

Abstract
The way in which spatially patterned cellular identities are generated is a central question of organogenesis. In the case of Drosophila heart formation, the cardiac progenitors are specified in precise mesodermal positions, giving rise to multiple cell types in a highly ordered arrangement. Here, we study the mechanisms by which positional information conveyed by signaling pathways and a combinatorial code of activating and repressing transcription factors work together to confine the expression of the homeobox gene even-skipped (eve) to a small region of the dorsal mesoderm. By manipulating both expression patterns and binding sites for transcription factors, we show that a complex combination of regulatory activities converge on a single enhancer of eve to generate precisely targeted gene expression within the cardiac mesoderm. In particular, ladybird early (lbe), a homeobox gene expressed adjacent to eve, restricts the positive actions of factors downstream of wingless, decapentaplegic, and ras to generate the eve pattern. Mutation of a Lbe binding site causes dramatic expansion of expression and abolishes the responsiveness to repression by lbe. Conversely, eliminating eve in the mesoderm expands lbe expression into the normal eve-expressing territory, suggesting that mutual repression between eve and lbe is essential for delineating the spatial patterns of gene expression that specify cell types within the cardiac mesoderm.
Keywords: even-skipped, heart, cell fate, tinman, ladybird, wingless, TGF-β, repression
INTRODUCTION
A recurring theme in studies of eukaryotic gene regulation has been the involvement of multiprotein complexes that bind DNA in a sequence-specific manner and either repress or activate transcription. This combinatorial control is thought to enable the function of the complex regulatory networks found in higher eukaryotes. However, little is known about mechanisms that orchestrate tissue assembly during organogenesis. In Drosophila, progenitor cells for a particular organ often come from a restricted region of the embryo under the influence of a group of factors with overlapping expression patterns, which continue to be expressed as the formation of tissue types progresses. Therefore, an efficient way to generate the diversity of cell types within a developing organ might be to continue to utilize those initiating factors along with other factors that integrate upstream the activities and also act combinatorially with them to directly regulate downstream genes.
The Drosophila heart is a highly organized linear tube located beneath the dorsal midline. The cardiac precursors are specified at the dorsal margin of the trunk mesoderm, giving rise to distinct cell types that are arranged in a segmentally repeated pattern. The molecular processes involved in cardiac mesoderm formation have been studied in some detail (reviewed in Bodmer and Frasch, 1999; Lockwood et al., 2001). A number of signaling pathways and transcription factors have been shown to function in these processes, leading to models of a hierarchical network of genetic interactions that govern mesoderm differentiation and heart development. The first zygotic mesoderm determinant, Twist, a bHLH protein (Thisse et al., 1988), is required for pan-mesodermal expression of the homeobox genes tinman and zfh-1, the MADS-box gene Dmef2, and the FGF-receptor Heartless (Bodmer et al., 1990; Lai et al., 1991; Shishido et al., 1993; Nguyen et al., 1994; Beiman et al., 1996; Gisselbrecht et al., 1996; Yin et al., 1997). After the mesoderm invaginates ventrally during gastrulation, it forms a monolayer of cells along the dorsal-ventral axis in close apposition to the overlaying ectoderm (Leptin and Grunewald, 1990). tinman is restricted to the dorsal portion of this monolayer and is required for specifying dorsal mesodermal fates, in conjunction with the TGF-β signal encoded by decapentaplegic (dpp; Frasch, 1995; Staehling-Hampton et al., 1994; Xu et al., 1998). The expression of tinman is further restricted to the cardiac mesoderm at the dorsal margin of the mesoderm by a combination of positional information provided by dpp and by a Wnt signal, encoded by wingless (wg), which is expressed in segmental stripes that are oriented orthogonally to dpp (Wu et al., 1995; Lockwood and Bodmer, 2002). During this process, receptor tyrosine kinase (RTK) action contributes to the subsequent subdivision of this region into small clusters of equivalent cells from which individual cardiac progenitors segregate (Halfon et al., 2000; Carmena et al., 2002).
By as yet unknown mechanisms, identity genes are expressed in distinct cardiac progenitors in different combinations, generating the diversity of cardiac cell types (Su et al., 1999; Lo and Frasch, 2001; Gajewski et al., 2000; Ward and Skeath, 2000; Jagla et al., 1997, 2002). By stage 14, the progenitor cells have generated at least eight different cardiac cell types (see Fig. 1J). Some of the earliest progenitors that emerge from the cardiogenic region express the pair-rule homeobox gene even-skipped (eve) in small, segmentally repeated clusters that later differentiate into Evepositive pericardial cells (EPCs) and the dorsal muscle DA1 (Frasch et al., 1987; see also Fig. 1A). eve has been proposed to act as an identity factor essential for EPCs to acquire and maintain their identity, as it appears to be essential for them to maintain normal gene expression patterns during subsequent differentiation (Su et al., 1999; Jagla et al., 2002).
FIG. 1.

Distinct combinations of transcription factors specify at least eight cardiac cell types in Drosophila embryos. Confocal laser scans of eve-expression (green) relative to other cardiac markers (red) in Drosophila embryos (except I, J). (A) At stage 11, mesodermal Eve cells are a subset of the tinman-expressing cardiogenic region (red) and thus appear yellow. (B) Stage 14. (C) Stage 16. The mesodermal Eve clusters differentiate into EPC pericardial cells (yellow) and DA1 muscle founders (green). In red are the Tinman myocardial and additional pericardial cells. Before stage 13, all myocardial cells express tinman, whereas from stage 15 on, only 4 out of 6 myocardial cells per hemisegment do so (see also J). (D) lbe is expressed in a subset of the late stage 11 tinman-expressing cardiogenic region (red) adjacent to Eve (green, compare with A). (E, F) Stage 14 Lbe clusters differentiate into two myocardial (TLMC) and two pericardial cells (LPC). Lbe is shown in red and either Eve or Tin in green as indicated. (G, H) svp-expressing heart cells (red) do not overlap with eve (late stage 11 and stage 13 are shown). (I) Zfh1 (red) marks all pericardial cells and is coexpressed with Odd-skipped (green) at stage 16. (J) Diagram of stage 16 cardiac cell types as identified by marker gene expression (see also Jagla et al., 1997, 2002; Su et al., 1999; Bodmer and Frasch, 1999; Ward and Skeath, 2000). MC, myocardial cells; PC, pericardial cells; TMC, Tinman myocardial cells; TLMC, Tinman and Lbe myocardial cells; SMC, Svp myocardial cells; EPC, Eve pericardial cells; ZPC, Zfh-1 pericardial cells; OPC, Odd-skipped pericardial cells; SOPC, Odd-skipped and Svp pericardial cells; LPC, Lbe pericardial cells; DA1, dorsal acute muscle 1; FB, fat body.
Regulatory information sufficient to target eve expression precisely to a small subset of cells within the cardiac mesoderm resides within a discrete genomic region located 3′ of the eve transcription unit (Fujioka et al., 1999; Su et al., 1999). All of the genetic inputs required for conferring a cardiac mesodermal competence regulate this enhancer region, which includes twist, tinman, wg, dpp, and RTK/ras (Halfon et al., 2000; Knirr and Frasch, 2001; this study). However, since these inputs do not distinguish EPC progenitors from other cardiac cell types, there must be additional inputs contributing to confer spatial specificity. Genetic data have suggested two additional candidates for delineating eve expression within the cardiac mesoderm (Jagla et al., 1997, 2002; Gajewski et al., 2000; Lo and Frasch, 2001): the homeobox gene ladybird early (lbe) and the COUP nuclear hormone receptor gene seven-up (svp). They are expressed adjacent to but not overlapping the Eve clusters (see Fig. 1). Although when overexpressed ubiquitously they can repress mesodermal eve, it is not known whether they act directly.
In this report, we explore the combinatorial mechanisms that lead to spatially controlled cell fate determination during cardiac development by examining the regulation of eve in vivo. Overexpression experiments and dissection of a minimal eve mesodermal enhancer, eme, suggest that integration of the genetic inputs that regulate mesodermal eve expression occurs directly on the enhancer. We find that Tinman and dTCF (mediating wg signaling) activate and maintain eme activity, and that lbe and at least one other factor restrict eve expression to a small subset of the cardiogenic region. Mesodermal eve expression appears to be directly restricted by lbe, since a mutated enhancer, unable to bind Lbe in vitro, generates dramatically expanded expression within the cardiac mesoderm and is no longer sensitive to inhibition by lbe. In contrast, we find that svp is unable to repress eve directly, but acts indirectly perhaps by inhibiting tinman expression in a subset of heart cells. By constructing an eve rescue transgene that lacks the eme enhancer, we find that eme is not only sufficient when assayed in isolation, but is also necessary within the context of the entire eve gene for conferring appropriate expression in the mesoderm. While subsets of this enhancer are apparently sufficient when assayed in isolation upstream of a reporter gene, the requirements seem to be more stringent within the normal context.
In addition to the direct restriction of eve expression by lbe, in the absence of mesodermal eve, lbe expression invades the eve-expressing territory. We conclude that mutual repression between eve and lbe within the region of cardiac competence (conferred by wg and dpp signaling in the mesodermal context of tinman) is essential for distinguishing cell type identities during cardiac organogenesis.
MATERIALS AND METHODS
Drosophila Strains
The following mutant stocks were used: Dmef2P520 (Bour et al., 1995), eve3 (Bloomington Stock Center). Overexpression of transgenes was accomplished by using the Gal4-UAS system (Brand and Perrimon, 1993). The following fly lines were used: twi-Gal4 and 24B-Gal4 (conferring early and late, respectively, pan-mesodermal expression; Greig and Akam, 1993; Brand and Perrimon, 1993), eme-Gal4 (see below), UAS-lbe (Jagla et al., 1997), UAS-Ricin (Sentry et al., 1993), UAS-DN-dTCF (van de Wetering et al., 1997), UAS-DN-tinman (see below), UAS-DN-thickvein (Haerry et al., 1998), and UAS-svp1 (obtained from M. Mlodzik). Oregon-R was used as the wild-type reference strain.
Generation of Transgenic Fly Lines
The various eve mesodermal enhancer (eme) fragments (see Fig. 3B) were PCR amplified and subcloned into P[lacZ,w+] transformation vectors, pWHL (Ip et al., 1992) or C4pLZ (Wharton and Crews, 1993) using SphI/XhoIor KpnI/NotI sites, respectively. The eme-PlacZ constructs (100 μg/ml) were injected according to standard procedures (Spradling, 1986). Germline transformed, transgenic flies were selected by red eye color (w+) and maintained as homozygotes. At least four independent transgenic lines were analyzed for each construct.
FIG. 3.

Dissecting cell fate decisions by targeting gene expression specifically to the mesodermal Eve lineage. (A) eme-Gal4 driving UAS-GFP (eme > GFP), which outlines the cellular morphologies of the EPCs and the DA1 muscles (and faintly DO2 in some segments). A living embryo at stage 16 is shown. (B-I) Double-labeling with Eve (red) and Dmef2 (green) of stage 14 embryos (except G, which is stage 12). (B) Wild-type embryo. EPCs (red) express Eve only, while DA1 (yellow) colabels with Eve and Dmef2. (C) Targeted expression (using eme-Gal4) of the toxic gene product Ricin selectively eliminates all of the eve-expressing cells (red) in the mesoderm (indicated by asterisks). (D-F) Targeted expression of a dominant-negative form of dTCF (DN-dTCF, D) of Tinman (DN-tin, E) and of Thickvein (DN-tkv, F) in the mesodermal Eve cell lineage reduces the level of eve expression. Asterisk indicates a missing EPC. (G, H) eme > lbe eliminates mesodermal eve expression after stage 12 (H) but not before (G). Asterisks indicate absence of eve expression. (I) eme > svp1 has no discernable effect.
The dominant-negative tinman (DN-Tin) was constructed by using a repressor domain from engrailed (EnR, amino acid 2-298; Jaynes and O’Farrell, 1991; Smith and Jaynes, 1996; Tolkunova et al., 1998) and the tinman homeodomain (TinHD, amino acid 291-370; Bodmer et al., 1990), according to the strategy described by Fu et al. (1998). EnR was PCR-amplified from the engrailed DNA and inserted into EcoRI- and StuI-digested pCS2+nls vector (Turner and Weinctraub, 1994). TinHD was PCR-amplified from the full-length tinman cDNA (5′ primers, CATCTCGAGATGAGCAACAGTGGTTCCACCAAGCCC; 3′ primer, GCTCTAGACAGATGCTTGGCGATGCCCTCGCA) and inserted into XhoI- and XbaI-digested pCS2+nls vector containing EnR (S. Evans, M.L., R.B., unpublished observations). The EnR-TinHD fusion construct was subcloned into EcoRI- and XbaI-digested pUAST vector, and transgenic fly lines were generated as previously described (Brand and Perrimon, 1993). EnR alone was also subcloned into the pUAST vector and used as a control.
The eme-GAL4 construct contains two tandem copies of the mesodermal enhancer region of eve, from +5.8 to +6.6 kb (Fujioka et al., 1999), upstream of the eve promoter from nt -275 to +11, followed by polylinker sequences and the eve 5′ untranslated leader from +91 to +99 nt, fused with the Gal4 coding region (resulting in replacement of the yeast translation initiation site, GAAAGATG, by that from eve, ATACCAAACATG), followed by the eve 3′ untranslated region from +1306 (BstUI site) to +1521 nt (KpnI). These modifications significantly increased Gal4 activity in the mesoderm. This was established in transgenic lines as previously described (Fujioka et al., 2000).
To generate eme-deficient eve rescue constructs, the region from +6.1 (EagI) to +6.6 kb (StuI), or from +6.3 (NcoI) to +6.6 kb (StuI), was deleted from an eve rescue construct from -6.4 to +9.2 kb, as described previously (Fujioka et al., 1999) to generate P[eve-emeΔES] and P[eve-emeΔNS], respectively (see also Fig. 7). Phenotypic analysis was done in either a Df(2R)eve or an eve3 (null) mutant background, both giving indistinguishable results.
FIG. 7.

Study of eve null mutants rescued by an eve transgene containing eme deletions. (A) Diagram of eme deletions in the eve transgene construct, P[eve+,emeΔES] and P[eve+,emeΔNS], as well as the P[eve+], which is the wild-type 16-kb genomic eve rescue construct. J48, J49, J43, and 86T indicate independent lines used. (B, C) Eve protein expression patterns in stage 11 embryos. Arrows indicate positions of mesodermal Eve clusters. (B) Wild-type embryo. (C) Df(2L)eve;P[eve+,emeΔES] (meso- rescued), J48, embryo. Note the selective loss of Eve protein in the dorsal mesoderm in (C) (arrows), while expression appears normal in all other tissues including the CNS. (D-G) Eve protein (green) and LacZ (red) in late stage 11 (D, E) and stage 13 embryos (F, G), containing emeA-LacZ in either a wild-type 16-kb eve gene rescued, Df(2L)eve;P[eve+], 86T (D,F) or a meso- rescued background, J48 (E, G). Note the absence of Eve but the presence of a near normal pattern of LacZ in the dorsal mesoderm in (E) and later largely reduced expression in (G). (H, I) Stage 12 embryos double-labeled for LacZ (green) and Lbe (red). emeA in either a wild-type rescued (H) or a meso- rescued background (I). Note that in the absence of Eve, lbe is derepressed in the territory normally expressing eve.
Immunohistochemistry and Microscopy
Antibody staining of embryos was carried out as previously described by Su et al. (1999), except that Cy3- or FITC-conjugated secondary antibodies (The Jackson Laboratory) were used for fluorescent confocal microscopy. For Lbe staining, Biotin-conjugated secondary antibodies were used, followed by incubation (30 min) with Streptavidin-Fluoresceine DTAF (1:300; The Jackson Laboratory). Embryos were mounted in VectaShield (Vector Laboratories). Fluorescent embryo staining was analyzed by using a Zeiss LSM510 confocal microscope, and the images were further processed by using Adobe Photoshop. The following primary antibodies were used: anti-β-galactosidase 1:300 (Promega); anti-Eve 1:10,000 (Frasch et al., 1987); anti-Tin 1:500 (Venkatesh et al., 2000); anti-Dmef2 1:1000 (Lilly et al., 1995); anti-Zfh-1c 1:1000 (Lai et al., 1991); and anti-Lbe 1:5 (Jagla et al., 1997).
Electrophoretic Mobility Shift Assays
Gene Inspector was used to find consensus binding sequences for known transcription factors allowing one or two mismatches in the eme900 enhancer. Oligonucleotides containing the putative binding sites were tested by gel electrophoretic mobility shift assays. Tinman: TNAAGTG (Gajewski et al., 1997); dTCF: CCTTTGATCTT (van de Wetering et al., 1997); Mad/Medea: CGCCGCGACG (Xu et al., 1998); Ladybird: CTAATTGA (K. Jagla, personal communication).
Tinman homeodomain protein was prepared by K. Occor (Venkatesh et al., 2000). Lbe protein was provided by C. Jagla. Su(H), dTCF, and Medea proteins were generated in vitro by using Promega’s TnT Transcription and Translation Kit. For electrophoretic mobility shift assays, oligonucleotides containing wild-type or mutated binding sites were end-labeled, protein-DNA binding assays were carried out as described previously (Su et al., 1999), and the products were electrophoresed in 7.5% nondenaturing polyacrylamide gels at 4°C. The following oligonucleotides were used: Tin3, GGATGCCCACACTTGAGGAG; Tin3*, GGATGCCTGCTGGAGGAG; Tin12, CTTCACAGTTCTCAGGCACTTAAGATA; Tin 12*, CTTCAGGCCACAGGCAGGCAAGATA; Lb1, GCCATCAATTAGCATACAATT; Lb1*, GCCATCAGCCAGCATACAATT; Lb2, CGCCTGCTAATTGAGATCGCGGCG; Lb2*, CGCCTGCTGCCTGAGATCGCG; dTCF, GGGCAGCAGATCAAAGCGACG; dTCF*, GGGCAGATTGCAGCGACG.
Site-Directed Mutagenesis
Site-directed mutagenesis of eme2 and emeA was performed by using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). The emeA fragment derived from eme900 was cloned into pT7Blue3, subjected to mutagenesis, and subcloned into the KpnI and NotI sites of C4pLZ. The following underlined base pair changes were made (see Fig. 3B): Tin1*, CTTCAGGCCACA; Tin2*, GGCAGGCAAGA; Tin3*, GCCTGCTGGAGG; dTCF*, GCAGATTGCAGCGA; Lb1*, CCATCAGCCAGCAT; Lb2*, GCCTGCTGCCTGAGA; Su(H)*, GTTGTAGGGCA; Medea*, GAGATC- - - -CGATCC.
RESULTS
eve Is Expressed within the Cardiogenic Region
As the pair-rule pattern of eve expression fades in the ectoderm at early stage 11, mesodermal eve expression begins in segmentally reiterated clusters of cells (Frasch et al., 1987). This is also the time when tinman expression is refined from the entire dorsal mesoderm to the cardiogenic region at the dorsal margin (Fig. 1A). Each of these clusters gives rise to two EPCs and a founder of the DA1 muscle. Although tinman is expressed in all of the Eve progenitor cells, expression persists at later stages only in the EPCs and not in the DA1 muscle founders (Figs. 1A-1C). After the completion of dorsal closure and heart tube assembly, tinman is segmentally expressed in a set of four of six myocardial cells per hemisegment, as well as in the EPCs and the other pericardial cells (Fig. 1C). The Lbe clusters are also confined within the tinman-expressing cardiac mesoderm but located immediately anterior (and later dorsal) to the Eve clusters (Figs. 1D-1F; Jagla et al., 1997). Lbe- and Eve-positive mesodermal cells never overlap. Similarly, svp expression does not coincide with Eve (Figs. 1G and 1H) or Lbe (data not shown). All mesodermal cells initially express zfh-1 and Dmef2, but later zfh-1 expression is restricted to the pericardial cells, including the EPCs and Odd-skipped positive pericardial cells (Fig. 1I), while Dmef2 persists only in cells destined to become myocardial heart cells and other muscles, including DA1 (see below; Bour et al., 1995; Lilly et al., 1995; Park et al., 1998; Su et al., 1999; Ward and Skeath, 2000). eve expression remains strong in the EPCs during heart tube formation, whereas the DA1 muscle founders fuse with surrounding myocytes to form a muscle syncytium (Figs. 1B and 1C; see below). A summary of all identified cell types is shown in Fig. 1J.
A Minimal Mesodermal eve Enhancer
The restricted expression of eve in the dorsal mesoderm suggests that, in addition to requiring the combination of the wg and dpp signals and the “cardiogenic context” of tinman, additional patterning mechanisms must contribute to the precise localization of the cell types that participate in cardiogenesis. In order to obtain a better understanding of the genetic mechanisms controlling mesodermal eve expression and EPC/DA1 specification, we examined the enhancer activity of a 900-bp element located 5.7-6.6 kb downstream of the eve transcription start site, which is sufficient to confer apparently normal mesodermal eve expression (Figs. 2A-2C; Fujioka et al., 1999; Su et al., 1999). This enhancer contains consensus binding sites for the transcription factors known to be regulated by the genetic inputs that give rise to the mesodermal eve pattern, including Tinman, dTCF (wg pathway), Mad/Medea (dpp pathway), and Lbe (Figs. 2A and 2O; see also Fig. 6). Gel-shift assays with Tinman, dTCF, and Lbe protein show that these consensus sites are indeed bound by these proteins in vitro (Fig. 2N).
FIG. 2.
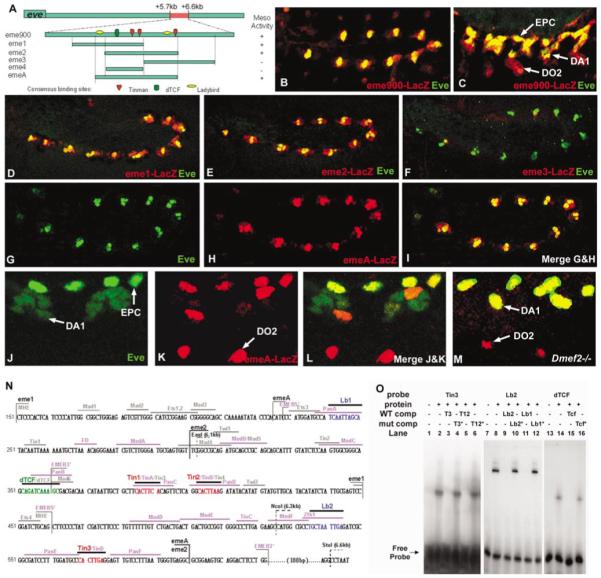
Dissection of the mesodermal eve enhancer. (A) Diagram of eme fragments and transcription factor consensus binding sites. The 900-bp eve mesodermal enhancer (eme900), located between +5.7 and +6.6 kb in the eve locus (Fujioka et al., 1999), 3′ of the coding region. This fragment was used to generate the eme-Gal4 driver used in Fig. 3. (B) Stage 11 and (C) stage 14 eme900-mediated cytoplasmic lacZ expression (red) and nuclear Eve (green) overlap in the EPCs and DA1 muscle. Note continued lacZ expression in an Eve-negative muscle (DO2), derived from an Eve-positive progenitor. (D-M) All embryos are double-stained for Eve (green) and/or LacZ (red), as indicated. (D-I) Late stage 11, and (J-M) high magnification (two hemisegments) of stage 14. (D) Overlap of cytoplasmic eme1-LacZ with nuclear Eve in the dorsal mesoderm (compare with eme-900 in Fig. 2B, which is more restricted). (E) Overlap of cytoplasmic eme2-LacZ with nuclear Eve in the dorsal mesoderm. Note that eme2-LacZ is slightly more restricted than eme1-LacZ (in D). (F) No expression is conferred by eme3-LacZ. (G-I) Overlap of nuclear emeA-LacZ with nuclear Eve in the dorsal mesoderm. Note the almost perfect coincidence of expression (in I). (J-L) emeA-LacZ coincides with Eve in EPCs and founders of the DA1 muscles. Note that myoblasts that have fused with the DA1 founder contain low levels of nuclear Eve but not LacZ, while LacZ but not Eve is present in the DO2 founders (see text). (M) emeA-LacZ expression in Dmef2 mutant embryo shows coincidence of LacZ and Eve in the DA1 founders, which fail to fuse with surrounding myoblasts in this mutant. (N) Nucleotide sequence of eme (numbering refers to eme900: +5.7 to +6.6 kb; Fujioka et al., 1999). MHE refers to the fragment used by Halfon et al. (2000); EMEB5′ and EMEB3′ refer to the fragments tested by Knirr and Frasch (2001). Identified transcription factor consensus binding sites are indicated by bars over the sequences. Sites studied by Halfon et al. are shown in gray, those studied by Knirr and Frasch in purple. All others are the focus of the present study. Restriction enzyme sites (EglI, NcoI, and StuI) were used to generate the deletions produced in the genomic -6.4 to +9.2kb eve rescue construct (see Fig. 7; Fujioka et al., 1999). (O) Gel-shift assays with the Tin, Lbe, and dTCF sites (see Materials and Methods). Lanes 1-6 show binding of the Tinman homeodomain (TinHD; Venkatesh et al., 2000) to a probe containing the Tin3 site, which is abolished when wild-type (Tin3 or Tin12) but not mutant (Tin3m or Tin12m) competitor is added. Lanes 7-12 show that binding of Lbe protein to the wild-type (Lb2) probe is reduced or abolished when wild-type (Lb1 or Lb2) but not mutant (Lb1m or Lb2m) competitor is added. Lanes 13-16 show that binding of dTCF protein to the dTCF site is abolished when wild-type (dTCF) but not mutant (dTCFm) competitor is added.
FIG. 6.

Combinatorial models of mesodermal Eve cell-type specification. (A) The eve mesodermal enhancer (eme) integrates both activation and repression from multiple signaling pathways and transcription factors to generate the highly restricted mesodermal Eve pattern. The data presented in this paper, together with other data (Halfon et al., 2000; Knirr and Frasch, 2001), suggest that the wg and dpp signals emanating in precise patterns from the ectoderm, together with the mesoderm-endogenous transcription factors Tinman and Twist, as well as augmenting contributions by Ras signaling, endow the eme enhancer with the competence to activate transcription within the cardiogenic region of the dorsal mesoderm. The highly restricted pattern of mesodermal Eve expression is achieved by repression: Ladybird represses the enhancer anterior to the normal Eve cluster position, while another repressor must exist (indicated by X) that acts through a site overlapping with that of Lbe, and inhibits expression posterior to the Eve clusters. Eve itself is also required for correct differentiation of cell types derived from the Eve clusters (for additionally details see Su et al., 1999). (B) Intersection of multiple competency information (wg, dpp, ras, twist, and tinman) is modulated by mutual repression between eve and lbe to generate cardiac cell-type specificity.
The high fidelity with which eme mimics endogenous mesodermal eve expression allowed us to pursue three complementary approaches to the combinatorial control of organogenesis: (1) Using eme together with the UAS-Gal4 system (Brand and Perrimon, 1993), we misexpressed the genetic factors of interest (or activated and dominantnegative versions thereof) specifically in the mesodermal Eve clusters, and thereby addressed whether or not they can act autonomously within the EPC/DA1 lineage; (2) By site-directed mutagenesis, we examined the requirements for the consensus sites within eme for mesodermal eve-specific expression, and thereby determined the extent to which these diverse genetic inputs are directly integrated by a single enhancer; (3) By eliminating eme within the context of the entire eve gene, we determined whether this element is required, as well as sufficient, for promoting mesodermal eve expression.
To examine the mechanisms of transcriptional control of the eme enhancer in vivo, we first tested the activity of three overlapping fragments, each containing different combinations of consensus sites (Fig. 2A): nt 150 - 450 (eme1), nt 300 - 600 (eme2), and nt 450 -750 (eme3). Both eme1 (6/7 lines) and eme2 (4/4 lines) show a pattern of (cytoplasmic) reporter expression similar to that of eme900 and to the endogenous Eve protein itself (nuclear), whereas eme3 (7/7 lines) has no activity (Figs 3D-3F and Figs. 2B and 2C). Thus, eme1 and eme2 contain a sufficient set of elements to produce a near normal mesodermal eve pattern. The overlap between eme1 and eme2 (nt 300 - 450) does not have any activity on its own, suggesting that the 5′ portion of eme1 and the 3′ portion of eme2 contain partially redundant information. Two other groups have independently reported minimal eve mesodermal enhancers: the MHE enhancer described by Halfon et al. (2000) is similar to eme1, whereas emeB described by Knirr and Frasch (2001) is similar to eme2 (Fig. 2O).
To compare the enhancer activity more precisely with endogenous Eve expression, we used a lacZ construct that confers nuclear localization to the reporter and contains both Lbe consensus sites (emeA; Fig. 2A, nt 225-600). The emeA reporter expression is virtually identical (6/6 lines) to endogenous eve (Fig. 2G-I), except for an additional nucleus corresponding to the Eve-negative DO2 muscle founder, a progeny of the Eve progenitor lineage that turns off eve expression but in which LacZ protein perdures (see Carmena et al., 2002). Nuclear LacZ labels only a single nucleus of the DA1 muscle (Figs. 2K and 2L), which may be the muscle founder nucleus. To test this, we examined emeA enhancer activity in Dmef2 mutant embryos, in which DA1 (and other) muscle founder cells are specified but remain mononucleate, failing to fuse with surrounding myoblasts (Bour et al., 1995). Indeed, Eve and nuclear LacZ now coincide in the position of the DA1 founder (Fig. 2M).
Dominant-Negative Forms of Tinman, dTCF, and Thickvein Repress eve Expression in the Mesoderm
To test whether Tinman and wg and dpp signaling are required within the Eve progenitor lineage itself, we used eme900 in conjunction with the UAS-Gal4 system (Brand and Perrimon, 1993) to specifically target gene expression. For this purpose, we generated eme900-Gal4 transgenic lines and crossed them first to UAS-GFP, generating an eme-specific GFP pattern in the progeny (eme > GFP; Fig. 3A). To further test the effectiveness of this expression system, we examined eve expression in the progeny of eme-Gal4 crossed to UAS-ricin, a cell toxin (eme > Ricin), and observed almost complete absence of both EPC and DA1 formation, without major effects on the surrounding tissues (Figs. 3B and 3C).
Next, we expressed dominant-negative forms of Tinman, dTCF and Thickvein (Tkv, a receptor kinase essential for transmission of the Dpp signal). DN-Tin contains the Tinman homeodomain fused to the Engrailed repressor domain (see Materials and Methods). DN-dTCF lacks the N-terminal Armadillo binding domain and is therefore unable to activate transcription (van de Wetering et al., 1997). In DN-Tkv, the GS box is deleted, which is thought reduce receptor signaling (Haerry et al., 1998). Mesodermal eve expression significantly but variably reduced when either DN-Tin, DN-dTCF, or DN-Tkv is expressed within the EPC/DA1 lineage (Figs. 3D-3F). The effect with DN-Tkv is less dramatic, probably because the available dominant-negative forms of Dpp-receptors are only of moderate strength (Haerry et al., 1998). These data suggest that the genetic functions that are prerequisite for establishing cardiac mesodermal competence also directly regulate gene expression in individual lineages, since their effectors continue to be required for mesodermal eve expression.
Ladybird Represses eve Expression in the Mesoderm
Since the functions of wg, dpp, and tinman function are required not only for Eve progenitor formation but also for the entire cardiac mesoderm, additional information is needed to restrict eve expression. lbe is a candidate to provide such an activity, because it is expressed in cell clusters adjacent to Eve (Fig. 1D), and ubiquitous expression reduces mesodermal eve expression (Jagla et al., 1997). To determine whether lbe function can repress eve expression within the mesodermal Eve lineage itself, we used the eme-Gal4 driver to overexpress lbe (eme > lbe). We observed that eve expression is initiated normally in the early Eve cell clusters, but then is lost entirely once Lbe has accumulated (Figs. 3G and 3H). These data, together with loss-of-function studies involving a deficiency for the lb genes (Jagla et al., 1997), suggest that lbe may have a direct role in repressing eve expression in cells located anterior to the Eve clusters, thereby confining EPC/DA1-specific differentiation to the appropriate segmental position. We do not know how the relative anterior-posterior position of Lbe versus Eve clusters is determined, except that both types of progenitors depend on the early and late pattern of wg activity (Wu et al., 1995; Jagla et al., 1997), which might provide additional cues for spatial subdivision along this axis.
Svp Is Not a Direct Repressor of Mesodermal Eve
Mesodermal cells located posterior to the Eve clusters express svp, and differentiate into two myocardial and two pericardial cells (Figs 1G, 1H, and 1J). tinman expression in these cells is initiated normally, but is subsequently lost (Gajewski et al., 2000; Ward et al., 2000; Lo and Frasch, 2001). Thus, as lbe is likely to repress eve in cells anterior to the normal Eve clusters, Svp (Svp1) may repress eve posteriorly. To test this hypothesis, we overexpressed Svp1 in the mesodermal Eve lineage. Although Svp1 is a potent inhibitor of tinman expression in myocardial cells (Gajewski et al., 2000; Lo and Frasch, 2001), eme > svp1 embryos exhibit neither an altered pattern of eve expression nor a change in EPC/DA1 differentiation (Fig. 3I). Thus, svp does not seem to contribute directly to the spatial restriction of eve expression.
Tinman and dTCF Directly Control the eme Enhancer
In order to determine whether the transcription factors necessary for mesodermal Eve cell specification directly regulate the eme enhancer, we specifically mutated each of the consensus sites and tested the effect on eme activity. First, we point-mutated one or more of the Tinman sites in emeA, based on data from gel shift assays (Fig. 2O). We found that reporter gene activity was progressively reduced as more Tinman binding sites were mutated (Figs. 4A-4D). Consistent with the robust reporter activity conferred by eme1 (Fig. 2D), eme-Tin3*-mediated expression was only slightly decreased (Fig. 4B). These findings, together with the effects of overexpressing DN-Tin (Fig. 3E), suggest that Tinman is a direct and essential activator of mesodermal eve expression, and that several Tinman sites cooperate in vivo to achieve full enhancer activity.
FIG. 4.

Mutations of Tinman and dTCF sites in eme reduce enhancer activity. Double-labeling of stage 11 embryos carrying either a wild-type or a mutated emeA-lacZ transgene for Eve (green) and LacZ (red). Asterisks indicate mutated positions (see Materials and Methods). (A) Wild-type emeA. (B) emeA with Tin3 site mutated. Note that LacZ is similar to that in (A). (C) emeA with Tin1 and Tin2 mutated. Note the decreased level of LacZ staining. (D) emeA with all three Tinman sites mutated. Note the absence of LacZ staining. (E) emeA with dTCF site mutated. Note the dramatic decrease in LacZ staining. (F) emeA with dTCF and Tin1&2 sites mutated. Note the complete loss of LacZ staining as in (D).
Altering the most conserved dTCF consensus binding site (Fig. 2N) resulted in reduced enhancer activity (Fig. 4E), similar to the reduction seen with two mutated Tinman sites (Fig. 4C). This finding, together with the effect of DN-dTCF expression (Fig. 3D), suggests that dTCF-mediated wg signaling directly activates mesodermal eve expression. When this dTCF site is mutated in conjunction with two Tinman sites, expression is completely abolished (Fig. 4F), similar to the mutation of all three Tinman sites (Fig. 4D), suggesting that there is synergy in the activation of mesodermal eve expression between Tinman and the Wg pathway. Multiple Smad sites are also needed for normal levels of eve expression in the mesoderm (Halfon et al., 2000; Knirr et al., 2001).
ladybird Directly Represses eme Enhancer Activity
As with Tinman and dTCF, Lbe might control eve expression directly, as suggested by the overexpression data (Figs. 3G and 3H) and by the presence of two Lb consensus binding sites in the emeA enhancer (Figs. 2A and 2N). Also, both sites are bound by the Lbe protein in vitro (Fig. 2O). Interestingly, a single Lb site is present in both eme1 and eme2, each of which gives a near-normal pattern, suggesting that these two sites are either functionally redundant or irrelevant. To examine the importance of these Lbe sites, we mutated the only Lb site in eme2. The mutant enhancer (eme2-Lb2*) exhibits dramatically expanded reporter gene expression as compared with emeA or eme2 (Fig. 5). At stage 11, reporter gene activity extends anteriorly and posteriorly from the normal Eve progenitor domain (Figs. 5A and 5B) and encompasses most if not all of the tinman-expressing region at the dorsal margin of the mesoderm (Fig. 1A). Later, the cells that express lacZ ectopically migrate dorsally to the Eve clusters and many of them go on to form the Dmef2-expressing myocardial heart tube, while others become pericardial cells (Figs. 5C-5H). Interestingly, some of the myocardial cells show lower expression than others (asterisks in Fig. 5H), perhaps because late tinman expression is repressed by Svp1 in these myocardial cells.
FIG. 5.

Mutation of Ladybird sites in eme expands the pattern of enhancer activity. Left-hand and middle panels (A, C, F, H, I): wild-type emeA-lacZ transgenic embryos. (D) wild-type transgenic for eme2-lacZ. Right-hand panels (B, E, G, J): similar embryos with Lb2 site mutated in eme2 (eme2-Lb*). (A-G) Eve (green) and LacZ (red) double-labeled embryos of stage 11 (A, B) and stage 13 (C-E). (B,E) Note the dramatic expansion of reporter gene expression into most if not all myocardial and pericardial cells and their precursors (indicated by arrowheads; compare to A,C,D and Figs. 1A and 1B). (F, G) Stage 16 embryos double-labeled for LacZ (red) and Dmef2 (green). (G) Note that most if not all cardiac cells, but not the body wall muscles (other than DA1) express the reporter gene, and that four of the six myocardial cells per hemisegment are expressing LacZ more strongly (in brackets) than the remaining two (indicated by asterisks, see text). Arrowhead indicates pericardial cell. (H-J) Eve and LacZ double-labeling of stage 13 embryos of the following genotype: emeA, eme > Lbe (H), emeA, twi > Lbe (I), and eme2-Lb*, twi-Lbe (J). Note the lack of repression of the reporter in the absence of a Lbe binding site in J.
If the Lbe site in eme2 indeed mediates repression, mutating this site might render reporter gene expression insensitive to repression by Lbe. We tested this hypothesis by crossing eme2-Lb* into a genetic background in which lbe is overexpressed in the entire mesoderm. In control embryos with a wild-type enhancer, we observed dramatic suppression of reporter expression using either eme-Gal4 or a pan-mesodermal driver (Figs. 5I and 5J). In contrast, pan-mesodermal lbe expression had little effect on reporter activity when the Lbe site was mutated in eme2 (Fig. 5K). Taken together, these data strongly suggest that Lbe directly represses eve in cells anterior to the Eve clusters.
While Lbe represses eve in cells anterior to the Eve clusters within the cardiogenic region, we do not know what represses eve posterior to the Eve clusters. However, the fact that expression driven by eme2 with a mutant Lbe site expands posteriorly as well as anteriorly suggests that a posterior repressor interacts with a site that overlaps with the Lbe site.
We conclude that a complex combination of activating and repressing signals, in conjunction with the activities of tissue-specific transcription factors, are integrated by eme to produce the highly stereotyped pattern of eve expression in the cardiogenic region (Fig. 6). The overall cardiac competence generated by the convergence of activators (wg, dpp and ras signaling, Tinman and Twist transcription factors) is modulated by mutual repression of identity genes to specify individual cardiac cell types (Fig. 6B).
eme Sequences Are Essential for Mesodermal Expression
Although eme900, eme1, eme2, and emeA are each sufficient for mesodermal eve-specific expression, this does not necessarily mean that the information contained in these fragments is required for expression within the context of the entire gene. To test this, we generated transgenic flies carrying the entire eve gene, including all of the regulatory region from -6.4 to +9.2 kb, except that sequences within eme900 were deleted. Two eve transgene constructs were tested (Fig. 7A), one missing +6.1 to +6.6kb (EagI to StuI, ΔES, independent lines J48, J49), the other missing +6.3 to +6.6kb (NcoI to StuI, ΔNS, line J43). It was previously shown that the wild-type eve transgene (-6.4 to either +8.4 or +9.2kb, without eme deletions) is able to rescue viable and fertile adult flies in a eve null mutant background (P[eve], eve-/-; Fujioka et al., 1999). These flies are also normal for mesodermal eve expression. In contrast, when we examined P[eve-emeΔES], eve-/-, mesodermal eve expression was undetectable, whereas all other aspects of eve expression were normal (Figs. 7B and 7C). Similar results were obtained with P[eve-emeΔNS], eve-/- (data not shown). This suggests that sequences within eme are not only sufficient but also necessary for activating eve expression within the dorsal mesoderm. Remarkably, however, P[eve-emeΔNS] does not delete any sequences within eme1, which by itself is sufficient for conferring a near normal mesodermal eve pattern (Figs. 2A and 2D). This indicates that information outside of eme1 and within eme3 is normally essential for mesodermal eve expression and that an element that is sufficient in isolation is not necessarily sufficient within the context of the entire gene (see Discussion).
To determine whether the eme reporter transgene is activated normally in the absence of mesodermal Eve, we combined the P[eve-emeΔES] transgene with P[emeA-lacZ] in an eve null background. In these embryos, emeA-mediated lacZ expression was initially present in segmental clusters in a near normal pattern (Fig. 7E), while at later stages of development, reporter activity was progressively lost (Fig. 7G). In contrast, in control embryos when an intact 16-kb eve transgene (86T) is used to rescue the eve null mutant, both the mesodermal eve and the emeA-lacZ expression are indistinguishable from wild-type (Figs. 7D and 7F). This suggests that mesodermal eve expression does not require its own activity to be initiated or maintained, at least through stage 11.
lbe Expression Expands into the Eve Territory in Mesodermal eve Mutants
It may be that not only does lbe repress eve, but conversely, eve may repress lbe. To test this, we took advantage of the fact that eme activity is initially normal in a mesodermal eve mutant background (P[eve-emeΔES], eve-/-), so that, like normal eve expression, it should not overlap with normal Lbe expression (Figs. 1D and 1E). However, when we compared eme reporter and lbe expression in mesodermal eve mutants, we observed an expansion of Lbe throughout the territory that normally expresses eve (Figs. 7H and 7I). This suggests that Eve normally represses Lbe in cells posterior to the Lbe clusters, a conclusion consistent with those of a recent study using a temperaturesensitive allele of eve (Jagla et al., 2002). Taken together, we conclude that mutual repression between eve and lbe is essential for regionally confining their territories of expression, which in turn is prerequisite for generating the correct patterns of cell type diversity within the cardiac mesoderm.
DISCUSSION
How eve Expression Is Confined to a Small Subset of Dorsal Mesodermal Cells
eve requires a combinatorial code of patterned gene activity for its highly restricted and stereotyped pattern of expression. As summarized in Fig. 6, a set of positive and negative inputs, some direct and some indirect, is required for generating the high fidelity of mesodermal eve expression (Halfon et al., 2000; Knirr and Frasch, 2001; this work): wg and dpp signaling from the ectoderm provides activating inputs, determining where within the mesoderm eve expression is initiated, whereas tinman, activated by twist, provides the mesodermal context in which these inputs are interpreted (Lockwood and Bodmer, 2002). wg- and dpp-dependent activation of the ras pathway also contributes to the level of eve activation (Carmena et al., 1998). The outputs of these signaling pathways are directly integrated along with the activities of transcription factors expressed at the dorsal mesodermal margin by a defined enhancer element (eme) within the regulatory region of eve.
Ladybird Is a Direct Repressor of Mesodermal Eve
Despite the complexity of this regulatory ensemble, it is unlikely to be sufficient for determining the highly restricted pattern of mesodermal eve expression, since the expression patterns of other factors within the cardiac mesoderm, such as tinman and lbe, require a similar combination of activating factors but the resulting patterns are different (Figs. 1A and 1G; Wu et al., 1995; Frasch, 1995; Park et al., 1996; Jagla et al., 1997, 2002; Lockwood and Bodmer, 2002).
Two additional mechanisms are likely to contribute to the restricted eve pattern. First, lateral inhibition mediated by Notch, along with cross talk between the Notch and ras pathways, results in the selection of two Eve progenitors per segmental cluster, which then divide asymmetrically (Park et al., 1998; Carmena et al., 1998, 2002). Second, to prevent eve transcription anterior and posterior to the Eve clusters, repressive mechanisms appear to be at work. We have shown that one of these mechanisms is direct repression of the eme enhancer by Lbe, which is expressed in cell clusters immediately anterior to the Eve clusters (Fig. 1D). Mutating the Lbe consensus binding sites in eme (eme2-Lb*) dramatically expands reporter gene expression (Fig. 5). Moreover, reporter expression is rendered insensitive to inhibition by Lbe overexpression (Fig. 5J), strongly suggesting that Lbe acts as a direct repressor of Eve.
Are There Additional Repressors of Mesodermal Eve?
An eme enhancer unable to bind Lbe causes expansion of reporter gene expression not only anteriorly into the lbe-expressing territory, but also posteriorly, such that nearly the entire tinman-expressing cardiogenic mesoderm is occupied (compare Fig. 1A with Fig. 5B). Therefore, it is necessary to invoke additional factors that interact with the Lbe sites in eme to restrict eve expression in cells posterior to the Eve clusters (Fig. 6A). The identity of this posterior repressor is unknown and is unlikely to be the product of svp1, since eme > svp1 overexpression has little or no effect on eve expression (Fig. 3I). Also, no recognizable consensus sites for the COUP-TF receptor family are present in eme (see Fig. 2N).
It was recently reported (Knirr and Frasch, 2001), that, as expected, when levels of wg activity were reduced, an eme enhancer similar to our emeA (or eve expression itself; Wu et al., 1995) failed to be activated. By contrast, when the enhancer activity was reduced by mutating additional putative dTCF sites, the remaining activity was unaffected by lowering wg activity. This led to the hypothesis that, while high levels of wg may cause activation of this enhancer, low levels of wg serve only to counteract the repressive activity of (unactivated) dTCF. Thus, dTCF probably serves to restrict eve expression in the absence of the wg signal, while also activating eve directly when converted to an activator by wg signal transduction.
Zfh-1 May Interact with the Same eme Sites as Ladybird
The consensus binding sites for Zfh-1 and Lbe are strikingly similar (Lai et al., 1991; K. Jagla, personal communication). Gel-shift assays show that these two proteins bind to the same sequences within the emeA enhancer, and mutation of the core binding sequence abolished the binding of both proteins (Fig. 2O; Su et al., 1999). Since zfh-1 is required for EPC differentiation without affecting DA1 formation (Su et al., 1999), it may act as a direct activator of eve transcription in the forming EPCs. Alternatively, it may act by inhibiting a repressor that can bind to the same sequence. Since Lbe acts as a repressor of eve transcription and can bind to the same site as Zfh-1, the latter hypothesis is particularly attractive. Consistent with this hypothesis, we observed that eme2-Lb2* is active in the EPCs, in addition to its expansion to other cell types, including those that express Lbe (Fig. 5E).
More Than One Way to Make a “Minimal” eve Mesodermal Enhancer?
Comparing our results with those of two other groups that have studied this enhancer region, we note that one element (MHE, nt 150 - 461 in Fig. 2N; Halfon et al., 2000) is similar to our eme1 (nt 150 - 450), while the other element (nt 222-622 in Fig. 2N; Knirr and Frasch, 2001) is similar to our emeA (nt 225-600). Interestingly, eme2 promotes essentially the same pattern of expression as eme1, but does not contain a number of the sites that were proposed to be crucial for eme1-like enhancer activity. In addition, many of these “missing” sites are not conserved in other Drosophila species. Thus, it is likely that additional (conserved) sites (Lb2, Tin3), not present in eme1, compensate for the ensemble of “missing” sites. Also, it appears that, even within discrete enhancers such as eme, there is significant redundancy, so that several overlapping “minimal” enhancers are capable of driving expression patterns that are nearly identical to that of endogenous eve, at least when assayed in isolation adjacent to a promoter in a reporter transgene. However, this redundancy may not apply to the endogenous locus (see below).
All of the sites we examined that affect the level or pattern of eme-mediated reporter gene expression when mutated, including both Lb sites, are conserved in at least four out of five Drosophila species (Z.H., S. Celniker, R.B., unpublished observations; the eve gene has been sequenced in five Drosophila species as part of a pilot comparative project to study conservation of non-coding regions: B. Pfeiffer, G. Rubin, and S. Celniker, unpublished observations). This observation, together with the present and previous experimental data, strongly suggests that eme (and thus mesodermal eve expression) is under the direct combinatorial regulation of the factors and pathways depicted in Fig. 6A. Among them, we have identified Lbe as an essential repressor that interacts directly with the eme enhancer, thus contributing to the spatial confinement of mesodermal eve expression, and corresponding cell fate specification within the cardiac mesoderm.
The Requirement for Enhancer Elements Is Context-Dependent
The fact that the combination of elements present in eme1 as well as those in eme2 are sufficient for conferring appropriate mesodermal eve-like expression when placed upstream of a reporter gene does not preclude the possibility that additional regions are essential for mesodermal expression within the context of the entire gene. Indeed, in P[eve-emeΔNS],eve-/- rescued embryos, only sequences outside of eme1 are deleted (corresponding approximately to eme3, see Figs. 2A and 2N), but no eve expression is observed in the mesoderm (Fig. 7). Thus, this deletion must contain elements that are needed in the normal context to initiate eve transcription from the normal start site, which is 6 kb upstream. Consistent with this hypothesis is the finding that there are at least two additional conserved regions within the deleted portion of P[eve-emeΔNS] (Z.H., S. Celniker, R.B., unpublished observations). Therefore, the redundancy in combinatorial information between eme1 and eme2, as determined in the isolated enhancer assay, apparently does not exist within the context of the entire gene. In addition to the impact of the additional enhancerpromoter distance in the normal context, there may be negative regulatory effects of other sequences within the eve locus. Such repressive elements might have a function in preventing ectopic expression, and might be tissuespecific, or they might have a more general repressive effect, such as that exerted on eve by Polycomb-G genes (Dura and Ingham, 1988; McKeon et al., 1994; Weigmann and Lehner, 1995). In either case, they might introduce a requirement for a stronger enhancer activity than that provided by any one of the “minimal” enhancers acting alone.
eve Acts as an Identity Gene of the Eve Progenitor Lineage
Previous previous data obtained with a temperature-sensitive allele of eve suggest that eve function is required for EPC differentiation at the time when Eve protein is accumulating in the early clusters (Su et al., 1999). In these conditional mutants, Ibe expression also expands to the Eve territory (Jagla et al., 2002), as in mesodermal eve mutants shown here (Fig. 7I), consistent with a switch in identity. Moreover, mesodermal eve function is necessary for normal cardiac physiology as well as for the formation of the DA1 and DO2 muscles (M.F., Z.H., G. L. Yusibova, R. J. Wessels, R.B., and J.B.J., unpublished observations). Therefore, eve expression in the Eve progenitors and their progeny is involved in directing their correct differentiation.
In conclusion, a complex combination of activating and repressing signals, in conjunction with the activities of tissue-specific transcription factors, is integrated by the eve mesodermal enhancer to produce the highly restricted and stereotyped pattern of eve expression (Fig. 6A). Reciprocal repression between lbe and eve (and others) in adjacent cardiogenic territories is crucial for achieving the precise confinement of identity gene expression (Fig. 6). Mutual repression among transcription factors that are activated by a common set of competence factors may represent a general mechanism for generating specificity in organ development.
ACKNOWLEDGMENTS
We thank Manfred Frasch, Corey Goodman, Christophe Jagla, Zhi-Chun Lai, Bruce Paterson, Nipam Patel, Gerd Technau, Lauren Yaich, and the Bloomington Stock Center for sending fly stocks and antibodies. We thank Galina L. Yusibova, Monika Deo, and Andrea Clark for technical assistance, and Krista Golden for imaging and art work. We also thank Susan Celniker for sharing data before publication. This work was supported by NIH (GM50231) and NSF (0110856) awards to J.B.J., by NIH (HL59875) and American Heart Association (Established Investigator) awards to R.B., and by a predoctoral fellowship award from the American Heart Association to Z.H.
REFERENCES
- Beiman M, Shilo BZ, Volk T. Heartless, a Drosophila FGF receptor homolog, is essential for cell migration and establishment of several mesodermal lineages. Genes Dev. 1996;10:2993–3002. doi: 10.1101/gad.10.23.2993. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Frasch M. Genetic determination of Drosophila heart development. In: Rosenthal N, Harvey R, editors. Heart Development. Academic Press; San Diego: 1999. pp. 65–90. [Google Scholar]
- Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–729. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Jan LY, Jan YN. A new homeobox-containing gene, msh-2 (tinman), is transiently expressed early during mesoderm formation in Drosophila. Development. 1990;110:661–669. doi: 10.1242/dev.110.3.661. [DOI] [PubMed] [Google Scholar]
- Bour BA, O’Brien MA, Lockwood WL, Goldstein ES, Bodmer R, Taghert PH, Abmayr SM, Nguyen HT. Drosophila mef2, a transcription factor that is essential for myogenesis. Genes Dev. 1995;9:730–741. doi: 10.1101/gad.9.6.730. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant pheno-types. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Carmena A, Gisselbrecht S, Harrison J, Jiménez F, Michelson AM. Combinatorial signaling codes for the pro-gressive determination of cell fates in the Drosophila embryonic mesoderm. Genes Dev. 1998;12:3910–3922. doi: 10.1101/gad.12.24.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena A, Buff E, Halfon MS, Gisselbrecht S, Jimenez F, Baylies MK, Michelson AM. Reciprocal regulatory interactions between the Notch and Ras signaling pathways in the Drosophila embryonic mesoderm. Dev. Biol. 2002;244:226–242. doi: 10.1006/dbio.2002.0606. [DOI] [PubMed] [Google Scholar]
- Dura JM, Ingham P. Tissue- and stage-specific control of homeotic and segmentation gene expression in Drosophila embryos by the polyhomeotic gene. Development. 1988;103:733–741. doi: 10.1242/dev.103.4.733. [DOI] [PubMed] [Google Scholar]
- Frasch M. Induction of visceral and cardiac mesoderm by ectodermal Dpp in the early Drosophila embryo. Nature. 1995;374:646–467. doi: 10.1038/374464a0. [DOI] [PubMed] [Google Scholar]
- Frasch M, Hoey T, Rushlow C, Doyle H, Levine M. Characterization and localization of the even-skipped protein of Drosophila. EMBO J. 1987;6:749–759. doi: 10.1002/j.1460-2075.1987.tb04817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Yan W, Mohun TJ, Evans SM. Vertebrate tinman homologues XNkx2-3 and XNkx2-5 are required for heart formation in a functionally redundant manner. Development. 1998;125:4439–4449. doi: 10.1242/dev.125.22.4439. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Emi-Sarker Y, Yusibova GL, Goto T, Jaynes JB. Analysis of an even-skipped rescue transgene reveals both composite and discrete neuronal and early blastoderm enhancers, and multi-stripe positioning by gap gene repressor gradients. Development. 1999;126:2527–2538. doi: 10.1242/dev.126.11.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka M, Jaynes JB, Bejsovec A, Weir M. Production of transgenic Drosophila. Methods Mol. Biol. 2000;136:353–363. doi: 10.1385/1-59259-065-9:353. [DOI] [PubMed] [Google Scholar]
- Gajewski K, Choi CY, Kim Y, Schulz RA. Genetically distinct cardial cells within the Drosophila heart. Genesis. 2000;28:36–43. doi: 10.1002/1526-968x(200009)28:1<36::aid-gene50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Gajewski K, Kim Y, Lee YM, Olson EN, Schulz RA. D-mef2 is a target for Tinman activation during Drosophila heart development. EMBO J. 1997;6:515–522. doi: 10.1093/emboj/16.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisselbrecht S, Skeath JB, Doe CQ, Michelson AM. heartless encodes a fibroblast growth factor receptor (DFR1/DFGF-R2) involved in the directional migration of early mesodermal cells in the Drosophila embryo. Genes Dev. 1996;10:3003–3017. doi: 10.1101/gad.10.23.3003. [DOI] [PubMed] [Google Scholar]
- Greig S, Akam M. Homeotic genes autonomously specify one aspect of pattern in the Drosophila mesoderm. Nature. 1993;362:630–632. doi: 10.1038/362630a0. [DOI] [PubMed] [Google Scholar]
- Haerry TE, Khalsa O, O’Connor MB, Wharton KA. Synergistic signaling by two BMP ligands through the SAX and TKV receptors controls wing growth and patterning in Drosophila. Development. 1998;125:3977–3987. doi: 10.1242/dev.125.20.3977. [DOI] [PubMed] [Google Scholar]
- Halfon MS, Carmena A, Gisselbrecht S, Sackerson CM, Jimenez F, Baylies MK, Michelson AM. Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell. 2000;103:63–74. doi: 10.1016/s0092-8674(00)00105-7. [DOI] [PubMed] [Google Scholar]
- Ip YT, Park RE, Kosman D, Yazdanbakhsh K, Levine M. dorsal-twist interactions establish snail expression in the presumptive mesoderm of the Drosophila embryo. Genes Dev. 1992;6:1518–1530. doi: 10.1101/gad.6.8.1518. [DOI] [PubMed] [Google Scholar]
- Jagla K, Frasch M, Jagla T, Dretzen G, Bellard F, Bellard M. ladybird, a new component of the cardiogenic pathway in Drosophila required for diversification of heart precursors. Development. 1997;124:3471–3479. doi: 10.1242/dev.124.18.3471. [DOI] [PubMed] [Google Scholar]
- Jagla T, Bidet Y, Da Ponte JP, Dastugue B, Jagla J. Cross-repression interactions of identity genes are essential for proper specification of cardiac and muscular fates in Drosophila. Development. 2002;129:1037–1047. doi: 10.1242/dev.129.4.1037. [DOI] [PubMed] [Google Scholar]
- Jaynes JB, O’Farrell PH. Active repression of transcription by the Engrailed homeodomain protein. EMBO J. 1991;10:1427–1433. doi: 10.1002/j.1460-2075.1991.tb07663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knirr S, Frasch M. Molecular integration of inductive and mesoderm-intrinsic inputs governs even-skipped enhancer activity in a subset of pericardial and dorsal muscle progenitors. Dev. Biol. 2001;238:13–26. doi: 10.1006/dbio.2001.0397. [DOI] [PubMed] [Google Scholar]
- Lai Z, Fortini ME, Rubin GM. The embryonic expression patterns of zfh-1 and zfh-2, two Drosophila genes encoding novel zinc-finger homeodomain proteins. Mech. Dev. 1991;34:123–134. doi: 10.1016/0925-4773(91)90049-c. [DOI] [PubMed] [Google Scholar]
- Leptin M, Grunewald B. Cell shape changes during gastrulation in Drosophila. Development. 1990;110:73–84. doi: 10.1242/dev.110.1.73. [DOI] [PubMed] [Google Scholar]
- Lilly B, Zhao B, Ranganayakulu G, Paterson BM, Schulz RA, Olson EN. Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science. 1995;267:688–693. doi: 10.1126/science.7839146. [DOI] [PubMed] [Google Scholar]
- Lo PC, Frasch M. A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identities in the dorsal vessel of Drosophila. Mech. Dev. 2001;104:49–60. doi: 10.1016/s0925-4773(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Lockwood WK, Liu M, Su MT, Bodmer R. A genetic model for cardiac pattern formation and cell fate determination. In: Haddad G, Xu T, editors. “Genetic Models In Cardiorespiratory Biology,” Lung Biology Series. Marel Dekker, Inc.; 2001. pp. 179–201. [Google Scholar]
- Lockwood WK, Bodmer R. The patterns of wingless, decapentaplegic, and tinman position the Drosophila heart. Mech. Dev. 2002;114:13–26. doi: 10.1016/s0925-4773(02)00044-8. [DOI] [PubMed] [Google Scholar]
- McKeon J, Slade E, Sinclair DA, Cheng N, Couling M, Brock HW. Mutations in some Polycomb group genes of Drosophila interfere with regulation of segmentation genes. Mol. Gen. Genet. 1994;244:474–483. doi: 10.1007/BF00583898. [DOI] [PubMed] [Google Scholar]
- Nguyen HT, Bodmer R, Abmayr SM, McDermott JC, Spoerel NA. D-mef2: A Drosophila mesoderm-specific MADS box-containing gene with a biphasic expression profile during embryogenesis. PNAS. 1994;91:7520–7524. doi: 10.1073/pnas.91.16.7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HT, Xu X. Drosophila mef2 expression during mesoderm development is controlled by a complex array of cis-acting regulatory modules. Dev. Biol. 1998;204:550–566. doi: 10.1006/dbio.1998.9081. [DOI] [PubMed] [Google Scholar]
- Park M, Yaich LE, Bodmer R. Mesodermal cell fate decisions in Drosophila are under the control of the lineage genes numb, Notch, and sanpodo. Mech. Dev. 1998;75:117–126. doi: 10.1016/s0925-4773(98)00098-7. [DOI] [PubMed] [Google Scholar]
- Park M, Wu X, Golden K, Bodmer R. The wingless signaling pathway is directly involved in Drosophila heart development. Dev. Biol. 1996;177:104–116. doi: 10.1006/dbio.1996.0149. [DOI] [PubMed] [Google Scholar]
- Sentry JW, Yang MM, Kaiser K. Conditional cell ablation in Drosophila. Bioessays. 1993;15:491–493. doi: 10.1002/bies.950150710. [DOI] [PubMed] [Google Scholar]
- Shishido E, Higashijima S, Emori Y, Saigo K. Two FGF-receptor homologues of Drosophila: one is expressed in mesodermal primordium in early embryos. Development. 1993;117:751–761. doi: 10.1242/dev.117.2.751. [DOI] [PubMed] [Google Scholar]
- Smith ST, Jaynes JB. A conserved region of engrailed, shared among all en-, gsc, Nk1-, Nk2-, and msh-class homeoproteins, mediates active transcriptional repression in vivo. Development. 1996;122:3141–3150. doi: 10.1242/dev.122.10.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling A. P-element mediated transformation. In: Roberts DB, editor. Drosophila: A Practical Approach. IRL Press; Oxford: 1986. pp. 175–197. [Google Scholar]
- Staehling-Hampton K, Hoffmann FM, Baylies MK, Rushton E, Bate M. dpp induces mesodermal gene expression in Drosophila. Nature. 1994;372:783–786. doi: 10.1038/372783a0. [DOI] [PubMed] [Google Scholar]
- Su MT, Fujioka M, Goto T, Bodmer R. The Drosophila homeobox genes zfh-1 and even-skipped are required for cardiac-specific differentiation of a numb-dependent lineage decision. Development. 1999;126:3241–3251. doi: 10.1242/dev.126.14.3241. [DOI] [PubMed] [Google Scholar]
- Thisse B, Stoetzel C, Gorositiza-Thisse C, Perrin-Schmitt F. Sequence of the twist gene and nuclear localization of its protein in endomesodermal cells of early Drosophila embryos. EMBO J. 1988;7:2175–2183. doi: 10.1002/j.1460-2075.1988.tb03056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkunova EN, Fujioka M, Kobayashi M, Deka D, Jaynes JB. Two distinct types of repression domain in engrailed: One interacts with the groucho corepressor and is preferentially active on integrated target genes. Mol. Cell Biol. 1998;18:2804–2814. doi: 10.1128/mcb.18.5.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, Weintraub H. Expression of achaetescute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–1447. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Cavallo R, Dooijes D, van Beest M, van Es J, Loureiro J, Ypma A, Hursh D, Jones T, Bejsovec A. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- Venkatesh TV, Park M, Ocorr K, Nemaceck J, Golden K, Wemple M, Bodmer R. Cardiac enhancer activity of the homeobox gene tinman depends on CREB consensus binding sites in Drosophila. Genesis. 2000;26:55–66. [PubMed] [Google Scholar]
- Ward EJ, Skeath JB. Characterization of a novel subset of cardiac cells and their progenitors in the Drosophila embryo. Development. 2000;127:4959–4969. doi: 10.1242/dev.127.22.4959. [DOI] [PubMed] [Google Scholar]
- Weigmann K, Lehner CF. Cell fate specification by even-skipped expression in the Drosophila nervous system is coupled to cell cycle progression. Development. 1995;121:3713–3721. doi: 10.1242/dev.121.11.3713. [DOI] [PubMed] [Google Scholar]
- Wharton KA, Jr., Crews ST. CNS midline enhancers of the Drosophila slit and Toll genes. Mech. Dev. 1993;40:141–154. doi: 10.1016/0925-4773(93)90072-6. [DOI] [PubMed] [Google Scholar]
- Wu X, Golden K, Bodmer R. Heart development in Drosophila requires the segment polarity gene wingless. Dev. Biol. 1995;169:619–628. doi: 10.1006/dbio.1995.1174. [DOI] [PubMed] [Google Scholar]
- Xu X, Yin Z, Hudson JB, Ferguson EL, Frasch M. Smad proteins act in combination with synergistic and antagonistic regulators to target Dpp responses to the Drosophila mesoderm. Genes Dev. 1998;12:2354–2370. doi: 10.1101/gad.12.15.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Xu X, Frasch M. Regulation of the Twist target gene tinman by modular cis-regulatory elements during early mesoderm development. Development. 1997;124:4971–4982. doi: 10.1242/dev.124.24.4971. [DOI] [PubMed] [Google Scholar]