

Abstract
Transient receptor potential vanilloid receptor 1 (TRPV1)-mediated release of neuroactive peptides and neurotransmitters from the peripheral and central terminals of primary sensory neurons can critically contribute to nociceptive processing at the periphery and in the CNS. However, the mechanisms that link TRPV1 activation with Ca2+ signaling at the release sites and neurosecretion are poorly understood. Here we demonstrate that a brief stimulation of the receptor using either capsaicin or the endogenous TRPV1 agonist N-arachidonoyl-dopamine induces a prolonged elevation of presynaptic [Ca2+]i and a concomitant enhancement of glutamate release at sensory synapses. Initiation of this response required Ca2+ entry, primarily via TRPV1. The sustained phase of the response was independent of extracellular Ca2+ and was prevented by inhibitors of mitochondrial Ca2+ uptake and release mechanisms. Measurements using a mitochondria-targeted Ca2+ indicator, mtPericam, revealed that TRPV1 activation elicits a long-lasting Ca2+ elevation in presynaptic mitochondria. The concentration of TRPV1 agonist determined the duration of mitochondrial and cytosolic Ca2+ signals in presynaptic boutons and, consequently, the period of enhanced glutamate release and action potential firing by postsynaptic neurons. These data suggest that mitochondria control vanilloid-induced neurotransmission by translating the strength of presynaptic TRPV1 stimulation into duration of the postsynaptic response.
Keywords: TRPV1, capsaicin, NADA, calcium, mitochondria, DRG neurons
Introduction
The transient receptor potential vanilloid receptor 1 (TRPV1) is a critical molecular detector of noxious signals in primary sensory neurons. A broad range of pain-producing stimuli either directly activate or modulate TRPV1. This includes noxious heat, protons, lipid-derived endovanilloids, and inflammatory mediators (Caterina and Julius, 2001; De Petrocellis and Di Marzo, 2005). Behavioral studies using TRPV1 knock-out mice or selective TRPV1 antagonists have indicated that the receptor is involved in the development of various chronic pain conditions, including those associated with cancer, arthritis, irritable bowel syndrome, and migraine (Caterina et al., 2000; Davis et al., 2000; Akerman et al., 2004; Ghilardi et al., 2005; Jones et al., 2005; Szabo et al., 2005). Accordingly, TRPV1 presents an attractive target for analgesics, and several TRPV1 antagonists are currently under preclinical investigation or clinical trials (Immke and Gavva, 2006; Szallasi et al., 2007).
TRPV1 is a nonselective cation channel with a high Ca2+ permeability, and robust Ca2+ entry into the cell is a hallmark of receptor activation (Caterina et al., 1997). At the peripheral terminals of primary nociceptors, TRPV1-mediated Ca2+ influx triggers the release of neuropeptides, which contributes to the development of neurogenic inflammation (Caterina and Julius, 2001). TRPV1 is also found in the central processes of primary afferent neurons (Guo et al., 1999; Hwang et al., 2004), and its expression in the dorsal horn of the spinal cord is upregulated during chronic inflammation (Tohda et al., 2001; Luo et al., 2004). Notably, intrathecal administration of selective TRPV1 antagonists significantly reduces pain associated with inflammation and nerve injury (Kanai et al., 2005; Christoph et al., 2006; Cui et al., 2006). Pharmacologic targeting of TRPV1 expressed in the CNS appears to be especially critical for alleviating pain mediated by central sensitization (Cui et al., 2006; Kanai et al., 2007).
Despite growing evidence for a significant role of TRPV1 in pain transduction in the spinal cord, the mechanisms contributing to this process remain unclear. It is likely that TRPV1-mediated effects in the spinal cord are presynaptic. Indeed, TRPV1 colocalizes with the presynaptic protein synaptophysin, and TRPV1-positive axonal boutons containing synaptic vesicles have been observed by electron microscopy (Guo et al., 1999; Hwang et al., 2004). Moreover, stimulation of the central terminals of primary afferents with endovanilloids or capsaicin triggers the release of calcitonin gene-related peptide (CGRP) and substance P and induces glutamatergic synaptic transmission (Yang et al., 1998; Tognetto et al., 2001; Huang et al., 2002; Nakatsuka et al., 2002; Labrakakis and MacDermott, 2003). Although Ca2+ dynamics at the sites of transmitter release are crucial for TRPV1-mediated responses, the mechanisms that shape presynaptic Ca2+ signals in primary sensory neurons remain unknown.
Here, we used fluorescent imaging of cytosolic and mitochondrial Ca2+ in the axonal boutons of dorsal root ganglion (DRG) neurons in conjunction with postsynaptic patch-clamp recording in spinal cord (SC) neurons to examine TRPV1-mediated signaling at sensory synapses formed between DRG and SC neurons in culture. Our data demonstrate that presynaptic mitochondria play an important role in regulating sensory synaptic transmission induced by TRPV1 activation.
Materials and Methods
Cell culture.
DRG/SC cocultures were prepared as described previously (Medvedeva et al., 2001). In brief, newborn (1–2 d old) Sprague Dawley rats were killed according to a protocol approved by the University of Iowa Institutional Animal Care and Use Committee. DRGs were dissected from the thoracic and lumbar segments and incubated in Pronase E dissolved in DMEM (1 mg/ml) for 7 min at 37°C in a 10% CO2 incubator. Ganglia were washed twice in cold DMEM containing HEPES (20 mm; pH 7.4) and then mechanically dissociated by trituration with flame-constricted Pasteur pipettes of decreasing diameter. The spinal cord was dissected in small segments (∼0.5 mm) and then digested in trypsin solution (0.025% in DMEM) for 8 min at 37°C in a 10% CO2 incubator. Spinal cord segments were then washed and dissociated using the procedure described above for DRG dissociation. Suspensions of spinal cord and DRG cells were plated onto 25 mm glass coverslips coated with poly-l-ornithine and laminin. Cells were grown in DMEM supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, and penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively) in a 10% CO2 incubator at 37°C. After 48 h, 5 μm cytosine-A-d-arabinofuranoside (AraC) was added to cultures for 24 h. Two days later, cells were again treated with 5 μm AraC for 24 h. Cultures were grown for 10–16 d before use; 50% of the culture medium was replaced every 5–6 d.
Lentivirus (feline immunodeficiency virus)-mediated gene transfer to neurons.
pSynaptophysin–EGFPN1 was a gift from Dr. Jane Sullivan (University of Washington, Seattle, WA). The entire Synaptophysin–enhanced green fluorescent protein (EGFP) coding sequence was PCR amplified (forward primer, 5′-GCTCTAGAGCCACCATGGACGTGGTG-3′; reverse primer, 5′-ACGCGTCGACCCGCTTTACTTGTACAGCTCG-3′), cut with XbaI and SalI, and inserted into the feline immunodeficiency virus (FIV) vector plasmid pVETL–Cmcs (Johnston et al., 1999). pCDNA3–mtPericam was a gift from Dr. Atsushi Miyawaki (RIKEN, Saitama, Japan). The mtPericam fragment was PCR amplified (forward primer, 5′-GCTCTAGAGCCACCATGCTGAGCCTG-3′; reverse primer, 5′-GCGAATTCTTACTTTGCTGTCATCATTTG-3′), released with XbaI and EcoRI, and ligated into the pVETL–Cmcs plasmid. EGFP, Synaptophysin–EGFP-, and mtPericam-containing FIV vectors were prepared by the University of Iowa Gene Transfer Vector Core (Iowa City, IA), as described previously (Johnston et al., 1999). After 5–6 d in culture, DRG/SC cocultures were incubated with the corresponding FIV vectors (0.5–1 × 104 pfu/ml) for 4 h. Cells were used within 4–8 d after infection. Transfection efficiency ranged from 10 to 30%.
Measurements of intracellular and mitochondrial Ca2+ concentration in axonal boutons.
For both sets of imaging experiments, cells were placed in a flow-through chamber mounted on the stage of an inverted epifluorescent IX-71 microscope (Olympus, Tokyo, Japan). For intracellular Ca2+ concentration ([Ca2+]i) measurements, DRG neurons were loaded with Bis-fura or fura-FF via patch pipettes by holding neurons in the whole-cell configuration for 3–5 min, as described previously (Usachev et al., 2002); the pipette was withdrawn and recordings began within 20–30 min. All measurements were performed at room temperature. Patch pipettes were filled with the following solution (in mm): 125 K-gluconate, 10 KCl, 3 Mg-ATP, 1 MgCl2, 0.2 Bis-fura or fura-FF, and 10 HEPES, pH 7.25 with KOH (290 mOsm/kg with sucrose). Fluorescence was alternately excited at 340 (12 nm bandpass) and 380 (12) nm using the Polychrome IV monochromator (T.I.L.L. Photonics, Martinsried, Germany). Excitation light was reflected off a dichroic mirror (410 nm; T.I.L.L. Photonics) and focused on the cells via a 40× oil-immersion objective [numerical aperture (NA) 1.35; Olympus]. Emitted fluorescence was collected at 510 (84) nm (single band filter; Semrock, Rochester, NY) using an IMAGO CCD camera (T.I.L.L. Photonics). Pairs of 340/380 nm images were sampled at 0.2 Hz (see Figs. 2, 3, 5, 6, 8) or 10 Hz (see Figs. 1, 4). The fluorescence ratio (R = F 340/F 380) was converted to [Ca2+]i according to the following formula: [Ca2+]i = K dβ(R − R min)/(R max − R) (Grynkiewicz et al., 1985). The dissociation constants (K d) used for Bis-fura-2 and fura-FF were 525 and 5500 nm, respectively (Molecular Probes Handbook; Invitrogen, Carlsbad, CA). R min, R max, and β were determined by applying 10 μm ionomycin in Ca2+-free buffer (1 mm EGTA) and saturating Ca2+ (1.3 mm Ca2+). For Bis-fura, calibration constants were as follows: R min = 0.15, R max = 1.20, and β = 5.85. For fura-FF, calibration constants were as follows: R min = 0.17, R max = 1.82, and β = 5.70. Fluorescence was corrected for background, as determined before loading of cells with the indicators. For each experiment, we present an image depicting Bis-fura (fura-FF) distribution (λex = 380 nm) within axons at rest, and the time course of [Ca2+]i changes in individual presynaptic boutons (indicated by white boxes). Individual boutons are identified by different numbers and colors. The criterion for scoring cells as capsaicin responding was capsaicin-elicited (1 μm, 30 s) mean presynaptic [Ca2+]i elevation ≥100% over the baseline, which was 0.11 ± 0.02 μm (n = 59 boutons/19 cells) in our studies. This criterion is similar to those that have been described previously by us and others (Stucky et al., 1996; Usachev et al., 2002) and has been chosen for the following reasons: (1) biologically significant [Ca2+]i elevations (e.g., those that activate intracellular Ca2+ targets) are generally considered to be those that exceed 100–200 nm (Baimbridge et al., 1992; Clapham, 1995; Berridge et al., 1998; Angleson and Betz, 2001); (2) the detection of [Ca2+]i changes of <50–100 nm in such small structures as presynaptic boutons was precluded by noise; and (3) the [Ca2+]i responses to capsaicin defined by using this proposed criterion were completely blocked pharmacologically by using the TRPV1 antagonist capsazepine (10 μm; n = 12 boutons/3 cells).
Figure 2.
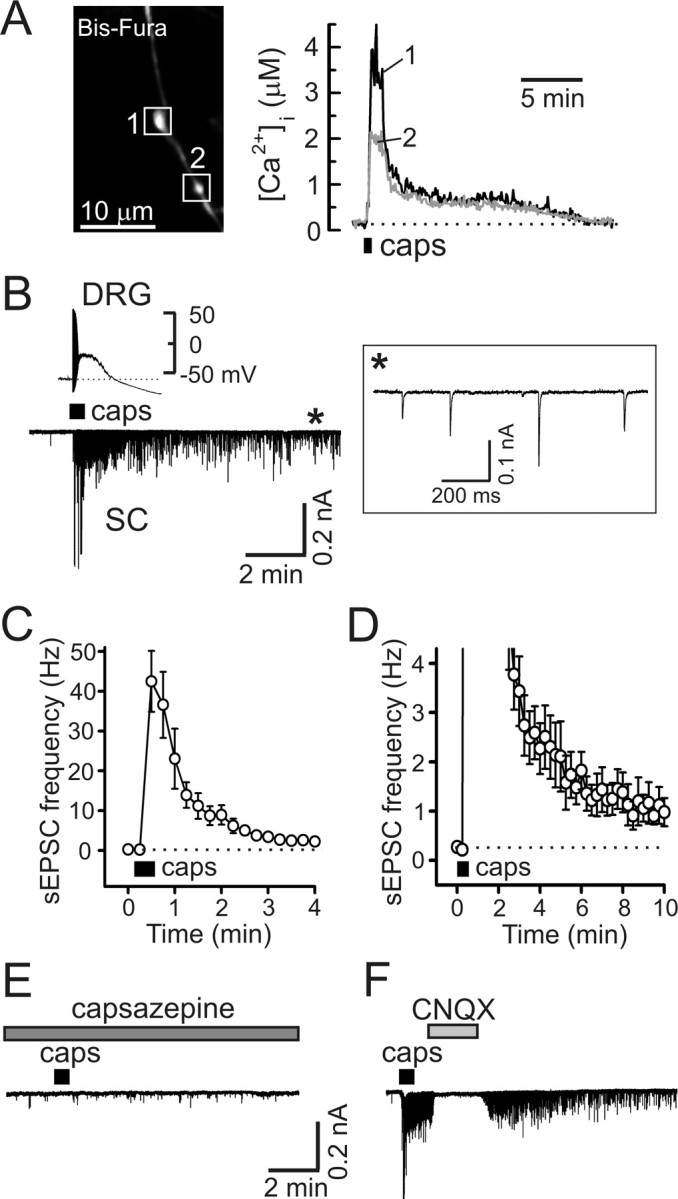
Capsaicin induces prolonged presynaptic [Ca2+]i elevation and enhancement of synaptic transmission in DRG/SC coculture. A, Capsaicin application (caps, 1 μm, 30 s) induced long-lasting [Ca2+]i elevation in presynaptic boutons (white boxes; left) of a DRG neuron loaded with Bis-fura. [Ca2+]i changes in individual boutons are indicated by different numbers and colors. The dotted line shows resting [Ca2+]i. B, Capsaicin (1 μm, 30 s) induced a transient depolarization and action potential firing in DRG neurons (top left) and a robust and long-lasting increase of excitatory synaptic activity (bottom left). Recordings were obtained in parallel experiments that used either the current-clamp (top left) or voltage-clamp (bottom left; V hold of −60 mV) techniques, and traces were aligned on the same timescale. The inset (right) shows sEPSC recordings on an expanded timescale for the timeframe indicated by the asterisk. C, D, Summary of the effects of capsaicin (1 μm, 30 s) on the frequency of sEPSCs derived from experiments like that shown in B. C and D depict the same set of data using different frequency and timescales for clarity. Each data point was obtained by averaging the sEPSC frequency during the 15 s of recording and is presented as mean ± SEM. E, The capsaicin-induced synaptic response was blocked by 10 μm capsazepine (5 min pretreatment). Recordings shown in B and E were obtained from the same cell (40 min between capsaicin applications). F, CNQX (10 μm) completely and reversibly blocked the capsaicin-induced synaptic response.
Figure 3.

The endovanilloid NADA elicits prolonged presynaptic [Ca2+]i elevation and a concomitant enhancement of synaptic transmission in DRG/SC coculture. A, [Ca2+]i changes (right) induced by NADA (5 μm, 30 s) were recorded from two boutons (white boxes, left) of a DRG neuron loaded with Bis-fura. B, The selective TRPV1 antagonist SB-366791 (10 μm, 5 min pretreatment) blocked NADA-evoked [Ca2+]i elevation in presynaptic boutons. A 30 min wash of the antagonist partially restored the NADA effect. C, NADA application (5 μm, 30 s) induced a strong and prolonged enhancement of excitatory synaptic activity. Postsynaptic currents were recorded from SC neurons voltage clamped at −60 mV. D, E, Summary of the NADA (5 μm, 30 s) effects on the frequency of sEPSC, as derived from experiments like that shown in C. D and E show the same set of data plotted using different frequency and timescales for clarity. Each data point was obtained by averaging sEPSC frequency during 15 s of recording and is presented as mean ± SEM. F, CNQX (10 μm) blocked the NADA (5 μm, 30 s)-induced response, indicating that non-NMDA glutamate (most likely AMPA) receptors mediated the postsynaptic currents. G, The effect of NADA (5 μm, 30 s) was almost completely blocked by the TRPV1 antagonist SB-366791 (10 μm; 5 min pretreatment). Postsynaptic recordings made after 4 and 10 min of antagonist washout show partial recovery of the NADA-evoked response.
Figure 5.

Properties of the vanilloid-induced presynaptic [Ca2+]i plateau in DRG neurons. A, Presynaptic [Ca2+]i recordings using fura-FF demonstrated a characteristic [Ca2+]i plateau phase in response to capsaicin (caps; n = 11 boutons/4 cells). All the recordings were done in low-Na+/TTX buffer. B, C, The [Ca2+]i plateau phase did not depend on extracellular Ca2+ (B), whereas the initiation of capsaicin-induced [Ca2+]i elevation required Ca2+ influx (C). In these experiments, DRG neurons were loaded with Bis-fura, as indicated on the images (left). D, Capsaicin-induced presynaptic [Ca2+]i changes were recorded in Bis-fura-loaded DRG neurons before (left) and after (right) the treatment with the SERCA inhibitor CPA (5 μm, 20 min pretreatment).
Figure 6.
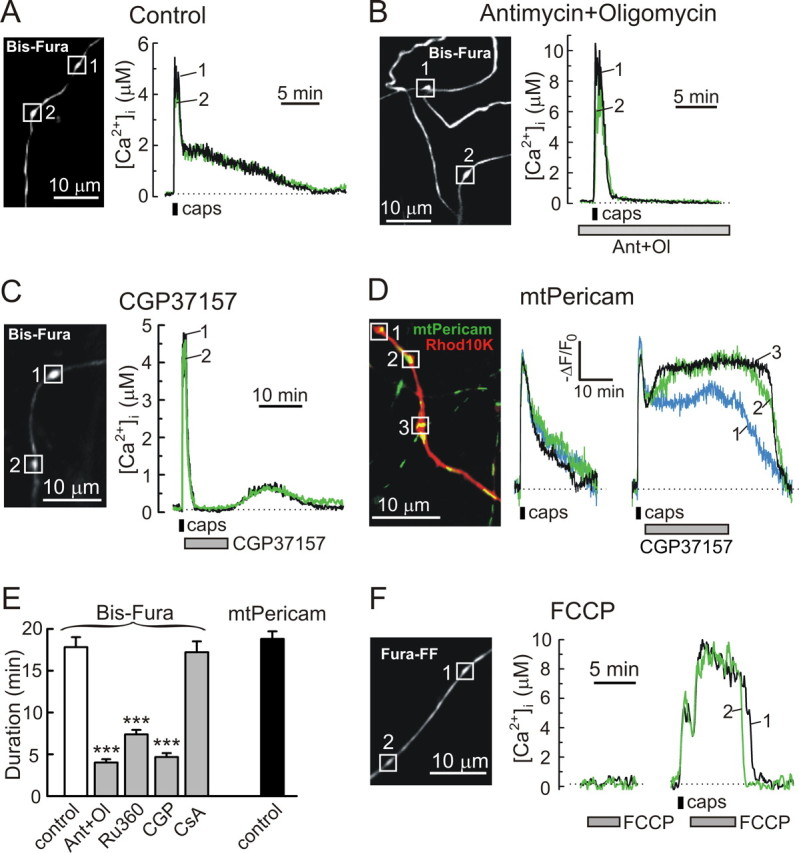
Mitochondria mediate vanilloid-induced presynaptic [Ca2+]i plateau in DRG neurons. A, B, Presynaptic [Ca2+]i changes induced by capsaicin application (caps; 1 μm, 30 s) in control cells (A) and in cells treated with 0.3 μm antimycin A1 and 1 μm oligomycin B (B). Dotted lines indicate presynaptic [Ca2+]i levels at rest. Treatment with antimycin and oligomycin (Ant+Ol) started 5 min before stimulation with capsaicin (B). All of the recordings were done in low-Na+/TTX buffer. C, The role of the mitochondrial Na+/Ca2+ exchange in the generation of the capsaicin-induced (1 μm, 30 s) presynaptic [Ca2+]i plateau was examined using the exchange inhibitor CGP37157 (10 μm). All of the recordings were done in low-Na+/TTX buffer. The drug was applied immediately after capsaicin removal. The washout of the drug was accompanied by a secondary [Ca2+]i elevation, which likely resulted from mitochondrial Ca2+ release. For these experiments, the total area under the [Ca2+]i trace, including the initial transient [Ca2+]i elevation and the second phase after the CGP37157 washout, was 14.4 ± 1.1 μm*min (n = 14 boutons/6 cells). For comparison, the area under the [Ca2+]i traces recorded in response to capsaicin under control conditions was 16.7 ± 1.4 μm*min (n = 48 boutons/17 cells; p = 0.27, Student's t test, CGP37157 vs control). D, DRG neurons transfected with mtPericam were subsequently loaded with Rhod10K via a patch pipette. Merged image on the left shows distribution of mtPericam (green) and Rhod10K (red). [Ca2+]mt responses to two sequential capsaicin applications (1 μm, 30 s, 30 min between applications) were studied in three boutons (white boxes). No additional treatment was applied during the first response (left trace), whereas the cell was treated with 10 μm CGP37157 during the second response (right trace). Drug applications are shown by horizontal bars under the traces. [Ca2+]mt changes in individual boutons are indicated by different colors and numbers. E, The duration of capsaicin-induced presynaptic cytosolic (white and gray bars) and mitochondrial (black bars) Ca2+ responses was obtained from experiments like those shown in A–D. The bars represent 48 boutons and 17 cells for the control, 17 boutons and 4 cells for the effects of 0.3 μm antimycin and 1 μm oligomycin (Ant+Ol), 26 boutons and 7 cells for treatment with Ru360 (100 μm), 26 boutons and 10 cells for treatment with CGP37157 (10 μm, CGP), 30 boutons and 4 cells for the treatment with 5 μm CsA, and 14 boutons and 5 cells for mtPericam (control) measurements. ***p < 0.001, one-way ANOVA with Bonferroni's post hoc test relative to the Bis-fura control. F, Effects of FCCP (5 μm) on presynaptic [Ca2+]i in unstimulated cell (left) and after stimulation with capsaicin (right; 1 μm, 30 s). FCCP applications were separated by 10 min, and Ca2+ was omitted from the extracellular solution (EGTA at 0.1 mm) during the FCCP treatments (n = 23 boutons/5 cells).
Figure 8.
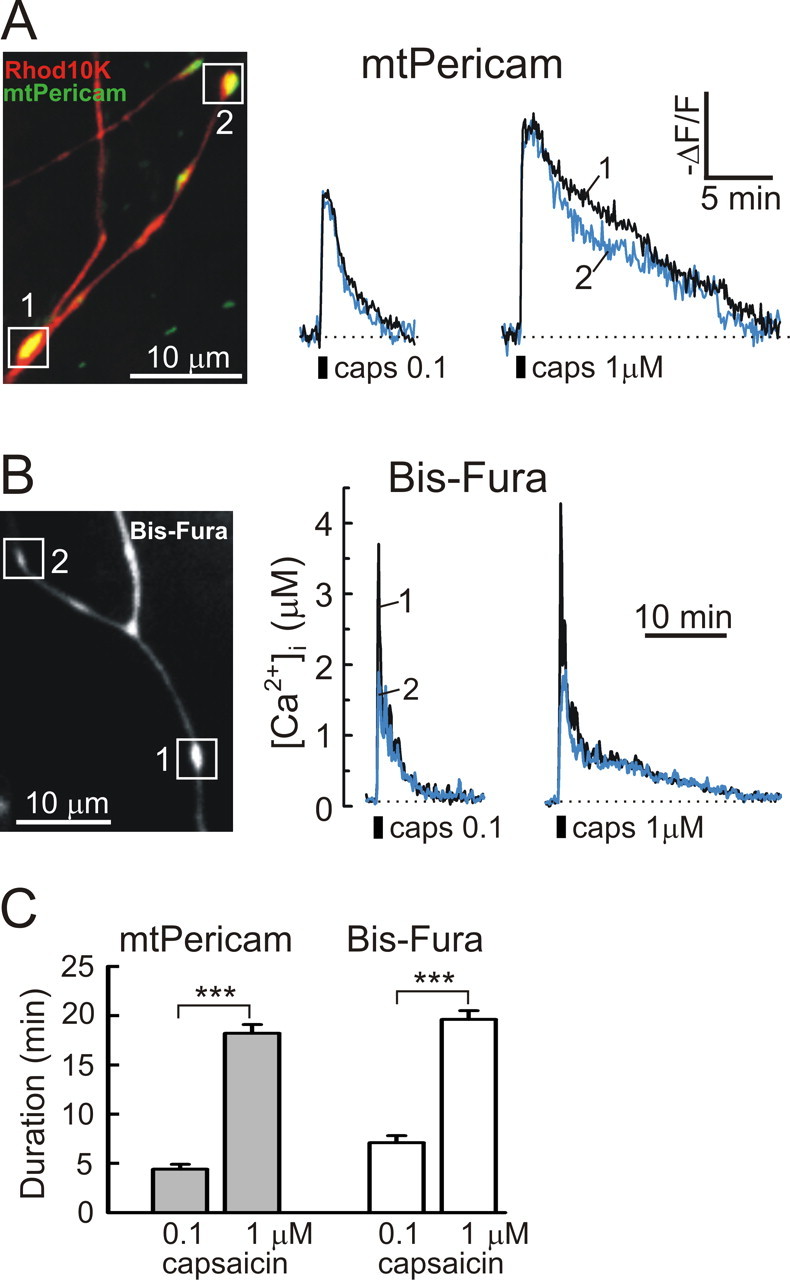
Raising capsaicin concentration increases the duration of the presynaptic [Ca2+]i plateau in DRG neurons. A, B, Presynaptic mitochondrial (A) and cytosolic (B) Ca2+ responses were elicited by 30 s applications of 0.1 and 1 μm capsaicin (caps; 30 min between applications), respectively. All of the experiments were done in low-Na+/TTX buffer. Recordings obtained from individual boutons are indicated by different colors and numbers. C, The durations of capsaicin-induced presynaptic mitochondrial (gray bars) and cytosolic (white bars) Ca2+ responses were obtained from experiments like those shown in A and B, and bars represent 18 boutons and 4 cells for the mtPericam measurements and 10 boutons and 3 cells for the Bis-fura experiments. Capsaicin concentration is indicated under the graph. ***p < 0.001, paired Student's t test.
Figure 1.
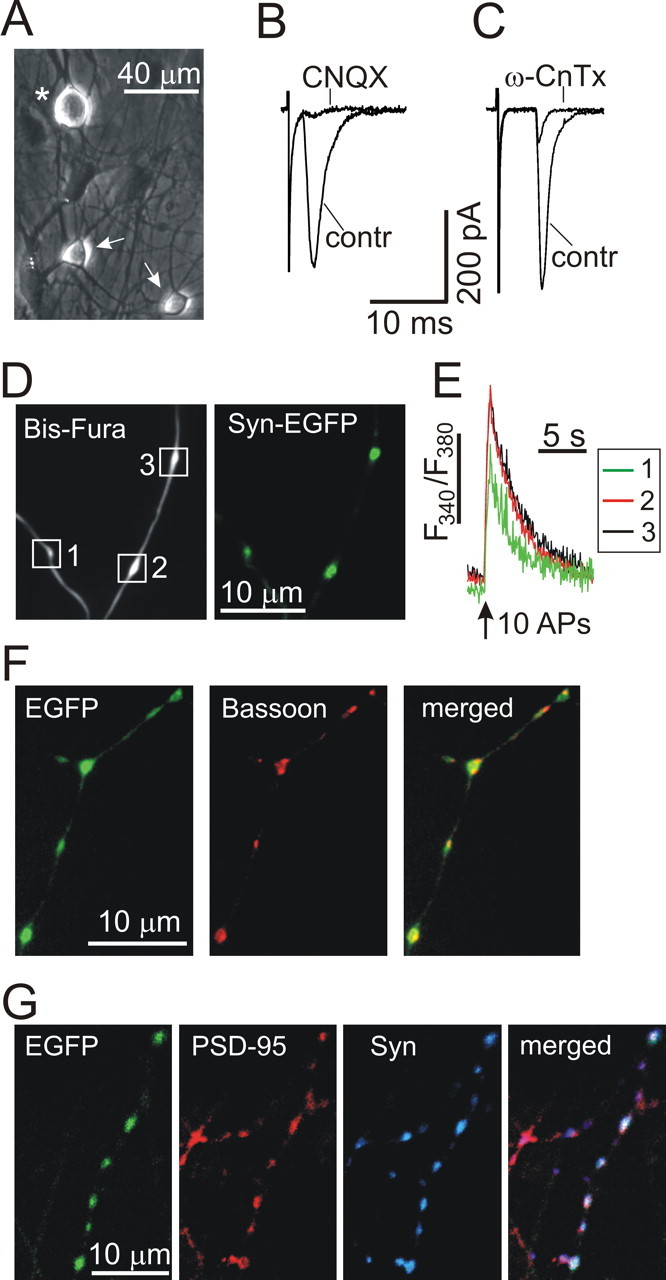
Excitatory synaptic transmission and presynaptic [Ca2+]i measurements in DRG/SC coculture. A, Phase-contrast image of a DRG neuron (asterisk) and two SC neurons (arrows) grown in culture for 12 d. B, C, The effects of CNQX (10 μm; B) and ω-conotoxin GVIA (ω-CnTx, 1 μm; C) on evoked EPSCs. Representative EPSC traces made before and during drug treatment are superimposed. D, E, Neurons were transfected with synaptophysin–EGFP (Syn-EGFP) using the lentivirus and subsequently loaded with Bis-fura via a patch pipette. D shows the distribution of Bis-fura (λex = 380 nm; left) and synaptophysin–EGFP (λex = 475 nm; right) in axons of DRG neurons. E depicts [Ca2+]i elevations in presynaptic boutons that were elicited by a train of action potentials (APs; 1 s at 10 Hz; vertical arrow) using extracellular field stimulation (Usachev et al., 2002). White boxes in D indicate boutons from which [Ca2+]i changes were recorded and plotted in E. [Ca2+]i changes in individual boutons are indicated by different colors. F, Neurons were transfected with EGFP using the lentivirus, fixed, and stained for the active zone protein bassoon (tom Dieck et al., 1998). The merged image shows bassoon localization in axonal varicosities of a DRG neuron. G, Neurons were transfected with EGFP using the lentivirus, fixed, and stained for the postsynaptic protein PSD-95 (red) and the presynaptic protein synaptophysin (Syn, blue). The merged image demonstrates the association of both proteins with axonal boutons.
Figure 4.
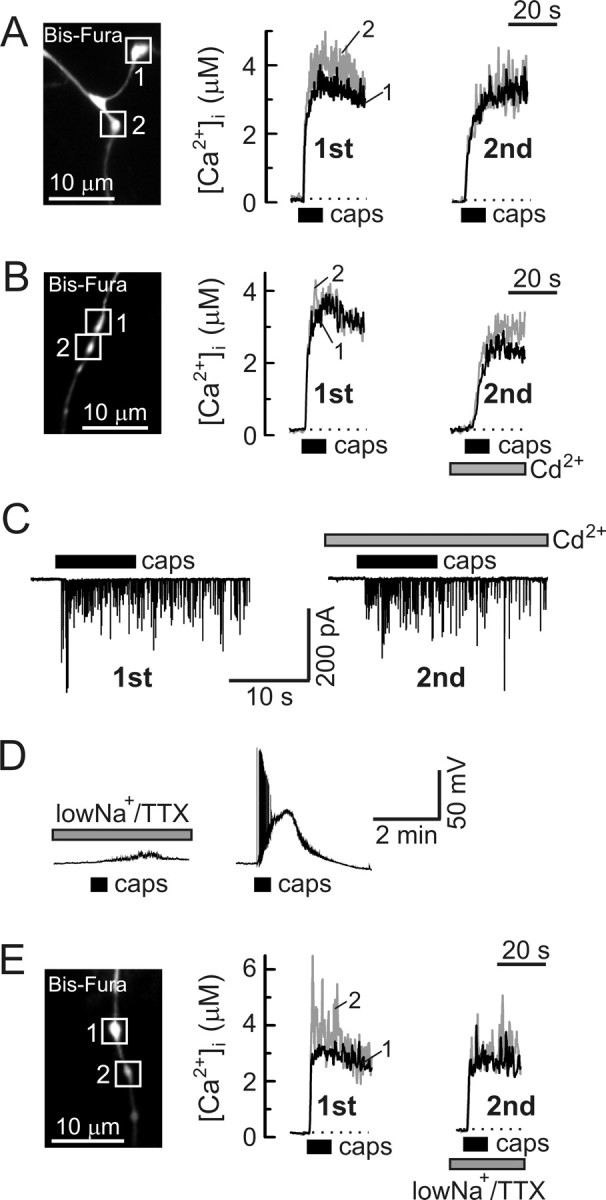
Voltage-gated Ca2+ channels are not required for capsaicin-induced presynaptic [Ca2+]i elevation and enhancement of synaptic activity. A, B, Presynaptic [Ca2+]i responses in DRG neurons were studied during two sequential applications of capsaicin (caps, 1 μm, 10 s, 20 min between applications). A shows an experiment in which no additional treatments were made, whereas the experiment shown in B involved the addition of 200 μm Cd2+ to the extracellular solution before and during the second application. [Ca2+]i traces obtained from individual boutons (white boxes) are indicated by different numbers and colors (black and gray). C, sEPSCs were recorded from an SC neuron voltage clamped at −60 mV during two sequential applications of capsaicin (1 μm, 10 s, 20 min between applications), either with no additional treatment (left) or in the presence of 200 μm Cd2+ (right). D, Reduction of extracellular Na+ concentration to 35 mm and addition of 1 μm TTX (low-Na+/TTX) completely blocked the generation of action potentials in response to capsaicin (1 μm, 30 s; left). This effect was reversible, as shown on the right. Both recordings are from the same DRG neuron (30 min between capsaicin applications). E, Exposure to low-Na+/TTX buffer did not affect capsaicin-evoked (1 μm, 10 s, 20 min between applications) [Ca2+]i responses in axonal boutons (white boxes) of DRG neurons.
Mitochondrial Ca2+ concentration ([Ca2+]mt) was measured using the mitochondria-targeted Ca2+ indicator mtPericam (Nagai et al., 2001; Filippin et al., 2003). To visualize axonal boutons, DRG neurons overexpressing mtPericam were loaded with tetramethylrhodamine 10,000 molecular weight dextran (Rhod10K) (Invitrogen) via patch pipettes (200 μm). mtPericam fluorescence was excited at 410 (12) nm using the Polychrome IV monochromator (T.I.L.L. Photonics). Excitation light was reflected off a dichroic mirror (505 nm; Chroma Technology, Rockingham, VT) and focused on the cells via a 60× oil-immersion objective (NA 1.40; Olympus). Emitted fluorescence was collected at 530 (50) nm (single band filter; Semrock) using an IMAGO CCD camera (T.I.L.L. Photonics) at the sampling rate of 0.2 Hz. At 410 nm, a decrease in mtPericam fluorescence corresponds to a [Ca2+]mt increase. [Ca2+]mt changes were presented as −ΔF/F 0 = −(F − F 0)/F 0, where F is current fluorescence intensity, and F 0 is fluorescence intensity in the resting cell.
The duration of the cytosolic (or mitochondrial) Ca2+ response was calculated at 5% of the net [Ca2+]i (or [Ca2+]mt) increase. Data are presented as mean ± SEM.
Electrophysiology.
Whole-cell patch-clamp recordings were obtained using a patch-clamp amplifier Axopatch 200B and an analog-to-digital converter Digidata 1322A (Molecular Devices, Union City, CA). Data were collected (filtered at 2 kHz and sampled at 5 kHz) and analyzed using the pClamp 9 software (Molecular Devices). Patch pipettes were pulled from borosilicate glass (3–5 mΩ; Narishige, Tokyo, Japan) on a Sutter Instruments (Novato, CA) P-87 micropipette puller and filled with the following solution (in mm): 125 K-gluconate, 10 KCl, 3 Mg-ATP, 1 MgCl2, 5 EGTA, and 10 HEPES, pH 7.25 adjusted with KOH (290 mOsm/kg with sucrose). The standard extracellular recording solution contained the following (in mm): 140 NaCl, 5 KCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 0.4 KH2PO4, 0.6 Na2HPO4, 3 NaHCO3, 10 glucose, and 10 HEPES, pH 7.35 with NaOH (310 mOsm/kg with sucrose). In a number of experiments (see Figs. 5 –8, 9 A–E), action potentials were blocked by adding 1 μm tetrodotoxin (TTX) and by replacing 105 mm NaCl with choline-Cl in the extracellular solution (low-Na+/TTX). Glutamate release from DRG neurons was studied by recording EPSCs from SC neurons voltage clamped at −60 mV in the presence of 10 μm bicuculline and 2 μm strychnine. For the analysis, frequencies of spontaneous EPSCs (sEPSCs) were averaged over 15 s intervals, unless indicated otherwise. Evoked EPSCs in SC neurons were elicited using a glass extracellular stimulating electrode (0.2–0.4 ms pulse) positioned near the cell body of a nearby DRG neuron. For Ca2+ current recordings, extracellular NaCl and KCl were replaced with tetraethylammonium-Cl, and 1 μm TTX was added to extracellular solution; K+ in the pipette solution was replaced with Cs+ (Cs-gluconate). Voltage-gated Ca2+ currents (VGCCs) were evoked by step depolarizations from −60 to 0 mV (50 ms, every 20 s).
Figure 7.
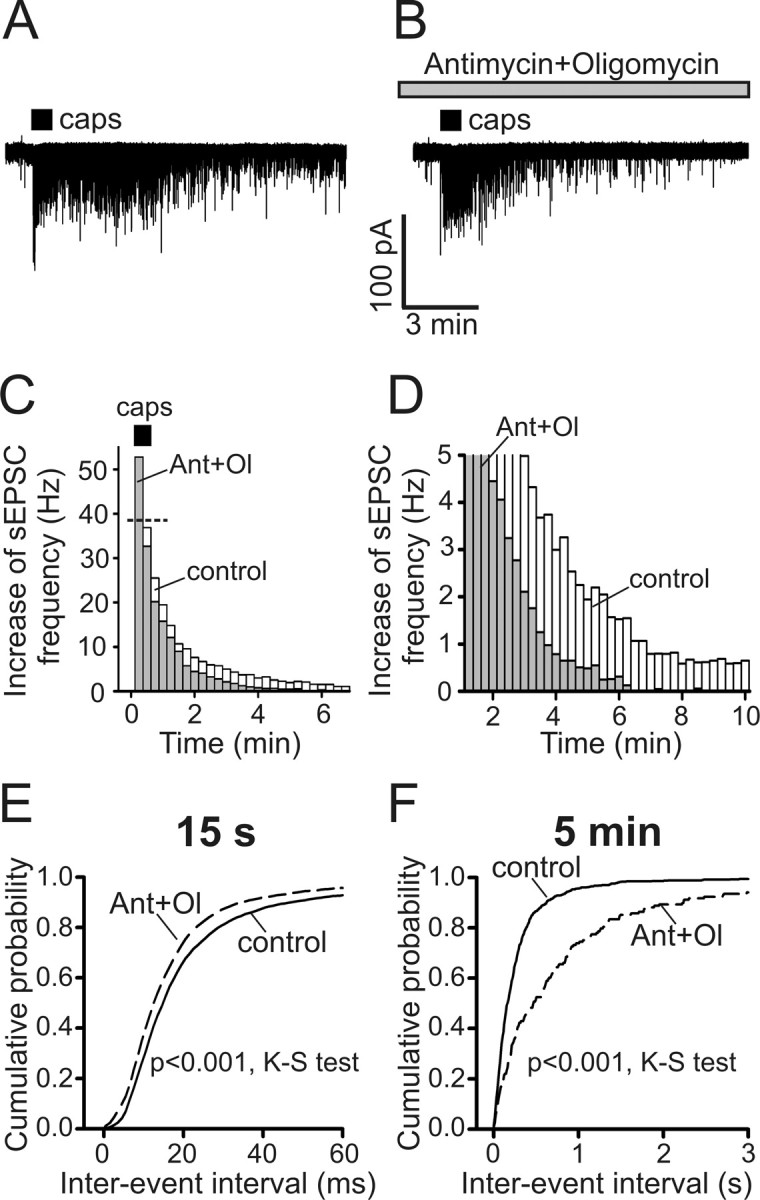
Inhibition of mitochondrial Ca2+ uptake results in shortening of vanilloid-induced synaptic activity. A, B, Representative postsynaptic responses to capsaicin (caps; 1 μm, 30 s) in the absence (A) or presence of 0.3 μm antimycin A1 and 1 μm oligomycin B (B) were recorded from SC neurons voltage clamped at −60 mV in low-Na+/TTX buffer. Antimycin and oligomycin were added 5 min before capsaicin application. C, D, Summary of the antimycin and oligomycin (Ant+Ol) effects on the sEPSC frequency obtained from experiments like those described in A and B. C and D display the same data sets using different frequency and timescales. Histograms representing the sEPSC frequency mean values for control conditions (n = 10; white bars) and for the antimycin plus oligomycin treatments (n = 7; gray bars) are superimposed. Each data point was obtained by averaging the sEPSC frequency during 15 s of recording and subtracting the resting sEPSC frequency. Capsaicin treatment (30 s) was started at t = 0 min (horizontal bar). The horizontal dotted line in C indicates the mean sEPSC frequency during the first 15 s of capsaicin application in controls. E, F, Cumulative probabilities of sEPSC interevent intervals were plotted for the control (n = 10; solid line) and antimycin plus oligomycin conditions (n = 7; dashed line) during the peak of the response (E; first 15 s of capsaicin application) and 5 min after capsaicin application (F; 30 s interval centered at 5 min). p < 0.001 for both time points, Kolmogorov–Smirnov test (K-S test).
Figure 9.
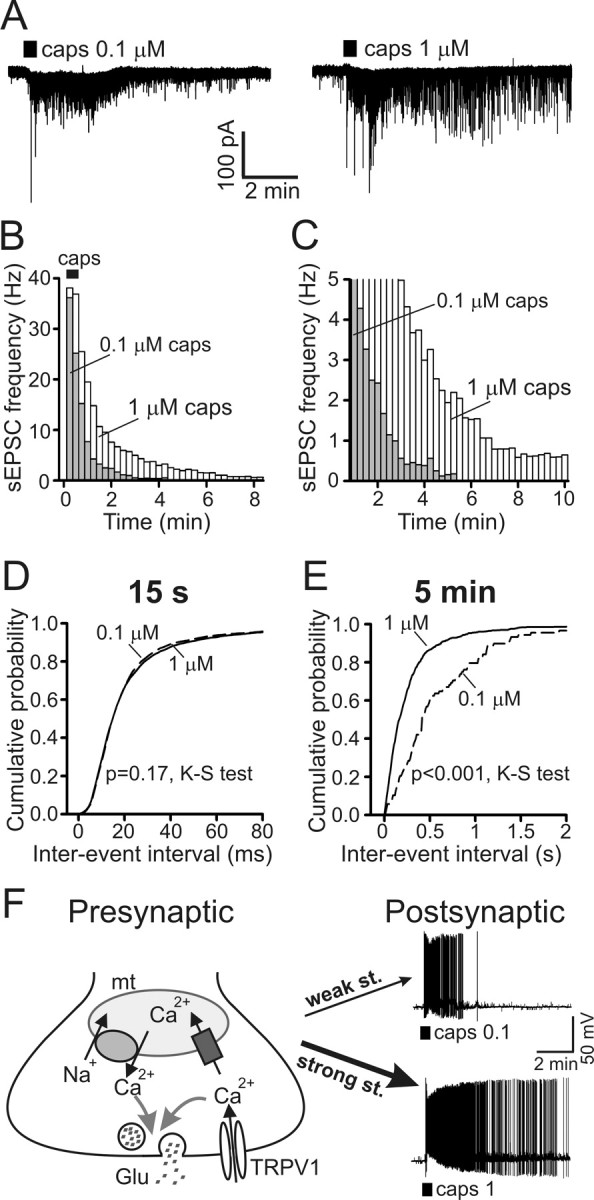
Duration of postsynaptic activity depends on the strength of presynaptic TRPV1 stimulation. A, Representative postsynaptic responses to 30 s applications of 0.1 and 1 μm capsaicin (caps; 30 min between applications). Both recordings were obtained from the same SC neuron (V hold of −60 mV) bathed in low-Na+/TTX buffer. B, C, Changes in frequency of sEPSCs evoked by 30 s applications of 0.1 and 1 μm capsaicin. B and C show the same data plotted on different frequency and timescales for clarity. Capsaicin treatment started at t = 0 min (horizontal bar in B). Histograms (sEPSC frequencies, mean values) for 0.1 μm capsaicin (n = 8; gray bars) and 1 μm capsaicin (n = 10; white bars) are superimposed. Data points were obtained by averaging sEPSC frequency during 15 s of recording and by subtracting the resting sEPSC frequency. The histograms for 1 μm capsaicin were replotted from Figure 7, C and D (control), for comparison with results obtained in 0.1 μm capsaicin experiments. D, E, Cumulative probabilities of sEPSC interevent intervals were calculated at the peak of the response (D; first 15 s of capsaicin application) and 5 min (E; 30 s interval centered at 5 min) after stimulation. Graphs for 1 μm capsaicin (solid lines; n = 10) were replotted from Figure 7, E and F (control), for comparison with data for 0.1 μm capsaicin (dashed lines; n = 8). p = 0.17 and p < 0.001 for 15 s and 5 min, respectively, Kolmogorov–Smirnov test. F, A model of presynaptic [Ca2+]i signaling mediated by TRPV1 and mitochondria in DRG neurons. The duration of the mitochondria-dependent [Ca2+]i plateau and of glutamate release depends on the strength of presynaptic stimulation. Consequently, 0.1 μm capsaicin (weak stimulus) produces only a short postsynaptic electrical discharge (top trace), whereas 1 μm capsaicin (strong stimulus) induces long-lasting firing of action potentials by postsynaptic SC neurons (bottom trace). In the presented experiments, both recordings of postsynaptic potentials were obtained from the same SC neuron using current clamp. Capsaicin applications (0.1 or 1 μm, 30 s; 30 min between applications) are indicated by the horizontal bars under the traces.
Immunocytochemistry.
DRG–SC cocultures overexpressing EGFP were stained with monoclonal mouse anti-bassoon (1:500; catalog #VAM-PS003; StressGen/Assay Designs, Ann Arbor, MI) and tetramethylrhodamine isothiocyanate (TRITC)-labeled donkey anti-mouse serum (1:200; Jackson ImmunoResearch, West Grove, PA), as described previously (Usachev et al., 2002). For the triple-labeling experiments, EGFP-transfected neurons were stained with monoclonal mouse anti-postsynaptic density-95 (PSD-95) (1:400–1:800; catalog #75-028; NeuroMab, Davis, CA) and polyclonal rabbit anti-Synaptophysin (1:200; catalog #sc-9116; Santa Cruz Biotechnology, Santa Cruz, CA); Alexa555-labeled goat anti-mouse serum and Alexa633-labeled goat anti-rabbit serum (1:200; Invitrogen) were used as secondary antibodies. EGFP [λex = 488 nm; λem = 515 (10) nm], TRITC [λex = 543 nm; λem = 580 (20) nm], Alexa555 [λex = 543 nm; λem = 580 (20) nm], and Alexa633 (λex = 633 nm; λem > 660 nm) fluorescence was observed using an Olympus BX61 microscope equipped with the Fluoview 300 laser-scanning confocal imaging system and a 60× oil-immersion objective (NA 1.40).
Reagents.
Bis-fura and fura-FF were obtained from Invitrogen; bicuculline, capsaicin, capsazepine, CGP37157 [7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one], CNQX, dl-AP-5, N-arachidonoyl-dopamine (NADA), SB-366791 (4′-chloro-3-methoxycinnamanilide), and tetrodotoxin were from Tocris Bioscience (Ellisville, MO); ionomycin and Ru360 [(μ)[(HCO2)(NH3)4Ru]2OCl3] were from Calbiochem (San Diego, CA); agatoxin IVA was from Bachem (Torrance, CA); and ω-conotoxin GVIA was from Alomone Labs (Jerusalem, Israel). All other reagents were purchased from Sigma (St. Louis, MO). Given that Ru360 rapidly loses its activity in solution, it was prepared daily and added to the patch pipette at supramaximal concentration (100 μm).
Results
We used a DRG/SC coculture system that has been widely used as a model for studying the first sensory synapse (Gu and Macdermott, 1997; Usachev et al., 2002; Tsuzuki et al., 2004; Sikand and Premkumar, 2007). This culture system (Fig. 1 allowed us to monitor cytosolic Ca2+ concentration ([Ca2+]i) in presynaptic boutons and to record EPSCs under highly defined conditions. As in vivo, DRG and SC neurons grown in coculture formed glutamatergic synaptic connections that were sensitive to the non-NMDA receptor antagonist CNQX (Fig. 1 B). Also, ω-conotoxin GVIA (1 μm) blocked the evoked EPSCs by 81 ± 2% (n = 5) (Fig. 1 C), consistent with the important role of N-type VGCCs in triggering glutamate release at the first sensory synapse in the dorsal horn of the spinal cord (Bao et al., 1998; Heinke et al., 2004). We also found that the selective inhibitor of the P/Q-type Ca2+ channels agatoxin IVA (200 nm) blocked the evoked EPSCs by 18 ± 5% (n = 5; data not shown), whereas the L-type channel inhibitor nimodipine (5 μm) was without effect (n = 4; data not shown).
The majority of presynaptic boutons on the central processes of primary afferent neurons are of the en passant type, with a diameter of 1–2 μm and a length of 1–4 μm (Semba et al., 1985; Maxwell and Rethelyi, 1987; Hwang et al., 2004). In our system, axonal boutons of similar size were identified in DRG neurons loaded with the Ca2+ indicator Bis-fura (Fig. 1 D). The presynaptic vesicular protein synaptophysin localized to these structures, as determined by imaging synaptophysin–EGFP fusion protein that was transferred to neurons using the lentivirus (Fig. 1 D) (102 of 105 boutons/11 cells analyzed contained synaptophysin–EGFP) or by staining cells with a synaptophysin-specific antibody (Usachev et al., 2002). Figure 1 E shows electrically evoked [Ca2+]i responses recorded from axonal boutons labeled with Bis-fura and synaptophysin–EGFP. Axonal boutons were also immunoreactive for another presynaptic protein, bassoon (Fig. 1 F) (76 of 78 boutons/10 cells analyzed stained positively for bassoon). Moreover, we found that the majority of axonal boutons were associated with the postsynaptic protein PSD-95 (Fig. 1 G) (70 of 76 boutons/8 cells analyzed were associated with PSD-95 immunofluorescence, and 68 of these boutons were associated with both PSD-95 and synaptophysin immunofluorescence). In these two latter cases, axonal boutons were visualized by overexpressing EGFP in DRG neurons, as described previously (Usachev et al., 2002).
Capsaicin induces prolonged presynaptic [Ca2+]i elevation and enhancement of synaptic activity
The resting [Ca2+]i in axonal boutons was 0.11 ± 0.02 μm (n = 59 boutons/19 cells). A 30 s application of 1 μm capsaicin elicited a robust elevation of presynaptic [Ca2+]i in 72% of the small-to-medium size DRG neurons (n = 19 cells). Only capsaicin responders (with a mean presynaptic [Ca2+]i elevation ≥100% above baseline) were analyzed throughout the study. In these cells, presynaptic [Ca2+]i peaked at 4.05 ± 0.31 μm (range, 0.4–7.5 μm; n = 47 boutons/14 cells) (Fig. 2 A). After capsaicin removal, [Ca2+]i rapidly recovered to a new steady-state level ranging between 0.5 and 1.5 μm. This delayed phase of the Ca2+ response ([Ca2+]i plateau) typically lasted >10–15 min. At 5 and 10 min after stimulation, the [Ca2+]i plateau was 0.78 ± 0.09 and 0.48 ± 0.07 μm, respectively, and the duration of the capsaicin-induced Ca2+ response was 15.7 ± 1.4 min (n = 47 boutons/14 cells; calculated at 5% of the net [Ca2+]i increase).
In parallel experiments, we monitored glutamate release by recording sEPSCs in SC neurons. These experiments were done in the presence of the GABAA receptor-selective antagonist bicuculline (10 μm) and the glycine receptor antagonist strychnine (2 μm). Capsaicin application (1 μm, 30 s) induced a >150-fold increase in the sEPSC frequency (from 0.27 ± 0.07 Hz at rest to 42.45 ± 7.64 Hz during the first 15 s of the stimulation; n = 6) (Fig. 2 B). Capsaicin induced no response in the cultures containing only spinal cord neurons (n = 4; data not shown). As in the case of presynaptic [Ca2+]i elevation, the enhanced release of glutamate was observed long after the capsaicin washout, with sEPSC frequencies 5 and 10 min after stimulation still at 1.74 ± 0.47 and 1.03 ± 0.42 Hz, respectively (n = 6) (Fig. 2 C,D). In contrast, capsaicin-induced action potential firing and depolarization in the DRG neurons rapidly diminished during drug washout (Fig. 2 B). Both the elevation in [Ca2+]i (n = 12 boutons/3 cells; data not shown) and the synaptic responses (n = 4) (Fig. 2 E) were blocked by the TRPV1-selective antagonist capsazepine (10 μm). Combined application of the NMDA receptor antagonist dl-AP-5 (50 μm) and the NK1 receptor antagonist L-703,606 [cis-2-(diphenylmethyl)-N-[(2-iodophenyl)methyl]-1 azabicyclo[2.2.2]octan-3-amine oxalate salt] (1 μm) did not affect the sEPSC frequency (n = 3; data not shown), whereas CNQX (10 μm) completely and reversibly inhibited sEPSCs (n = 6) (Fig. 2 F), suggesting that the synaptic response was mediated by non-NMDA glutamate (most likely AMPA) receptors.
Long-lasting presynaptic [Ca2+]i signaling and synaptic activity induced by the endovanilloid NADA
We next examined the ability of the endogenous TRPV1 agonist NADA to elevate presynaptic [Ca2+]i and modulate sensory synaptic transmission. NADA is found in various regions of the CNS and is thought to be a product of the condensation of arachidonic acid with dopamine or tyrosine (Huang et al., 2002; Starowicz et al., 2007). NADA activates both cloned and native TRPV1 with a potency that is slightly lower than that of capsaicin and induces the release of CGRP from cultured trigeminal sensory neurons and from dorsal spinal cord slices (Huang et al., 2002; Price et al., 2004; Millns et al., 2006).
In the DRG/SC coculture system, NADA (5 μm, 30 s) evoked a robust biphasic presynaptic [Ca2+]i response (Fig. 3 A) similar to that triggered by capsaicin (Fig. 2 A). The initial rapid [Ca2+]i rise from the resting level of 0.14 ± 0.06 μm to the peak concentration of 4.41 ± 0.38 μm (n = 31 boutons/6 cells) was followed by a [Ca2+]i plateau phase that often lasted for >15 min. The overall duration of the NADA-induced [Ca2+]i elevation in axonal boutons was 17.58 ± 1.16 min (n = 31 boutons/6 cells). A selective TRPV1 antagonist, SB-366791 (10 μm) (Gunthorpe et al., 2004), blocked presynaptic [Ca2+]i elevation elicited by NADA (Fig. 3 B) (n = 19 boutons/3 cells). Another TRPV1 antagonist, capsazepine (10 μm), had similar effect (n = 14 boutons/3 cells; data not shown).
In parallel experiments, NADA (5 μm, 30 s) dramatically enhanced excitatory synaptic activity, yielding an increase of sEPSC frequency from 0.36 ± 0.12 Hz at rest to 33.51 ± 7.65 Hz (Fig. 3 C–E) (n = 7). As in the case of capsaicin (Fig. 2 B–D), the sEPSC frequency remained elevated long after the removal of agonist from the extracellular medium. For example, the frequencies were still at 2.83 ± 0.66 and 1.95 ± 0.72 Hz when measured at 5 and 10 min, respectively, after NADA application (n = 7) (Fig. 3 D,E). The NADA effect was blocked by the non-NMDA receptor antagonist CNQX (10 μm; n = 3) (Fig. 3 F) and by the TRPV1 antagonists SB-366791 (10 μm; n = 6) (Fig. 3 G) and capsazepine (10 μm; n = 4; data not shown). Thus, like capsaicin, NADA induces long-lasting presynaptic [Ca2+]i elevation in DRG neurons and a concomitant enhancement of glutamatergic synaptic activity at the sensory synapse.
Vanilloid-induced presynaptic Ca2+ entry does not require VGCCs
Vanilloids could potentially induce Ca2+ entry into axonal boutons both directly, via the TRPV1 channel, and indirectly, by triggering depolarization and activation of voltage-gated Ca2+ channels. To examine the contribution of VGCCs to the TRPV1-mediated response, we compared presynaptic [Ca2+]i elevations elicited by two sequential capsaicin applications (1 μm, 10 s, with 20 min between applications) in control (no additional treatment) (Fig. 4 A) and after blockade of VGCCs by 200 μm Cd2+ during the second capsaicin application (Fig. 4 B). At 200 μm, Cd2+ completely blocked whole-cell voltage-gated Ca2+ currents (n = 7; data not shown) but had no effect on capsaicin-induced currents in DRG neurons (n = 4; data not shown). In control experiments, the amplitude of the second Ca2+ response (A 2) was slightly smaller than that of the first one (A 1), with an A 2/A 1 ratio of 86 ± 9% (n = 14 boutons/4 cells). This decrease could be a consequence of incomplete TRPV1 recovery from desensitization (Koplas et al., 1997). A comparable decrease in the amplitude of [Ca2+]i elevation was observed when cells were treated with Cd2+ (200 μm) during the second capsaicin application (A 2/A 1 = 81 ± 7%; n = 12 boutons/5 cells; p = 0.68, Student's t test), indicating that VGCCs play a negligible role in capsaicin-induced Ca2+ entry. Accordingly, treatment with Cd2+ did not prevent a pronounced increase in synaptic activity induced by capsaicin (Fig. 4 C); the frequency of sEPSC reached 41 ± 13 and 37 ± 13 Hz (n = 3) during the initial 15 s of the response in the absence or presence of 200 μm Cd2+, respectively.
In an additional series of experiments, the capsaicin-induced propagation of electrical activity and activation of VGCCs was prevented by blocking neuronal excitation. This block was achieved by adding 1 μm TTX to the extracellular solution and simultaneously reducing external Na+ concentration to 35 mm by equimolar substitution with choline (this solution will henceforth be termed low-Na+/TTX). Under these conditions, action potentials were completely blocked, and only a negligible depolarization developed in response to capsaicin (3 ± 1 mV; n = 7) (Fig. 4 D). The addition of TTX (1 μm) alone did not prevent the capsaicin-induced firing of action potentials (7 of 10 cells tested), consistent with the expression of TTX-resistant voltage-gated Na+ channels in a large proportion of capsaicin-responsive DRG neurons (Gold et al., 1996). As shown in Figure 4 E, the suppression of electrical activity and depolarization by incubation in low-Na+/TTX did not prevent a rapid, capsaicin-induced [Ca2+]i rise in the presynaptic boutons: A 2/A 1 ratio was 80 ± 7% (n = 12 boutons/4 cells; p = 0.62, Student's t test relative to control in Fig. 4 A). Together with the results of the Cd2+ experiments, these findings suggest that Ca2+ influx via TRPV1 is the major route of presynaptic Ca2+ entry in response to stimulation with vanilloids.
The sustained phase of the presynaptic [Ca2+]i response does not depend on either extracellular Ca2+ or the endoplasmic reticulum Ca2+ stores
The initial vanilloid-induced rapid [Ca2+]i elevation was followed by a sustained [Ca2+]i plateau (Figs. 2 A, 3 A). A distinct presynaptic [Ca2+]i plateau was also observed in response to either depolarization or intense stimulation using action potentials (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This phenomenon did not depend on the type of Ca2+ indicator used, because similar responses were observed using a low-affinity (fura-FF, K d of ∼5.5 μm) and intermediate-affinity (Bis-fura, K d of ∼525 nm) Ca2+ indicators (Fig. 5, compare A with B–D). We wanted to further explore the nature of the [Ca2+]i plateau in presynaptic boutons. To prevent the spread of electrical excitation induced by vanilloids and, thus, to minimize the influence of extrasynaptic TRPV1, all of the following experiments were performed using low-Na+/TTX solution, unless indicated otherwise.
We found that, once initiated, the presynaptic Ca2+ response was not affected by the removal of extracellular Ca2+ (Fig. 5 B) (n = 25 boutons/4 cells), suggesting the involvement of an intracellular Ca2+ source during the plateau phase. In soma of DRG neurons, at least two intracellular Ca2+ compartments could potentially contribute to the capsaicin-induced [Ca2+]i responses. These are the endoplasmic reticulum (ER) Ca2+ stores (Liu et al., 2003; Karai et al., 2004) and mitochondria (Dedov and Roufogalis, 2000). Capsaicin has been reported to release Ca2+ from the ER Ca2+ stores in DRG neurons, and this response persisted in the absence of extracellular Ca2+ (Liu et al., 2003; Karai et al., 2004). However, we found that capsaicin failed to trigger presynaptic [Ca2+]i elevation in Ca2+-free medium, whereas subsequent readdition of extracellular Ca2+ restored the response (Fig. 5 C) (n = 35 boutons/7 cells). Cyclopiazonic acid (CPA) (5 μm) is a selective inhibitor of the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA); it depletes Ca2+ stores and essentially eliminates any additional Ca2+ release from or uptake by the stores (Thomas and Hanley, 1994; Verkhratsky, 2005). Although CPA modulates electrically evoked [Ca2+]i transients in DRG neurons (Usachev and Thayer, 1999; Lu et al., 2006), capsaicin-induced presynaptic [Ca2+]i plateau was not significantly affected by the drug (Fig. 5 D) (n = 15 boutons/4 cells). CPA did, however, slow the clearance of presynaptic [Ca2+]i transients induced by trains of action potentials (8 Hz, 1–3 s), indicating that functional ER Ca2+ stores are present in axonal boutons. In these experiments, [Ca2+]i decay was fitted with a monoexponential function, and the time constants were found to be 2.4 ± 0.2 s in control and 4.4 ± 0.7 s after treating cells with 5 μm CPA for 20 min (p < 0.01, paired Student's t test; n = 18 boutons/4 cells; data not shown). Thus, CPA-sensitive Ca2+ stores are present in the axonal boutons of DRG neurons in which, together with the plasma membrane Ca2+-ATPase (Usachev et al., 2002; Gover et al., 2007), the stores are involved in clearing small presynaptic [Ca2+]i loads (<400–500 nm) induced by short trains of action potentials. However, the Ca2+ stores are not likely to mediate the sustained presynaptic [Ca2+]i elevation (plateau) that follows large Ca2+ loads induced by intense TRPV1 stimulation.
Mitochondria control the presynaptic [Ca2+]i plateau and sustained synaptic activity
Mitochondria are highly concentrated in presynaptic terminals in which, in addition to producing ATP, these organelles can regulate presynaptic [Ca2+]i and synaptic plasticity (Zucker and Regehr, 2002; Hollenbeck, 2005; Wimmer et al., 2006). The function and mechanisms of Ca2+ transport by presynaptic mitochondria vary substantially among synapses and are likely specialized according to the signaling needs at a particular synapse (Tang and Zucker, 1997; Billups and Forsythe, 2002; Jonas et al., 2003; Levy et al., 2003; Yang et al., 2003; Garcia-Chacon et al., 2006). Factors contributing to such specialization include the number and the architecture of presynaptic mitochondria, their proximity to the active zones and Ca2+ channels, and the presence of other Ca2+ clearance mechanisms that compete with mitochondria for Ca2+ (Nguyen et al., 1997; Zenisek and Matthews, 2000; Guo et al., 2005).
Mitochondria buffer and release Ca2+ via distinct mechanisms causing Ca2+ to cycle across the mitochondrial membranes (Babcock and Hille, 1998; Friel, 2000; Thayer et al., 2002). Ca2+ uptake is mediated by the mitochondrial uniporter and is driven by the mitochondrial membrane potential ΔΨmt (−150 ÷ −180 mV) (Nicholls and Ward, 2000). Thus, we blocked Ca2+ uptake by either depolarizing mitochondria with the electron chain inhibitor antimycin A1 or applying the selective uniporter inhibitor Ru360. Antimycin A1 dissipates the proton gradient, which can reverse the mode of operation of F 1, F 0 ATP-synthase, so that it pumps H+ out of the matrix while consuming ATP (Nicholls and Budd, 2000; Toescu and Verkhratsky, 2003). To prevent ATP depletion, the selective inhibitor of the ATP synthase oligomycin B was added in these experiments. The combined treatment with 0.3 μm antimycin and 1 μm oligomycin virtually eliminated the presynaptic [Ca2+]i plateau (Fig. 6, compare A, B). Another consequence of mitochondrial membrane depolarization was a significant increase in the amplitude of the Ca2+ response, from 5.02 ± 0.32 μm in control cells (n = 48 boutons/17 cells) to 7.82 ± 0.83 μm in cells pretreated with antimycin plus oligomycin (n = 17 boutons/4 cells; p < 0.001, Student's t test). Ru360 is membrane impermeable and thus was applied via the patch pipette along with the Ca2+ indicator. Blockade of the Ca2+ uniporter with Ru360 also markedly reduced the duration of the Ca2+ signal (Fig. 6 E) and increased the amplitude of the Ca2+ response to 6.74 ± 0.64 μm (n = 26 boutons/7 cells; p < 0.05, Student's t test relative to control). These observations suggest that mitochondria limit the amplitude and prolong the duration of the capsaicin-induced presynaptic [Ca2+]i elevation. Given that Ca2+ buffering by mitochondria could potentially interfere with the analysis of presynaptic Ca2+ influx mechanisms, we repeated the experiments with Cd2+ (Fig. 4 A,B) under the conditions in which mitochondrial Ca2+ uptake was blocked by the combined treatment with 0.3 μm antimycin and 1 μm oligomycin. As shown in supplemental Figure 2 (available at www.jneurosci.org as supplemental material), treatment with Cd2+ had no effect on the capsaicin-induced presynaptic [Ca2+]i elevation recorded in the presence of these mitochondrial inhibitors, which is consistent with the results described in Figure 4.
Under physiological conditions, Ca2+ efflux from mitochondria in neurons is mediated primarily by the mitochondrial Na+/Ca2+ exchanger (Baron and Thayer, 1997; David, 1999; Colegrove et al., 2000; Garcia-Chacon et al., 2006). We found that a selective inhibitor of the mitochondrial Na+/Ca2+ exchanger, CGP37157 (10 μm), abolished the capsaicin-induced presynaptic [Ca2+]i plateau in DRG neurons (Fig. 6 C,E). Because this drug can inhibit Ca2+ influx (Baron and Thayer, 1997), it was applied after the washout of capsaicin. Interestingly, the washout of CGP37157 was accompanied by a secondary [Ca2+]i elevation (Fig. 6 C). This secondary presynaptic [Ca2+]i rise likely results from the release of Ca2+ that was trapped previously within mitochondria by the CGP37157 treatment. A similar secondary [Ca2+]i transient during the CGP37157 removal has been also reported in the cell body of DRG neurons (Baron and Thayer, 1997).
Ca2+ release from the matrix can also be mediated by the mitochondrial permeability transition pore (MPT or PTP) (Nicholls and Budd, 2000; Thayer et al., 2002; Newmeyer and Ferguson-Miller, 2003), and the voltage-dependent anion channel, which is the principal molecular component of the MPT, has been implicated in synaptic plasticity (Jonas et al., 1999; Levy et al., 2003; Jonas, 2004). We therefore examined the effect of the MPT inhibitor cyclosporin A (CsA) on the capsaicin-induced [Ca2+]i elevation. As summarized in Figure 6 E, treatment with 5 μm CsA did not change the duration of the presynaptic [Ca2+]i response (17.2 ± 1.3 min; n = 30 boutons/4 cells) compared with that of the control (17.8 ± 1.2 min; n = 48 boutons/17 cells). Combined with a complete elimination of the plateau phase by CGP37157 (Fig. 6 C,E), these data suggest that the mitochondrial Na+/Ca2+ exchanger is the major route of Ca2+ efflux from presynaptic mitochondria in sensory neurons.
Capsaicin-induced Ca2+ uptake by and its subsequent release from presynaptic mitochondria should be accompanied by corresponding changes in [Ca2+]mt. To monitor [Ca2+]mt, we expressed the mitochondrial Ca2+ indicator mtPericam (Nagai et al., 2001; Filippin et al., 2003) in DRG neurons using lentivirus as a vector. Before the experiments, transfected DRG neurons were loaded with Rhod10K via the patch pipette to enable visualization of axonal boutons. Imaging using mtPericam and Rhod10K revealed clusters of mitochondria in the presynaptic boutons (Fig. 6 D). Application of 1 μm capsaicin (30 s) elicited a rapid increase in [Ca2+]mt, followed by a slow decay to the prestimulus level. The mitochondrial and cytosolic Ca2+ responses were of similar duration (Fig. 6 E). CGP37157 treatment blocked Ca2+ efflux from the mitochondrial matrix, and [Ca2+]mt remained elevated as long as the agent was present (Fig. 6 D). Washout of CGP37157 restored Ca2+ release from mitochondria until [Ca2+]mt fully recovered to the resting level.
We further probed the capability of mitochondria to accumulate Ca2+ after TRPV1 activation by using the protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (Brocard et al., 2001). This agent rapidly and reversibly dissipates the proton gradient, causing the mobilization of mitochondrial Ca2+ into the cytosol. To isolate mitochondrial Ca2+ release from potential Ca2+ influx, we used Ca2+-free (EGTA at 0.1 mm) extracellular buffer during FCCP application. Virtually no changes in [Ca2+]i were observed when the mitochondrial Ca2+ content was probed with 5 μm FCCP in resting cells (Fig. 6 F). In contrast, FCCP treatment immediately after capsaicin application induced a robust [Ca2+]i elevation. Thus, presynaptic mitochondria are nearly empty at rest but rapidly accumulate Ca2+ in response to TRPV1 activation.
We next examined how the disruption of mitochondrial Ca2+ cycling affects vanilloid-induced synaptic activity (Fig. 7 A,B). We found that the blockade of mitochondrial Ca2+ uptake by combined treatment with 0.3 μm antimycin A1 and 1 μm oligomycin B markedly reduced the duration of the capsaicin-induced postsynaptic response, causing it to return to the prestimulus level (0.31 ± 0.07 Hz; n = 7) within 5–6 min after capsaicin washout (Fig. 7 B–D,F). In contrast, the sEPSC frequency in controls remained elevated long after capsaicin application was terminated (Fig. 7 A,C,D,F). For example, the sEPSC frequencies were 2.45 ± 0.45 and 0.96 ± 0.20 Hz at 5 and 10 min after stimulation, respectively, compared with 0.23 ± 0.07 Hz at rest (n = 10). Another consequence of inhibiting mitochondrial Ca2+ buffering was a more frequent release of glutamate during the first 15 s of stimulation (Fig. 7 E). Overall, our data indicate that mitochondria play an important role in the regulation of neurotransmission at sensory synapses after TRPV1 activation.
Mitochondria set the duration of synaptic activity in a manner that depends on the strength of presynaptic TRPV1 stimulation
The data described above suggest that stimulation of presynaptic TRPV1 induces a biphasic response. During the initial phase, Ca2+ entry mediated primarily by TRPV1 results in the elevation of presynaptic [Ca2+]i and glutamate release. A significant portion of Ca2+ entering the cell is then taken up by presynaptic mitochondria. Subsequently, this Ca2+ is gradually transported from the matrix back to the cytosol, providing a long-lasting supply of presynaptic Ca2+ for glutamate release during the sustained phase of the response, until Ca2+ is eventually extruded from the cell by the plasma membrane Na+/Ca2+ exchangers and Ca2+-ATPases (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Given that the rate of Ca2+ efflux from mitochondria is essentially constant and independent of Ca2+ load (Nicholls, 2005), this model predicts that the duration of the synaptic response is determined by stimulus strength and the amount of Ca2+ initially captured by mitochondria.
To test this idea, we examined [Ca2+]i, [Ca2+]mt, and sEPSC responses induced by low (0.1 μm) and high (1 μm) concentrations of capsaicin [K d of ∼0.7 μm (Caterina et al., 1997)]. First, we compared presynaptic [Ca2+]mt changes induced by the low and high capsaicin concentrations. Somewhat surprisingly, we found that capsaicin was able to induce a pronounced [Ca2+]mt elevation even at low concentration (0.1 μm) (Fig. 8 A). Both the amplitude and duration of mitochondrial Ca2+ responses were significantly increased by raising capsaicin concentration from 0.1 to 1 μm (Fig. 8 A,C). The relative changes in mtPericam fluorescence evoked by 0.1 and 1 μm capsaicin were 26 ± 1 and 35 ± 2%, respectively (p < 0.001, paired Student's t test; n = 18 boutons/4 cells). The durations of mitochondrial Ca2+ elevations induced by 0.1 and 1 μm capsaicin were 4.4 ± 0.5 and 18.2 ± 0.9 min, respectively (p < 0.001, paired Student's t test; n = 18 boutons/4 cells). Raising capsaicin concentration also markedly increased the duration of the cytosolic Ca2+ responses in axonal boutons (7.1 ± 0.7 min for 0.1 μm capsaicin and 19.6 ± 0.9 min for 1 μm capsaicin; p < 0.001, paired Student's t test; n = 10 boutons/3 cells), and the plateau phase became much more pronounced (Fig. 8 B,C). We found no significant differences in the amplitudes of the presynaptic [Ca2+]i elevations that were elicited by 0.1 μm capsaicin (4.59 ± 1.08 μm) and 1 μm capsaicin (4.87 ± 0.89 μm; n = 10 boutons/3 cells; p = 0.83, paired Student's t test). This suggests that Ca2+ buffering by mitochondria sets the limit of the presynaptic [Ca2+]i increase. We additionally verified these findings using a low-affinity Ca2+ dye, fura-FF. Again, no significant differences were found among the [Ca2+]i responses: the amplitudes were 4.99 ± 0.52 and 5.66 ± 0.48 μm for 0.1 and 1 μm capsaicin, respectively (n = 19 boutons/3 cells; p = 0.07, paired Student's t test).
Comparison of the postsynaptic responses induced by 0.1 and 1 μm capsaicin revealed that the sEPSC frequencies during the initial phase of the response were similar for both capsaicin concentrations (Fig. 9 B,D). However, synaptic activity induced by 0.1 μm capsaicin returned to the prestimulus level within 3–4 min, whereas the synaptic response induced by 1 μm capsaicin lasted >10 min (Fig. 9 A–C,E). These results are consistent with the [Ca2+]i imaging data (Fig. 8) and suggest that stronger TRPV1 activation enables greater calcium loading of presynaptic mitochondria and, consequently, a more prolonged mitochondrial Ca2+ release that maintains presynaptic [Ca2+]i elevation and synaptic activity.
How do TRPV1- and mitochondria-dependent presynaptic [Ca2+]i elevation and glutamate release translate into the electrical activity exhibited by postsynaptic SC neurons under physiological conditions? To address this question, we used current clamp to record postsynaptic potentials during stimulation with 0.1 or 1 μm capsaicin. The standard extracellular recording solution ([Na+] of 140 mm, no TTX added) was used in these experiments. Treatment with capsaicin (30 s, 0.1 or 1 μm) triggered intense firing of action potentials by the postsynaptic cell (Fig. 9 F), and this was blocked by 10 μm CNQX (n = 3; data not shown). The response to 0.1 μm capsaicin subsided within 3–5 min after stimulation (n = 4), whereas stimulation with 1 μm capsaicin resulted in postsynaptic neurons firing action potentials for >10 min (n = 8). This prolonged activity depended on mitochondria and was markedly shortened by treatment with 0.3 μm antimycin plus 1 μm oligomycin (n = 6; data not shown).
Discussion
TRPV1-mediated release of neurotransmitters and peptides from nociceptive terminals is thought to contribute to the development of pain hypersensitivity. However, signaling pathways that link TRPV1 activation with neurosecretion remain poorly understood. Here, we have identified the mechanisms that shape presynaptic [Ca2+]i signals and regulate glutamate release in primary sensory neurons in response to TRPV1 activation. Based on our data, we propose a model that is summarized in Figure 9 F. Initiation, but not maintenance, of the response requires Ca2+ influx, primarily through TRPV1. Ca2+ entry elevates presynaptic [Ca2+]i and triggers glutamate release. Ca2+ is rapidly sequestered by presynaptic mitochondria and then gradually transported back to the cytosol via Na+/Ca2+ exchange. This process is responsible for the prolonged presence of Ca2+ at the site of transmitter release, and the more Ca2+ is taken up by mitochondria during the stimulus, the longer the duration of the presynaptic [Ca2+]i plateau, glutamate release, and synaptic activity. Thus, mitochondria store information about the strength of presynaptic stimulation and translate it into the duration of postsynaptic activity.
TRPV1 mediates presynaptic Ca2+ entry during onset of the vanilloid-induced response
Activation of TRPV1 causes depolarization and action potential firing by primary afferents. Therefore, at least two routes for Ca2+ entry should be considered: voltage-gated Ca2+ channels and TRPV1. Presynaptic N-type VGCCs are responsible for evoked EPSCs at these synapses (Fig. 1 C) (Heinke et al., 2004). In contrast, VGCCs contributed little to capsaicin-induced presynaptic [Ca2+]i elevation and glutamate release (Fig. 4), possibly because of Ca2+/calcineurin-dependent inhibition of VGCCs (Wu et al., 2005). Thus, the TRPV1 channel is likely the major route mediating vanilloid-induced presynaptic Ca2+ influx at sensory synapses. Our data are in agreement with earlier findings showing that capsaicin-induced [Ca2+]i transients in peripheral terminals of the cornea and vanilloid-activated glutamatergic synaptic transmission in brainstem slices are independent of VGCCs (Marinelli et al., 2002; Gover et al., 2003; Jin et al., 2004).
Mitochondria control the sustained phase of the vanilloid-induced response
The rapid initial elevation of presynaptic [Ca2+]i was followed by a prolonged [Ca2+]i plateau accompanied by an enhancement of glutamate release. Presynaptic [Ca2+]i levels of ∼0.5–1 μm during the plateau phase are consistent with the [Ca2+]i requirements for asynchronous transmitter release at other synapses, a process that is likely controlled by a high-affinity (K d of ∼1 μm) Ca2+ sensor (Angleson and Betz, 2001). The sustained [Ca2+]i increase was independent of extracellular Ca2+ and was not mediated by the ER Ca2+ stores. Instead, our results strongly suggest that this phase of the TRPV1-mediated response was controlled by presynaptic mitochondria. The proposed presynaptic function of mitochondria (Fig. 9 F) may also help to explain the previous observations by several groups that brief TRPV1 activation triggers long-lasting release of glutamate and prolonged synaptic activity in the spinal cord and brainstem slices (Ueda et al., 1993; Yang et al., 1998; Marinelli et al., 2002, 2003; Nakatsuka et al., 2002).
Previous studies showed that mitochondria speed up recovery from synaptic depression and contribute to the long-term potentiation at central synapses (Billups and Forsythe, 2002; Levy et al., 2003) and control posttetanic potentiation at neuromuscular synapses (Tang and Zucker, 1997; Yang et al., 2003; Garcia-Chacon et al., 2006). It is generally agreed that Ca2+ uptake by presynaptic mitochondria is mediated by the mitochondrial Ca2+ uniporter (Tang and Zucker, 1997; David et al., 1998; Billups and Forsythe, 2002). Which mechanisms are responsible for Ca2+ efflux from mitochondria is less clear. MPT can mediate Ca2+ release from mitochondria (Ichas et al., 1997) and has been implicated in synaptic plasticity in the hippocampus (Levy et al., 2003). In motor nerve terminals, the mitochondrial Na+/Ca2+ exchanger is likely the predominant pathway for Ca2+ release (David, 1999; Yang et al., 2003; Garcia-Chacon et al., 2006), although Na+-independent Ca2+ release from mitochondria has also been reported (Zhong et al., 2001). Our work identifies the mitochondrial Ca2+ uniporter and Na+/Ca2+ exchanger as the principal mediators of mitochondrial Ca2+ cycling in the axonal boutons of DRG neurons. Disruption of Ca2+ uptake by mitochondrial inhibitors eliminated the [Ca2+]i plateau and significantly shortened capsaicin-induced synaptic activity. The arrest of Ca2+ efflux from mitochondria by CGP37157 (Fig. 6 D) and the absence of a cyclosporin A effect (Fig. 6 E) indicate that Na+/Ca2+ exchange is the major route of Ca2+ mobilization from mitochondria at this synapse.
Mitochondria markedly prolonged the presynaptic Ca2+ signal and limited the presynaptic [Ca2+]i peak elevation (Fig. 6, compare A, B). Accordingly, raising the capsaicin concentration from 0.1 to 1 μm resulted in little change to the amplitude of the [Ca2+]i response but led to a significant augmentation of the mitochondrial Ca2+ signal (Fig. 8 A). A >300% increase in the duration of [Ca2+]mt elevation appears somewhat disproportionate to the observed 25% increase in [Ca2+]mt amplitude. However, one must consider the complexity of the relationship between the total and free mitochondrial Ca2+ concentration. In the matrix, Ca2+ is heavily buffered through the reversible formation of insoluble Ca-phosphate salts (Babcock and Hille, 1998; Pivovarova et al., 1999; Nicholls, 2005), which limits [Ca2+]mt elevation to ∼1–2 μm, whereas the total mitochondrial calcium content continues to rise (David, 1999; Chalmers and Nicholls, 2003). Another consequence of calcium precipitation within mitochondria is a marked slowing of [Ca2+]mt recovery attributable to the continuous release of Ca2+ from phosphate precipitates as Ca2+ is extruded from the matrix into the cytosol (David, 1999). Thus, the stimulation intensity and the magnitude of Ca2+ entry strongly affect the duration of mitochondrial Ca2+ elevation and, in the context of the sensory synapse, the duration of both the presynaptic [Ca2+]i response and synaptic activity.
Presynaptic TRPV1, mitochondria, and nociceptive processing
TRPV1 is found in the spinal cord and in other CNS regions (Szallasi et al., 2006). Putative TRPV1 agonists in the CNS include protons, anandamide, NADA, and lipoxygenase products, and the effects of these endogenous compounds depend on the phosphorylation state of the receptor (Tognetto et al., 2001; Huang et al., 2002; Bhave and Gereau, 2004; Premkumar et al., 2004; De Petrocellis and Di Marzo, 2005). We found that, like capsaicin, NADA induced a prolonged presynaptic [Ca2+]i elevation and synaptic activity at sensory synapses (Fig. 3). Although the question of which signals activate TRPV1 in vivo remains a matter of debate, numerous studies imply the importance of spinal cord TRPV1 for pain processing. Indeed, the receptor is significantly upregulated in the dorsal horn during chronic inflammation (Tohda et al., 2001; Luo et al., 2004), and intrathecal administration of either selective TRPV1 antagonists or small interfering RNAs that target TRPV1 produced strong analgesic effects in animal models of inflammatory, neuropathic, and visceral pain (Kelly and Chapman, 2002; Kanai et al., 2005; Christoph et al., 2006). Moreover, significant CNS penetration of orally administered TRPV1 antagonists was required to attenuate pain mediated by central sensitization (Cui et al., 2006).
The identity of the signaling pathways that are invoked by TRPV1 in the spinal cord remains unclear. The fact that the receptor is found presynaptically suggests that it is involved in the regulation of synaptic transmission (Guo et al., 1999; Hwang et al., 2004). Here we establish a mechanistic link between TRPV1 activation, presynaptic Ca2+ signaling, and prolonged synaptic activity at sensory synapses. The proposed role of presynaptic mitochondria (Fig. 9 F) may become especially prominent when TRPV1 is sensitized by prostaglandin E2, bradykinin, and other pain mediators produced in the spinal cord after injury or inflammation (Svensson and Yaksh, 2002; Wang et al., 2005; Sikand and Premkumar, 2007). TRPV1- and mitochondria-mediated release of glutamate may contribute to the temporal and spatial summation of multiple synaptic inputs and, importantly, may trigger action potential firing by SC neurons (Fig. 9 F). TRPV1 activation also induces the release of substance P and CGRP in the dorsal spinal cord (Tognetto et al., 2001; Huang et al., 2002), and both these peptides may contribute to central sensitization (Khasabov et al., 2002; Sun et al., 2004). According to our model, mitochondria would sustain the accumulation of these peptides in the spinal cord.
The proposed mechanisms may also apply to TRPV1-mediated signaling at the periphery. Like the central terminals, peripheral terminals are rich in mitochondria, express TRPV1, and can release glutamate, substance P, and CGRP (Heppelmann et al., 1990; Carlton, 2001; Richardson and Vasko, 2002). It is therefore conceivable that mitochondria prolong the release of these mediators from peripheral terminals to facilitate neurogenic inflammation in response to noxious stimulation or injury. Indeed, intradermal administration of various electron transport inhibitors, including antimycin, have been shown to attenuate hyperalgesia in several pain models (Joseph and Levine, 2006).
Finally, it is likely that the role of mitochondria is not limited to the responses associated with TRPV1. Intensified activity of primary nociceptors triggered by a broad range of pain mediators after tissue or nerve injury would almost inevitably lead to a large accumulation of Ca2+ in the central and peripheral terminals and to the recruitment of mitochondria. Thus, the function of presynaptic mitochondria that is described here may have an impact on multiple aspects of pain processing in the spinal cord and at the periphery.
Footnotes
This work was supported by National Institutes of Health Grant NS054614 and American Heart Association National Scientist Development Grant (Y.M.U.). We thank Donna Hammond, Stanley Thayer, Andriy Yeromin, and Katrin Schnizler for helpful comments on this manuscript, Jane Sullivan for the gift of the synaptophysin–EGFP plasmid, and Atsushi Miyawaki for the gift of the mtPericam plasmid.
References
- Akerman S, Kaube H, Goadsby PJ. Anandamide acts as a vasodilator of dural blood vessels in vivo by activating TRPV1 receptors. Br J Pharmacol. 2004;142:1354–1360. doi: 10.1038/sj.bjp.0705896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angleson JK, Betz WJ. Intraterminal Ca2+ and spontaneous transmitter release at the frog neuromuscular junction. J Neurophysiol. 2001;85:287–294. doi: 10.1152/jn.2001.85.1.287. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Curr Opin Neurobiol. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992;15:303–307. doi: 10.1016/0166-2236(92)90081-i. [DOI] [PubMed] [Google Scholar]
- Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron KT, Thayer SA. CGP37157 modulates mitochondrial Ca2+ homeostasis in cultured rat dorsal root ganglion neurons. Eur J Pharmacol. 1997;340:295–300. doi: 10.1016/s0014-2999(97)01433-7. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P. Calcium—a life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- Bhave G, Gereau RW., IV Posttranslational mechanisms of peripheral sensitization. J Neurobiol. 2004;61:88–106. doi: 10.1002/neu.20083. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocard JB, Tassetto M, Reynolds IJ. Quantitative evaluation of mitochondrial calcium content in rat cortical neurones following a glutamate stimulus. J Physiol (Lond) 2001;531:793–805. doi: 10.1111/j.1469-7793.2001.0793h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM. Peripheral excitatory amino acids. Curr Opin Pharmacol. 2001;1:52–56. doi: 10.1016/s1471-4892(01)00002-9. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem. 2003;278:19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Christoph T, Grunweller A, Mika J, Schafer MK, Wade EJ, Weihe E, Erdmann VA, Frank R, Gillen C, Kurreck J. Silencing of vanilloid receptor TRPV1 by RNAi reduces neuropathic and visceral pain in vivo. Biochem Biophys Res Commun. 2006;350:238–243. doi: 10.1016/j.bbrc.2006.09.037. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD. Dissection of mitochondrial Ca2+ uptake and release fluxes in situ after depolarization-evoked [Ca2+]i elevations in sympathetic neurons. J Gen Physiol. 2000;115:351–369. doi: 10.1085/jgp.115.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Honore P, Zhong C, Gauvin D, Mikusa J, Hernandez G, Chandran P, Gomtsyan A, Brown B, Bayburt EK, Marsh K, Bianchi B, McDonald H, Niforatos W, Neelands TR, Moreland RB, Decker MW, Lee CH, Sullivan JP, Faltynek CR. TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J Neurosci. 2006;26:9385–9393. doi: 10.1523/JNEUROSCI.1246-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G. Mitochondrial clearance of cytosolic Ca2+ in stimulated lizard motor nerve terminals proceeds without progressive elevation of mitochondrial matrix [Ca2+] J Neurosci. 1999;19:7495–7506. doi: 10.1523/JNEUROSCI.19-17-07495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett JN, Barrett EF. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol (Lond) 1998;509:59–65. doi: 10.1111/j.1469-7793.1998.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Di Marzo V. Lipids as regulators of the activity of transient receptor potential type V1 (TRPV1) channels. Life Sci. 2005;77:1651–1666. doi: 10.1016/j.lfs.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Dedov VN, Roufogalis BD. Mitochondrial calcium accumulation following activation of vanilloid (VR1) receptors by capsaicin in dorsal root ganglion neurons. Neuroscience. 2000;95:183–188. doi: 10.1016/s0306-4522(99)00423-6. [DOI] [PubMed] [Google Scholar]
- Filippin L, Magalhaes PJ, Di Benedetto G, Colella M, Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J Biol Chem. 2003;278:39224–39234. doi: 10.1074/jbc.M302301200. [DOI] [PubMed] [Google Scholar]
- Friel DD. Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium. 2000;28:307–316. doi: 10.1054/ceca.2000.0172. [DOI] [PubMed] [Google Scholar]
- Garcia-Chacon LE, Nguyen KT, David G, Barrett EF. Extrusion of Ca2+ from mouse motor terminal mitochondria via a Na+-Ca2+ exchanger increases post-tetanic evoked release. J Physiol (Lond) 2006;574:663–675. doi: 10.1113/jphysiol.2006.110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilardi JR, Rohrich H, Lindsay TH, Sevcik MA, Schwei MJ, Kubota K, Halvorson KG, Poblete J, Chaplan SR, Dubin AE, Carruthers NI, Swanson D, Kuskowski M, Flores CM, Julius D, Mantyh PW. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci. 2005;25:3126–3131. doi: 10.1523/JNEUROSCI.3815-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gover TD, Kao JP, Weinreich D. Calcium signaling in single peripheral sensory nerve terminals. J Neurosci. 2003;23:4793–4797. doi: 10.1523/JNEUROSCI.23-12-04793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gover TD, Moreira TH, Kao JP, Weinreich D. Calcium regulation in individual peripheral sensory nerve terminals of the rat. J Physiol (Lond) 2007;578:481–490. doi: 10.1113/jphysiol.2006.119008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gu JGG, Macdermott AB. Activation of Atp P2x receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, Luis Hannan S, Lappin SC, Egerton J, Smith GD, Worby A, Howett L, Owen D, Nasir S, Davies CH, Thompson M, Wyman PA, Randall AD, Davis JB. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology. 2004;46:133–149. doi: 10.1016/s0028-3908(03)00305-8. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Heinke B, Balzer E, Sandkuhler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci. 2004;19:103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- Heppelmann B, Messlinger K, Neiss WF, Schmidt RF. Ultrastructural three-dimensional reconstruction of group III and group IV sensory nerve endings (“free nerve endings”) in the knee joint capsule of the cat: evidence for multiple receptive sites. J Comp Neurol. 1990;292:103–116. doi: 10.1002/cne.902920107. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. Mitochondria and neurotransmission: evacuating the synapse. Neuron. 2005;47:331–333. doi: 10.1016/j.neuron.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Burette A, Rustioni A, Valtschanoff JG. Vanilloid receptor VR1-positive primary afferents are glutamatergic and contact spinal neurons that co-express neurokinin receptor NK1 and glutamate receptors. J Neurocytol. 2004;33:321–329. doi: 10.1023/B:NEUR.0000044193.31523.a1. [DOI] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Immke DC, Gavva NR. The TRPV1 receptor and nociception. Semin Cell Dev Biol. 2006;17:582–591. doi: 10.1016/j.semcdb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Jin YH, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals in nucleus tractus solitarius. J Neurosci. 2004;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JC, Gasmi M, Lim LE, Elder JH, Yee JK, Jolly DJ, Campbell KP, Davidson BL, Sauter SL. Minimum requirements for efficient transduction of dividing and nondividing cells by feline immunodeficiency virus vectors. J Virol. 1999;73:4991–5000. doi: 10.1128/jvi.73.6.4991-5000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas E. Regulation of synaptic transmission by mitochondrial ion channels. J Bioenerg Biomembr. 2004;36:357–361. doi: 10.1023/B:JOBB.0000041768.11006.90. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Buchanan J, Kaczmarek LK. Prolonged activation of mitochondrial conductances during synaptic transmission. Science. 1999;286:1347–1350. doi: 10.1126/science.286.5443.1347. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Hoit D, Hickman JA, Brandt TA, Polster BM, Fannjiang Y, McCarthy E, Montanez MK, Hardwick JM, Kaczmarek LK. Modulation of synaptic transmission by the BCL-2 family protein BCL-xL. J Neurosci. 2003;23:8423–8431. doi: 10.1523/JNEUROSCI.23-23-08423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RC, III, Xu L, Gebhart GF. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J Neurosci. 2005;25:10981–10989. doi: 10.1523/JNEUROSCI.0703-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain. 2006;121:105–114. doi: 10.1016/j.pain.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Nakazato E, Fujiuchi A, Hara T, Imai A. Involvement of an increased spinal TRPV1 sensitization through its up-regulation in mechanical allodynia of CCI rats. Neuropharmacology. 2005;49:977–984. doi: 10.1016/j.neuropharm.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hara T, Imai A, Sakakibara A. Differential involvement of TRPV1 receptors at the central and peripheral nerves in CFA-induced mechanical and thermal hyperalgesia. J Pharm Pharmacol. 2007;59:733–738. doi: 10.1211/jpp.59.5.0015. [DOI] [PubMed] [Google Scholar]
- Karai LJ, Russell JT, Iadarola MJ, Olah Z. Vanilloid receptor 1 regulates multiple calcium compartments and contributes to Ca2+-induced Ca2+ release in sensory neurons. J Biol Chem. 2004;279:16377–16387. doi: 10.1074/jbc.M310891200. [DOI] [PubMed] [Google Scholar]
- Kelly S, Chapman V. Spinal administration of capsazepine inhibits noxious evoked responses of dorsal horn neurons in non-inflamed and carrageenan inflamed rats. Brain Res. 2002;935:103–108. doi: 10.1016/s0006-8993(02)02552-0. [DOI] [PubMed] [Google Scholar]
- Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neurosci. 2002;22:9086–9098. doi: 10.1523/JNEUROSCI.22-20-09086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrakakis C, MacDermott AB. Neurokinin receptor 1-expressing spinal cord neurons in lamina I and III/IV of postnatal rats receive inputs from capsaicin sensitive fibers. Neurosci Lett. 2003;352:121–124. doi: 10.1016/j.neulet.2003.08.042. [DOI] [PubMed] [Google Scholar]
- Levy M, Faas GC, Saggau P, Craigen WJ, Sweatt JD. Mitochondrial regulation of synaptic plasticity in the hippocampus. J Biol Chem. 2003;278:17727–17734. doi: 10.1074/jbc.M212878200. [DOI] [PubMed] [Google Scholar]
- Liu M, Liu MC, Magoulas C, Priestley JV, Willmott NJ. Versatile regulation of cytosolic Ca2+ by vanilloid receptor I in rat dorsal root ganglion neurons. J Biol Chem. 2003;278:5462–5472. doi: 10.1074/jbc.M209111200. [DOI] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol (Lond) 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Cheng J, Han JS, Wan Y. Change of vanilloid receptor 1 expression in dorsal root ganglion and spinal dorsal horn during inflammatory nociception induced by complete Freund's adjuvant in rats. NeuroReport. 2004;15:655–658. doi: 10.1097/00001756-200403220-00016. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Christie MJ, Connor M. Capsaicin activation of glutamatergic synaptic transmission in the rat locus coeruleus in vitro. J Physiol (Lond) 2002;543:531–540. doi: 10.1113/jphysiol.2002.022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Berretta N, Matias I, Maccarrone M, Bernardi G, Mercuri NB. Presynaptic facilitation of glutamatergic synapses to dopaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J Neurosci. 2003;23:3136–3144. doi: 10.1523/JNEUROSCI.23-08-03136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell D, Rethelyi M. Ultrastructure and synaptic connections of cutaneous afferent fibers in the spinal cord. Trends Neurosci. 1987;10:117–123. [Google Scholar]
- Medvedeva YV, Veselovsky N, Fedulova SA, Kostyuk PG. Postsynaptic currents in dorsal root ganglion neurons co-cultured with spinal cord neurons. Neurophysiology. 2001;33:1–4. [Google Scholar]
- Millns PJ, Chimenti M, Ali N, Ryland E, de Lago E, Fernandez-Ruiz J, Chapman V, Kendall DA. Effects of inhibition of fatty acid amide hydrolase vs. the anandamide membrane transporter on TRPV1-mediated calcium responses in adult DRG neurons; the role of CB receptors. Eur J Neurosci. 2006;24:3489–3495. doi: 10.1111/j.1460-9568.2006.05236.x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ . Proc Natl Acad Sci USA. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuka T, Furue H, Yoshimura M, Gu JG. Activation of central terminal vanilloid receptor-1 receptors and αβ-methylene-ATP-sensitive P2X receptors reveals a converged synaptic activity onto the deep dorsal horn neurons of the spinal cord. J Neurosci. 2002;22:1228–1237. doi: 10.1523/JNEUROSCI.22-04-01228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Marin L, Atwood HL. Synaptic physiology and mitochondrial function in crayfish tonic and phasic motor neurons. J Neurophysiol. 1997;78:281–294. doi: 10.1152/jn.1997.78.1.281. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar LS, Qi ZH, Van Buren J, Raisinghani M. Enhancement of potency and efficacy of NADA by PKC-mediated phosphorylation of vanilloid receptor. J Neurophysiol. 2004;91:1442–1449. doi: 10.1152/jn.00745.2003. [DOI] [PubMed] [Google Scholar]
- Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM. Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyl-dopamine and arachidonyl-2-chloroethylamide. Br J Pharmacol. 2004;141:1118–1130. doi: 10.1038/sj.bjp.0705711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–845. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- Semba K, Masarachia P, Malamed S, Jacquin M, Harris S, Yang G, Egger MD. An electron microscopic study of terminals of rapidly adapting mechanoreceptive afferent fibers in the cat spinal cord. J Comp Neurol. 1985;232:229–240. doi: 10.1002/cne.902320208. [DOI] [PubMed] [Google Scholar]
- Sikand P, Premkumar LS. Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol (Lond) 2007;581:631–647. doi: 10.1113/jphysiol.2006.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starowicz K, Nigam S, Di Marzo V. Biochemistry and pharmacology of endovanilloids. Pharmacol Ther. 2007;114:13–33. doi: 10.1016/j.pharmthera.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Stucky CL, Thayer SA, Seybold VS. Prostaglandin E2 increases the proportion of neonatal rat dorsal root ganglion neurons that respond to bradykinin. Neuroscience. 1996;74:1111–1123. doi: 10.1016/0306-4522(96)00264-3. [DOI] [PubMed] [Google Scholar]
- Sun RQ, Tu YJ, Lawand NB, Yan JY, Lin Q, Willis WD. Calcitonin gene-related peptide receptor activation produces PKA- and PKC-dependent mechanical hyperalgesia and central sensitization. J Neurophysiol. 2004;92:2859–2866. doi: 10.1152/jn.00339.2004. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Yaksh TL. The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Annu Rev Pharmacol Toxicol. 2002;42:553–583. doi: 10.1146/annurev.pharmtox.42.092401.143905. [DOI] [PubMed] [Google Scholar]
- Szabo A, Helyes Z, Sandor K, Bite A, Pinter E, Nemeth J, Banvolgyi A, Bolcskei K, Elekes K, Szolcsanyi J. Role of transient receptor potential vanilloid 1 receptors in adjuvant-induced chronic arthritis: in vivo study using gene-deficient mice. J Pharmacol Exp Ther. 2005;314:111–119. doi: 10.1124/jpet.104.082487. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cruz F, Geppetti P. TRPV1: a therapeutic target for novel analgesic drugs? Trends Mol Med. 2006;12:545–554. doi: 10.1016/j.molmed.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6:357–372. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- Tang YG, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Thayer SA, Usachev YM, Pottorf WJ. Modulating Ca2+ clearance from neurons. Front Biosci. 2002;7:D1255–D1279. doi: 10.2741/A838. [DOI] [PubMed] [Google Scholar]
- Thomas D, Hanley MR. Pharmacological tools for perturbing intracellular calcium storage. Methods Cell Biol. 1994;40:65–89. doi: 10.1016/s0091-679x(08)61110-3. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. Neuronal ageing from an intraneuronal perspective: roles of endoplasmic reticulum and mitochondria. Cell Calcium. 2003;34:311–323. doi: 10.1016/s0143-4160(03)00142-8. [DOI] [PubMed] [Google Scholar]
- Tognetto M, Amadesi S, Harrison S, Creminon C, Trevisani M, Carreras M, Matera M, Geppetti P, Bianchi A. Anandamide excites central terminals of dorsal root ganglion neurons via vanilloid receptor-1 activation. J Neurosci. 2001;21:1104–1109. doi: 10.1523/JNEUROSCI.21-04-01104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohda C, Sasaki M, Konemura T, Sasamura T, Itoh M, Kuraishi Y. Axonal transport of VR1 capsaicin receptor mRNA in primary afferents and its participation in inflammation-induced increase in capsaicin sensitivity. J Neurochem. 2001;76:1628–1635. doi: 10.1046/j.1471-4159.2001.00193.x. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Sanmarti-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kampf U, Franzer JT, Stumm M, Garner CC, Gundelfinger ED. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol. 1998;142:499–509. doi: 10.1083/jcb.142.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki K, Xing H, Ling J, Gu JG. Menthol-induced Ca2+ release from presynaptic Ca2+ stores potentiates sensory synaptic transmission. J Neurosci. 2004;24:762–771. doi: 10.1523/JNEUROSCI.4658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M, Kuraishi Y, Satoh M. Detection of capsaicin-evoked release of glutamate from spinal dorsal horn slices of rat with on-line monitoring system. Neurosci Lett. 1993;155:179–182. doi: 10.1016/0304-3940(93)90702-m. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol (Lond) 1999;519:115–130. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachev YM, DeMarco SJ, Campbell C, Strehler EE, Thayer SA. Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron. 2002;33:113–122. doi: 10.1016/s0896-6273(01)00557-8. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- Wang H, Kohno T, Amaya F, Brenner GJ, Ito N, Allchorne A, Ji RR, Woolf CJ. Bradykinin produces pain hypersensitivity by potentiating spinal cord glutamatergic synaptic transmission. J Neurosci. 2005;25:7986–7992. doi: 10.1523/JNEUROSCI.2393-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer VC, Horstmann H, Groh A, Kuner T. Donut-like topology of synaptic vesicles with a central cluster of mitochondria wrapped into membrane protrusions: a novel structure-function module of the adult calyx of Held. J Neurosci. 2006;26:109–116. doi: 10.1523/JNEUROSCI.3268-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Transient receptor potential vanilloid type 1 activation down-regulates voltage-gated calcium channels through calcium-dependent calcineurin in sensory neurons. J Biol Chem. 2005;280:18142–18151. doi: 10.1074/jbc.M501229200. [DOI] [PubMed] [Google Scholar]
- Yang F, He XP, Russell J, Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na+-Ca2+ exchanger and protein kinase C. J Cell Biol. 2003;163:511–523. doi: 10.1083/jcb.200307027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Yoshimura M. Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neurosci Lett. 1998;255:135–138. doi: 10.1016/s0304-3940(98)00730-7. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Matthews G. The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron. 2000;25:229–237. doi: 10.1016/s0896-6273(00)80885-5. [DOI] [PubMed] [Google Scholar]
- Zhong N, Beaumont V, Zucker RS. Roles for mitochondrial and reverse mode Na+/Ca2+ exchange and the plasmalemma Ca2+ ATPase in post-tetanic potentiation at crayfish neuromuscular junctions. J Neurosci. 2001;21:9598–9607. doi: 10.1523/JNEUROSCI.21-24-09598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]