|
| AD | Increased levels highest in very early AD, but declined with progressive cognitive impairment | [81] |
| Increased 5-fold in early AD | [82] | |
| ALS | Increased 6-fold | [83,84] |
| Increased 2 to 10 fold | [83,84] | |
| No increase | [72] | |
| CJD | Increased 6-fold in patients with sporadic or familial forms of CJD | [85] |
| Increased 6-fold in patients with variant CJD | [86] | |
| Ischemic stroke | Increased 2-fold during initial 72 hr | [87] |
| HAD | Increased 40% in all HIV-seropositive patients and positively correlated with degree of cognitive impairment | [88] |
Alzheimer's disease
AD is an age-related neurodegenerative disease with the pathological hallmarks of amyloid plaques, neurofibrillary tangles (NFTs), and neuroinflammation. Transgenic mouse models of Familial Alzheimer's disease (FAD) overexpress versions of mutant human amyloid precursor protein (APP) with or without expression of mutant presenilin 1 (PS1), such as the APPSwe-PS1ΔE9 mouse. These transgenic mice develop age-dependent accumulation of Aβ along with significant microglial activation. In these mice, NSAID treatment can reduce Aβ peptide deposition and reverse mild behavioral deficits [45-48]. COX inhibition in the PG pathway is the likely mechanism by which NSAIDs suppress Aβ deposition in aged transgenic mouse brain. A recently proposed alternative hypothesis based on cell culture studies asserts that high, supraphysiologic concentrations of some NSAIDs can alter γ-secretase (an APP cleaving enzyme) activity and reduce the ratio of the most toxic Aβ peptide species (Aβ1-42) [49-51]. However, in vivo pharmacological dosing of these NSAIDs challenges this alternative hypothesis [52,53].
Deletion of the EP2 receptor in aged APPSwe-PS1ΔE9 mice results in lower total levels of Aβ peptides, fewer accumulated amyloid deposits, and a significant decrease in neuronal oxidative damage [54]. However, it is not clear which happens first in vivo, formation of toxic aggregated Aβ species or neuronal oxidative damage. One group showed that neuronal oxidative damage is secondary to Aβ activation of microglial, while another group suggests that oxidative damage precedes, and possibly stimulates, Aβ deposition [37]. There is additional data suggesting a reinforcing cycle between the production of oxidative damage and formation of neurotoxic Aβ deposition [55]. It is possible that decreased Aβ deposition decrease in the brains of EP2-/- mice is due to enhanced microglial internalization of Aβ species. Regardless of the mechanism in these transgenic mouse models of FAD, these data highlight the highly desirable dual phenotype of EP2-/- microglia of increased Aβ clearance with decreased neurotoxicity.
Parkinson's disease
PD is another age-related neurodegenerative disease that prominently involves the dopaminergic neurons within the substantia nigra pars compacta, and has the pathological hallmarks of Lewy bodies and neuroinflammation. Paralelling AD, there is significant microglial and complement activation in autopsy specimens from PD patients (reviewed in [56]). COX2 and PG signaling play a significant role in disease progression in rodent models of PD utilizing 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Inhibiting COX2 either pharmacologically or with genetic ablation partially protects against dopaminergic neurodegeneration and development of the related motor system deficits [57,58]. Recent in vitro experiments show that EP1 antagonists confer protection against 6-Hydroxydopamine (6-OHDA) mediated selective toxicity of rat primary dopaminergic neurons [59]. As in the above experiments using AD brain and transgenic mouse models of FAD, EP2-/- microglia also display enhanced ex vivo clearance of aggregated β-synuclein in mesocortex of Lewy body disease patients while attenuating neurotoxicity and β-synuclein aggregation in the MPTP mouse model [60].
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive loss of both upper and lower motor neurons. As with AD and PD, pathologic findings in ALS include an inflammatory component involving activated microglia and astrocytes along with increased expression of COX2 [61-63]. Suppressing COX2 activity shows neuroprotection in an organotypic model of ALS [57]. Mutations in the copper/zinc superoxide dismutase 1 (SOD1) gene cause a familial form of ALS [58-60]. In mutant SOD1 mice, levels of COX2 mRNA and PGE2 increased [68,69]. Treatment with COX2 inhibitors before onset of hindlimb paralysis in SOD1 mice reduces CSF PGE2 levels and extends lifespan [69-71]. This indicates that, at least in the SOD1 model of familial ALS, PGs promote motor neuron injury. Despite the successful preclinical studies using COX2 inhibitors, a clinical trial of the selective COX2 inhibitor celecoxib at 800 mg/day in ALS patients demonstrated no benefit to morbidity or mortality measures [72]. In addition, PGE2 levels in the CNS did not decrease with treatment. The mechanism of COX2 inhibition in SOD1 mutant mice has also been postulated to occur independent of PG signaling [73]. In fact, physiological levels of PGE2 in an organotypic model of ALS paradoxically protect motor neurons [31]. These effects were mediated through the receptors EP2 and EP3. The role of specific EP receptors in ALS needs to be examined further.
Ischemia
Ischemic injury pathophysiology is complex and depends on severity of blood flow loss, the mechanism of ischemia, and the extent and timing of reperfusion. However, there is evidence for an underlying role of excitotoxicity and immune mediated damage in ischemic pathophysiology. Rapidly induced COX2 expression and slower induction of COX1 expression occurs in neurons, followed by glial cells and infiltrating mononuclear cells, in response to ischemic injury induced massive glutamate release in the brain [27]. Inhibition of COX2 activity either pharmacologically or with genetic ablation reduces infarct size in rodent models of cerebral ischemia [74,75]. Similarly, inhibition of EP1 activity either pharmacologically or with genetic ablation rescues brain tissue in a model of transient focal ischemia [12,76,77]. One interpretation of these findings is that NMDA receptor activation increases COX2 activity, which in turn increases PGE2 levels, which then activate neuronal EP1, and ultimately this leads to neuronal injury. Recently, neuroprotection via EP1 inhibition was shown to occur through increased activation of the PI3K/AKT survival signaling pathway [78]. EP2-/- mice also demonstrate reduced infarct size in a model of focal cerebral ischemia [30,79]. The similarities between EP1 and EP2 mediated rescue from brain ischemic injury is paradoxical because EP1 and EP2 have opposite effects on excitotoxicity. This paradox may be partially explained by the prominent contribution of EP2 to glial cell pathophysiology discussed above, which is included in the innate immune response component found in CNS ischemic injury. Stimulating EP3 pharmacologically also increases infarct size in the middle cerebral artery occlusion model of ischemia [80].
CONCLUSION
PG signaling plays a central role in physiologic and pathophysiologic states in the CNS. The complex dual neuroprotective and neurotoxic role of PG receptor activation in human neurological disease leaves many pathogenic issues to be resolved. Our review clearly demonstrates the utility and promise of therapeutically targeting the PG signaling pathway, especially PGE2, EP1 and EP2, in neurodegenerative disease. Further studies of differential EP receptor activation in the CNS are needed not only to provide insight into the pathogenesis of neurodegenerative disease, but also to guide the development of selective and efficacious therapies that target neurotoxic mechanisms while maintaining neuroprotective actions.
Acknowledgments
Supported by grants from the National Institutes of Health (AG05136 and GM015431) and by the Nancy and Buster Alvord Endowment.
REFERENCES
- [1].Kudo I, Murakami M. Prostaglandins Other Lipid Mediat. 2002;68-69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- [2].Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Prostaglandins. 1997;54:601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- [3].Hata AN, Breyer RM. Pharmacol. Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- [4].Difranco E, Subbanagounder G, Kim S, Murthi K, Taneda S, Monnier V, Salomon R. Chem. Res. Toxicol. 1994;8:61–67. doi: 10.1021/tx00043a008. [DOI] [PubMed] [Google Scholar]
- [5].Boutaud O, Ou JJ, Chaurand P, Caprioli RM, Montine TJ, Oates JA. J Neurochem. 2002;82:1003–1006. doi: 10.1046/j.1471-4159.2002.01064.x. [DOI] [PubMed] [Google Scholar]
- [6].Hayden KM, Zandi PP, Khachaturian AS, Szekely CA, Fotuhi M, Norton MC, Tschanz JT, Pieper CF, Corcoran C, Lyketsos CG, Breitner JC, Welsh-Bohmer KA. Neurology. 2007;69:275–282. doi: 10.1212/01.wnl.0000265223.25679.2a. [DOI] [PubMed] [Google Scholar]
- [7].Breitner JC, Martin BK, Meinert CL. PLoS Clin. Trials. 2006;1:e41. doi: 10.1371/journal.pctr.0010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Salpeter SR, Gregor P, Ormiston TM, Whitlock R, Raina P, Thabane L, Topol EJ. Am. J. Med. 2006;119:552–559. doi: 10.1016/j.amjmed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- [9].Bresalier RS, Friedewald VE, Jr., Rakel RE, Roberts WC, Williams GW. Am. J. Cardiol. 2005;96:1589–1604. doi: 10.1016/j.amjcard.2005.09.069. [DOI] [PubMed] [Google Scholar]
- [10].Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Ann. Rev. Pharmacol. Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- [11].Nakamura K, Li YQ, Kaneko T, Katoh H, Negishi M. Neuroscience. 2001;103:763–775. doi: 10.1016/s0306-4522(01)00027-6. [DOI] [PubMed] [Google Scholar]
- [12].Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Nat. Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- [13].Batshake B, Nilsson C, Sundelin J. Eur. J. Biochem. 1995;231:809–814. doi: 10.1111/j.1432-1033.1995.tb20765.x. [DOI] [PubMed] [Google Scholar]
- [14].Candelario-Jalil E, Slawik H, Ridelis I, Waschbisch A, Akundi RS, Hull M, Fiebich BL. J. Mol. Neurosci. 2005;27:303–310. doi: 10.1385/JMN:27:3:303. [DOI] [PubMed] [Google Scholar]
- [15].Kitanaka J, Hashimoto H, Gotoh M, Kondo K, Sakata K, Hirasawa Y, Sawada M, Suzumura A, Marunouchi T, Matsuda T, Baba A. Brain Res. 1996;707:282–287. doi: 10.1016/0006-8993(95)01256-7. [DOI] [PubMed] [Google Scholar]
- [16].Caggiano AO, Kraig RP. J. Neurochem. 1999;72:565–575. doi: 10.1046/j.1471-4159.1999.0720565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Slawik H, Volk B, Fiebich B, Hull M. Neurochem. Int. 2004;45:653–660. doi: 10.1016/j.neuint.2004.04.007. [DOI] [PubMed] [Google Scholar]
- [18].Ek M, Arias C, Sawchenko P, Ericsson-Dahlstrand A. J. Comp. Neurol. 2000;428:5–20. doi: 10.1002/1096-9861(20001204)428:1<5::aid-cne2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- [19].Bhattacharya M, Peri K, Almazan G, Ribeiro-da-Silva A, Shichi H, Durocher Y, Abramovitz M, Hou X, Varma D, Chemtob S. Proc. Natl. Acad. Sci. USA. 1998;95:15792–15727. doi: 10.1073/pnas.95.26.15792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Negishi M. J Comp. Neurol. 2000;421:543–569. doi: 10.1002/(sici)1096-9861(20000612)421:4<543::aid-cne6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- [21].Oka T, Oka K, Schammell TE, Lee C, Kelly JF, Nantel F, Elmquist JK, Saper CB. J. Comp. Neurol. 2000;428:20–32. doi: 10.1002/1096-9861(20001204)428:1<20::aid-cne3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- [22].Sugimoto Y, Shigemoto R, Namba T, Negishi M, Mizuno N, Narumiya S, Ichikawa A. Neuroscience. 1994;62:919–928. doi: 10.1016/0306-4522(94)90483-9. [DOI] [PubMed] [Google Scholar]
- [23].Milatovic D, Zaja-Milatovic S, Montine KS, Nivison M, Montine TJ. Current Medicinal Chemistry. 2005;5:151–156. [Google Scholar]
- [24].Borda ES, Tenenbaum A, Sales ME, Rumi L, Sterin-Borda L. Prostaglandins Leukot. Essent. Fatty Acids. 1998;58:85–90. doi: 10.1016/s0952-3278(98)90145-4. [DOI] [PubMed] [Google Scholar]
- [25].Breyer MD, Breyer RM. Annu. Rev. Physiol. 2001;63:579–605. doi: 10.1146/annurev.physiol.63.1.579. [DOI] [PubMed] [Google Scholar]
- [26].Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- [27].Pepicelli O, Fedele E, Berardi M, Raiteri M, Levi G, Greco A, Ajmone-Cat MA, Minghetti L. J. Neurochem. 2005;93:1561–1567. doi: 10.1111/j.1471-4159.2005.03150.x. [DOI] [PubMed] [Google Scholar]
- [28].Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. J. Pharmacol. Exp. Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- [29].Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Proc. Natl. Acad. Sci. USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Ann. Neurol. 2005;57:758–761. doi: 10.1002/ana.20461. Therapeutic Targets in Prostaglandin E2 Signaling for Neurologic Disease Current Medicinal Chemistry, 2008 Vol. 15, No. 14 7. [DOI] [PubMed] [Google Scholar]
- [31].Bilak M, Wu L, Wang Q, Haughey N, Conant K, Hillaire C, Andreasson K. Ann. Neurol. 2004;56:240–248. doi: 10.1002/ana.20179. [DOI] [PubMed] [Google Scholar]
- [32].Shie FS, Breyer RM, Montine TJ. Am. J. Pathol. 2005;166:1163–1172. doi: 10.1016/s0002-9440(10)62336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Echeverria V, Clerman A, Dore S. Eur. J. Neurosci. 2005;22:2199–2206. doi: 10.1111/j.1460-9568.2005.04427.x. [DOI] [PubMed] [Google Scholar]
- [34].Imler JL, Hoffmann JA. Trends Cell Biol. 2001;11:304–311. doi: 10.1016/s0962-8924(01)02004-9. [DOI] [PubMed] [Google Scholar]
- [35].Akira S. J. Biol. Chem. 2003;278:38105–38108. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- [36].Johnson GB, Brunn GJ, Platt JL. Crit. Rev. Immunol. 2003;23:15–44. doi: 10.1615/critrevimmunol.v23.i12.20. [DOI] [PubMed] [Google Scholar]
- [37].Fassbender K, Walter S, Kuhl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ, Rivest S, Lentschat A, Gulbins E, Jucker M, Staufenbiel M, Brechtel K, Walter J, Multhaup G, Penke B, Adachi Y, Hartmann T, Beyreuther K. FASEB J. 2004;18:203–205. doi: 10.1096/fj.03-0364fje. [DOI] [PubMed] [Google Scholar]
- [38].Moffatt OD, Devitt A, Bell ED, Simmons DL, Gregory CD. J. Immunol. 1999;162:6800–6810. [PubMed] [Google Scholar]
- [39].Nicolle MM, Gonzalez J, Sugaya K, Baskerville KA, Bryan D, Lund K, Gallagher M, McKinney M. Neuroscience. 2001;107:415–431. doi: 10.1016/s0306-4522(01)00374-8. [DOI] [PubMed] [Google Scholar]
- [40].Milatovic D, Zaja-Milatovic S, Montine KS, Shie FS, Montine TJ. J. Neuroinflammation. 2004;1:20. doi: 10.1186/1742-2094-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. J. Neurochem. 2003;87:1518–1526. doi: 10.1046/j.1471-4159.2003.02120.x. [DOI] [PubMed] [Google Scholar]
- [42].Shie FS, Montine KS, Breyer RM, Montine TJ. Glia. 2005;52:70–77. doi: 10.1002/glia.20220. [DOI] [PubMed] [Google Scholar]
- [43].Dringen R. Antioxid. Redox Signal. 2005;7:1223–1233. doi: 10.1089/ars.2005.7.1223. [DOI] [PubMed] [Google Scholar]
- [44].Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Nat. Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- [45].Jantzen PT, Connor KE, DiCarlo G, Wenk G, Wallace J, Rojiani A, Coppola D, Morgan D, Gordon M. J. Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM. Neurobiol. Aging. 2001;22:983–991. doi: 10.1016/s0197-4580(01)00299-8. [DOI] [PubMed] [Google Scholar]
- [47].Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. J. Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. J. Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- [50].Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH. J. Biol. Chem. 2003;278:30748–30754. doi: 10.1074/jbc.M304824200. [DOI] [PubMed] [Google Scholar]
- [51].Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH. J. Biol. Chem. 2003;278:31831–31837. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]
- [52].Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. J. Clin. Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lanz TA, Fici GJ, Merchant KM. J. Pharmacol. Exp. Ther. 2005;312:399–406. doi: 10.1124/jpet.104.073965. [DOI] [PubMed] [Google Scholar]
- [54].Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. J. Neurosci. 2005;25:10180–10187. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Woltjer RL, McMahan W, Milatovic D, Kjerulf JD, Shie FS, Rung LG, Montine KS, Montine TJ. Neurobiol. Dis. 2007;25:427–437. doi: 10.1016/j.nbd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- [56].McGeer PL, McGeer EG. Parkinsonism Relat. Disord. 2004;10(Suppl 1):S3–7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- [57].Feng ZH, Wang TG, Li DD, Fung P, Wilson BC, Liu B, Ali SF, Langenbach R, Hong JS. Neurosci. Lett. 2002;329:354–358. doi: 10.1016/s0304-3940(02)00704-8. [DOI] [PubMed] [Google Scholar]
- [58].Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S. Proc. Natl. Acad. Sci. USA. 2003;100:5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Carrasco E, Casper D, Werner P. J. Neurosci. Res. 2007;85:3109–3117. doi: 10.1002/jnr.21425. [DOI] [PubMed] [Google Scholar]
- [60].Jin J, Shie FS, Liu J, Wang Y, Davis J, Schantz AM, Montine KS, Montine TJ, Zhang J. J. Neuroinflammation. 2007;4:2. doi: 10.1186/1742-2094-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Maihofner C, Probst-Cousin S, Bergmann M, Neuhuber W, Neundorfer B, Heuss D. Eur. J. Neurosci. 2003;18:1527–1534. doi: 10.1046/j.1460-9568.2003.02879.x. [DOI] [PubMed] [Google Scholar]
- [62].Yasojima K, Tourtellotte WW, McGeer EG, McGeer PL. Neurology. 2001;57:952–956. doi: 10.1212/wnl.57.6.952. [DOI] [PubMed] [Google Scholar]
- [63].Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Drachman DB, Rothstein JD. Ann. Neurol. 2000;48:792–795. [PubMed] [Google Scholar]
- [65].Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- [66].Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- [67].Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- [68].Almer G, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP, Chen C, Przedborski S. Ann. Neurol. 2001;49:176–185. [PubMed] [Google Scholar]
- [69].Klivenyi P, Kiaei M, Gardian G, Calingasan NY, Beal MF. J. Neurochem. 2004;88:576–582. doi: 10.1046/j.1471-4159.2003.02160.x. [DOI] [PubMed] [Google Scholar]
- [70].Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, Rothstein JD. Ann. Neurol. 2002;52:771–778. doi: 10.1002/ana.10374. [DOI] [PubMed] [Google Scholar]
- [71].Pompl PN, Ho L, Bianchi M, McManus T, Qin W, Pasinetti GM. FASEB J. 2003;17:725–727. doi: 10.1096/fj.02-0876fje. [DOI] [PubMed] [Google Scholar]
- [72].Cudkowicz ME, Shefner JM, Schoenfeld DA, Zhang H, Andreasson KI, Rothstein JD, Drachman DB. Ann. Neurol. 2006;60:22–31. doi: 10.1002/ana.20903. [DOI] [PubMed] [Google Scholar]
- [73].Almer G, Kikuchi H, Teismann P, Przedborski S. Ann. Neurol. 2006;59:980–983. doi: 10.1002/ana.20847. [DOI] [PubMed] [Google Scholar]
- [74].Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Proc. Natl. Acad. Sci. USA. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Nogawa S, Zhang F, Ross ME, Iadecola C. J. Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ahmad AS, Saleem S, Ahmad M, Dore S. Toxicol. Sci. 2006;89:265–270. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- [77].Saleem S, Li RC, Wei G, Dore S. J. Neurosci. Res. 2007;85:2433–2440. doi: 10.1002/jnr.21399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhou P, Qian L, Chou T, Iadecola C. Neurobiol. Dis. 2008;29:543–551. doi: 10.1016/j.nbd.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. J. Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ahmad M, Ahmad AS, Zhuang H, Maruyama T, Narumiya S, Dore S. J. Neuroimmunol. 2007;184:172–179. doi: 10.1016/j.jneuroim.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Combrinck M, Williams J, De Berardinis MA, Warden D, Puopolo M, Smith AD, Minghetti L. J. Neurol. Neurosurg. Psychiatry. 2006;77:85–88. doi: 10.1136/jnnp.2005.063131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ, 2nd, Morrow JD. Neurology. 1999;53:1495–1498. doi: 10.1212/wnl.53.7.1495. [DOI] [PubMed] [Google Scholar]
- [83].Ilzecka J. Acta Neurol. Scand. 2003;108:125–129. doi: 10.1034/j.1600-0404.2003.00102.x. [DOI] [PubMed] [Google Scholar]
- [84].Almer G, Teismann P, Stevic Z, Halaschek-Wiener J, Deecke L, Kostic V, Przedborski S. Neurology. 2002;58:1277–1279. doi: 10.1212/wnl.58.8.1277. [DOI] [PubMed] [Google Scholar]
- [85].Minghetti L, Greco A, Cardone F, Puopolo M, Ladogana A, Almonti S, Cunningham C, Perry VH, Pocchiari M, Levi G. J. Neuropathol. Exp. Neurol. 2000;59:866–871. doi: 10.1093/jnen/59.10.866. [DOI] [PubMed] [Google Scholar]
- [86].Minghetti L, Cardone F, Greco A, Puopolo M, Levi G, Green AJ, Knight R, Pocchiari M. Neurology. 2002;58:127–129. doi: 10.1212/wnl.58.1.127. [DOI] [PubMed] [Google Scholar]
- [87].Aktan S, Aykut C, Ercan S. Prostaglandins Leukot. Essent. Fatty Acids. 1991;43:247–249. doi: 10.1016/0952-3278(91)90037-6. [DOI] [PubMed] [Google Scholar]
- [88].Griffin DE, Wesselingh SL, McArthur JC. Ann. Neurol. 1994;35:592–597. doi: 10.1002/ana.410350513. [DOI] [PubMed] [Google Scholar]