

Abstract
In proteomic research, it is often necessary to screen a large number of polypeptides for the presence of stable structure. Described here is a technique (referred to as SUPREX, stability of unpurified proteins from rates of H/D exchange) for measuring the stability of proteins in a rapid, high-throughput fashion. The method uses hydrogen exchange to estimate the stability of microgram quantities of unpurified protein extracts by using matrix-assisted laser desorption/ionization MS. The stabilities of maltose binding protein and monomeric λ repressor variants determined by SUPREX agree well with stability data obtained from conventional CD denaturation of purified protein. The method also can detect the change in stability caused by the binding of maltose to maltose binding protein. The results demonstrate the precision of the method over a wide range of stabilities.
The function of a protein is contingent on the stability of its native conformation. Consequently, in the field of protein biochemistry, stability measurements frequently are performed to establish a polypeptide as a stably folded protein and to study the physical forces that lead to its folding (1). Stability measurements also provide important biological information; a decrease in stability can be a sign of misfolding, which in some proteins leads to disease (2), whereas an increase in stability can be indicative of ligand binding (3). Despite their utility, stability measurements currently necessitate time-consuming experiments with pure protein samples. In proteomic experiments (4), where a large number of polypeptides often need to be analyzed, stability measurements are not practical. We have developed the SUPREX (stability of unpurified proteins from rates of H/D exchange) technique to rapidly screen a large number of protein samples for the presence of stable structure. The method uses hydrogen exchange coupled with matrix-assisted laser desorption/ionization (MALDI) MS to obtain quantitative measurements of stability from crude extracts of recombinant Escherichia coli cultures grown in 96-well microtiter plates.
Within a polypeptide, certain labile hydrogen atoms can exchange freely with the surrounding solvent. Native structure protects a subset of these hydrogens from exchange (5), and some of these protected protons exchange only if the protein globally unfolds (6). The stability of a protein can be analyzed by monitoring the exchange rates of these “globally protected” hydrogens (7). Protein hydrogen exchange rates typically are measured by allowing labile protons to exchange with D2O. The proton/deuteron exchange reaction can be monitored by NMR (6) or MS (8). MS is experimentally more convenient than NMR for several reasons: it requires less protein, is faster and simpler to use, does not require complicated spectral assignments, and does not require pure protein samples. These advantages come at the expense of the residue-specific information NMR affords, but that information is not necessary for assessing global stability.
Recent studies have demonstrated that hydrogen exchange coupled with electrospray ionization MS can qualitatively distinguish native-like proteins from unfolded polypeptides in partially purified samples (9) and can be used to study the kinetics and thermodynamics of folding (8, 10). In contrast, the experiments described here use MALDI MS to detect hydrogen exchange, an approach previously described by Komives and coworkers (11). MALDI is ideally suited for fast, high-throughput screening because a large number of samples can be analyzed in a short period. More importantly, the MALDI technique is tolerant of impure samples that contain moderate levels of salts and other small molecule contaminants (12). This feature allows the measurement of hydrogen exchange as a function of denaturant concentration to give a quantitative measurement of the global protein stability. To demonstrate the accuracy and utility of the method, the analysis was performed on maltose binding protein (MBP) and eight variants of monomeric λ repressor (λ6–85) expressed in bacterial cultures grown in microtiter plates. The change in stability resulting from the mutations in λ6–85 and maltose binding to MBP were measured. The stability measurements are verified by comparison with CD denaturation curves obtained with purified proteins.
Materials and Methods
Sample Preparation.
The gene encoding the protein of interest was cloned into a T7 expression vector and expressed in E. coli strain BL21-(DE3). LB cultures (200 μl) of the recombinant E. coli were grown in 96-well plates and induced by the addition of 0.4 mM isopropylthio-β-d-galactosidase. The cells subsequently were pelleted and lysed by suspension in 10 μl Bug Buster solution (Novagen). The lysates were centrifuged, and the supernatant was used for hydrogen exchange experiments without any further manipulation. SDS/PAGE indicated that the crude samples consisted of ≈10–50% expressed protein in a background of cellular impurities. From the gel, the concentrations of the proteins were estimated to range between 50 and 500 μM.
Hydrogen Exchange.
Hydrogen exchange was initiated by adding 10-fold excess deuterated exchange buffer (20 mM sodium phosphate/20 mM sodium acetate/100 mM NaCl, pH 6.3 for MBP or pH 6.7 for λ6–85) to the lysed cultures. The exchange buffers contained different concentrations of guanidinium chloride (GdmCl). At a given exchange time, 0.5 μl of the exchange reaction was added to 50 μl of matrix solution. The matrix used for these experiments was sinapinic acid. It was prepared as a saturated aqueous solution containing 45% acetonitrile/0.1% trifluoroacetic acid (pH 3.0) and was kept on ice (2°C) before the addition of the protein. The choice of sinapinic acid as the MALDI matrix was critical because it was more tolerant than other MALDI matrices to the GdmCl used in our experiments. To obtain highly accurate mass measurements, a reference protein was added to the matrix solution. The reference proteins used were BSA for the MBP experiments and a 10-kDa biosynthetic non-natural polypeptide for the λ6–85 experiment. Once the sample protein was added to the matrix, 2 μl of the solution was immediately placed on a MALDI target and rapidly dried under an air stream. Typically, the samples were placed in the matrix and dried within 40 sec. For a given data point five MALDI spectra were analyzed and the results were averaged. No more than 10 min passed between matrix formation and data collection. It is estimated that the amount of protein in each spot was approximately 1 pmol, based on the estimated protein concentration in cell lysates.
Measurement of Back Exchange in the Protonated Matrix.
Control experiments were performed to measure the rate of exchange of protons from the matrix back into deuterated protein sites, both in the matrix solution and in the solid matrix crystals. To examine the exchange in the matrix solution, fully deuterated MBP was placed in a prechilled (2°C) matrix solution. At given time intervals the sample was dried on a MALDI target and processed as described below. To examine the exchange in the solid matrix, fully deuterated MBP was placed in a prechilled (2°C) matrix solution and immediately dried on a MALDI target at room temperature. At given time intervals mass spectra were collected and processed as described below.
Data Collection.
Mass spectra were collected on a Voyager Biospectrometry Workstation from PerSeptive Biosystems (Framingham, MA) by using the autosampler mode. All spectra were obtained in the positive ion mode and summed over 32 laser shots.
Analysis of SUPREX Data to Determine Protein Stability.
According to the classical hydrogen exchange model (5):
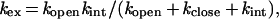 |
1 |
where kex is the observed exchange rate constant for each hydrogen, kopen and kclose are the rate constants for the conformational changes leading to exchange competent and exchange incompetent states, respectively, and kint is the exchange rate for the unprotected hydrogen. Under EX2 conditions where kclose (or kopen) are much greater than kint:
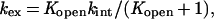 |
2 |
where Kopen is the equilibrium constant between the exchange competent and exchange incompetent conformations of the protein (kopen/kclose). For the hydrogens that are exchanging through a global unfolding mechanism (see Results and Discussion),
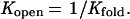 |
3 |
Substituting Eq. 3 into Eq. 2:
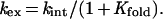 |
4 |
Because kint is similar among the majority of the backbone amide hydrogens (13), the total exchange of the hydrogens that exchange through global unfolding can be estimated by a single rate constant. The increase in mass due to the exchange of labile protons (ΔMass) as a function of time can thus be estimated by the following first-order rate equation:
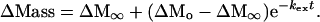 |
5 |
ΔMo is ΔMass before global exchange, ΔM∞ is ΔMass after complete exchange, t is the exchange time. Substitution of Eq. 4 into Eq. 5 gives an equation for ΔMass vs. [GdmCl]:
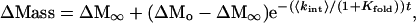 |
6 |
where (14):
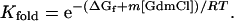 |
7 |
ΔGf is the free energy of folding in the absence of GdmCl, [GdmCl] is the GdmCl concentration, m is δΔGf/δ[GdmCl], R is the gas constant, and T is the temperature in kelvin. The m value determines the sharpness of the transition in the ΔMass vs. [GdmCl] plot. Myers et al. (15) have shown that m values can be estimated from the size of the protein. The average GdmCl m value per residue for the 34 proteins in table 1 of ref. 15 is 26 (σ = 7.2) cal⋅mol−1⋅M−1 per residue. According to this analysis, MBP and λ6–85 are predicted to have m values of 9.7 ± 2.6 and 2.1 ± 0.6 kcal⋅mol−1⋅M−1. These measurements are close to the previously reported experimental m values (12 ± 1 and 2.1 ± 0.1 kcal⋅mol−1⋅M−1, respectively) measured from CD denaturation curves (16, 17). To assess the error involved in using estimated m values, as would be the case in a high-throughput screen of proteins, the calculated m values were used in this analysis. In Eq. 6, <kint> is the average exchange rate of unprotected amide hydrogens and is a function of pH and temperature. In principle, <kint> can be estimated by averaging the values for all of the backbone amide hydrogens using the measurements of Englander and coworkers (13). However, for these studies, the simple relationship <kint> = 10pH-5 min-1 was used to estimate the rate at room temperature and pH > 4.
Results and Discussion
In the SUPREX method, 200 μl recombinant E. coli cultures overexpressing the protein of interest are pelleted in a 96-well plate. The cells are lysed by the addition of a nonionic detergent, and hydrogen exchange is initiated by the addition of deuterated exchange buffer containing GdmCl at concentrations ranging from 0 to 8 M. After exchange occurs for a fixed period the protein extract is diluted into a MALDI matrix solution that has been prechilled to 2°C. This addition unfolds the protein and causes a fixed number of deuterons to re-exchange immediately with protons. Control experiments show that the remaining deuterons are relatively stable and re-exchange at a slower rate of 0.001 s−1 (Fig. 1A). Once the matrix forms a solid crystal, the exchange slows to a rate of 0.001 min−1 (Fig. 1B). By rapidly drying the sample on a MALDI target, the slowly exchanging deuterons are trapped in the time frame of the experiment and their number can be determined from a mass spectrum (Fig. 2A). To obtain highly accurate mass measurements, a reference protein is added to the sample/matrix solution. The mass of the sample protein is determined by using the reference protein as an internal standard.
Figure 1.

Exchange of a fully deuterated MBP with the MALDI matrix as a function of time. ΔMass is the increase in mass relative to the protonated sample. The arrow is the ΔMass of the fully deuterated protein. (A) The exchange in the matrix solution at 2°C. The bar represents the range of time required for the sample to form a solid matrix and trap the deuterons. The plot is fitted to a single exponential equation with a rate of 0.001 s−1. (B) The exchange in the crystallized solid matrix at room temperature. The plot is fitted to a single exponential equation with a rate of 0.001 min−1.
Figure 2.

Hydrogen exchange of MBP detected by MALDI. (A) The MALDI-TOF mass spectrum of a nonexchanged (all protonated) sample. Peak b is the signal from the singly charged molecular ion of MBP, peaks c and a are the signals from the singly and doubly charged molecular ions of BSA, which is added as an internal mass reference. (B) The change in mass (ΔMass) of MBP as a function of exchange time in the presence of different GdmCl concentrations: ●, 0 M; ▴, 1 M; ■, 2 M; and ▿, 6 M.
As deuterium atoms replace protons during the hydrogen exchange period, the mass of the protein increases. The extent of exchange is determined by monitoring the change in mass relative to a fully protonated sample (ΔMass). The observed ΔMass is caused by deuterated hydrogens that do not re-exchange in the protonated matrix. This set is restricted to the backbone amide hydrogens because they have the slowest intrinsic exchange rates among the exchangeable hydrogens.
Under native conditions, most of these observable amide hydrogens do not provide global stability information because they exchange by local (partial) unfolding processes. However, the addition of denaturant enhances the relative contribution of global unfolding to the exchange rates because local unfolding mechanisms are less denaturant dependent (18, 19). For this reason, many amide hydrogens exchange through a global unfolding mechanism at moderate denaturant concentrations below that required to significantly denature the protein. Fig. 2B shows the exchange of MBP as a function of time in the presence of different GdmCl concentrations. The amplitude of the 6 M GdmCl curve shown in Fig. 2B represents the total number of exchangeable hydrogens observable by SUPREX for MBP. Of these ≈230 hydrogens, ≈100 exchange within 1 h in the absence of GdmCl. These ≈100 protons are not globally protected under these conditions and exchange with deuterons relatively quickly. The remaining mass increase is caused by backbone amide hydrogens that are more protected and exchange at a slower rate. The addition of GdmCl dramatically increases the exchange rate of these hydrogens through global unfolding (18, 19). Fig. 3A shows the mass spectra and Fig. 3B depicts the ΔMass of MBP after 60 min of exchange as a function of GdmCl concentration in the presence and absence of 100 μM maltose. Both curves in Fig. 3B indicate the cooperative unfolding of the protein induced by the addition of GdmCl. The presence of maltose increases the stability by binding to the native state and the protein unfolds at higher concentrations of GdmCl. These data were fit to Eq. 6 to determine the stability under native conditions (Table 1). This analysis involves the extrapolation of the linear free energy versus denaturant concentration curves to 0 M. The presence of 100 μM maltose stabilizes the protein by 3.0 kcal/mol, a value that is consistent with the stabilization expected from the previously published binding constant (20).
Figure 3.

Stability of MBP obtained by SUPREX. (A) MALDI-TOF mass spectra after 60 min of exchange in the presence of different [GdmCl] with no maltose present. The spectra have been calibrated by using an internal reference (BSA). The arrow indicates the population averaged mass of the singly charged state of MBP. The small peak at m/z MBP + 224 is due to a sinapinic acid matrix adduct of MBP. (B) ΔMass as a function of [GdmCl]. □, No maltose present; ●, in the presence of 100 μM maltose. The lines represent fits to Eq. 6. Hydrogen exchange was conducted at room temperature at pH 6.3.
Table 1.
Stability of proteins by SUPREX and CD denaturation
| Protein | ΔGf, kcal/mol
|
ΔΔGf, kcal/mol
|
||
|---|---|---|---|---|
| SUPREXa | CD | SUPREXa | CD | |
| λ6–85 | −5.0 ± 0.4 | −4.4 ± 0.2b | — | — |
| λ6–85 (A66G) | >−4.0 | >1.0 | 1.5c | |
| λ6–85 (A63G) | −4.8 ± 0.4 | 0.2 | 0.4c | |
| λ6–85 (G46A/G48A/A66G) | −5.2 ± 0.4 | −0.2 | −0.4c | |
| λ6–85 (G46A/G48A/A49G) | −5.6 ± 0.4 | −0.6 | −1.0c | |
| λ6–85 (Q33Y) | −6.5 ± 0.4 | −1.5 | −1.5d | |
| λ6–85 (G46A/G48A) | −6.7 ± 0.4 | −6.1 ± 0.2b | −1.7 | −1.7 |
| λ6–85 (G46A/G48A/Q33Y) | −7.9 ± 0.7 | −2.9 | −3.4d | |
| MBP | −16 ± 3 | −14.5 ± 0.4e | ||
| −12.5 ± 0.2f | ||||
| MBP + 100 μM maltose | −19 ± 3 | — | −3.0 | −2.9g |
To demonstrate that the method is capable of accurately measuring stability perturbations caused by mutations, SUPREX assays were performed on a series of λ6–85 variants known to have different stabilities (21–23). The exchange was performed at room temperature and pH 6.7 for 60 min. Fig. 4 depicts the stability curves for this set of proteins. Stabilizing mutations shift the titration curves to higher GdmCl concentrations. Table 1 compares the calculated ΔGf values obtained by the SUPREX analysis of the crude samples of wild-type λ6–85, the G46A/G48 variant, and MBP with published values obtained by conventional CD denaturation curves of the purified proteins under similar conditions. Table 1 also lists the calculated change in ΔGf values relative to wild-type proteins (ΔΔGf) for a series of λ6–85 variants whose stabilities were determined at either 25°C or 37°C by conventional methods (23). Protein-dependent variations in <kint> values (24) and the uncertainty involved in estimating m values introduces systematic errors in the SUPREX ΔGf estimates. However, because variants of a protein have nearly identical <kint> and m values, this error does not affect the ΔΔGf measurements. Thus, even in the absence of exact <kint> and m values, SUPREX can accurately determine the change in ΔGf. The results in Table 1 show a good correlation between the stability changes measured by CD denaturation and SUPREX.
Figure 4.

Stability plots for eight mutants of λ6–85 obtained by SUPREX. The curves are arranged in order of the stability of the mutant, from the least to the most stable: (A) A66G, (B) A63G, (C) wild type, (D) G46A/G48A/A66G, (E) G46A/G48A/A49G, (F) Q33Y, (G) G46A/G48A, and (H) Q33Y/G46A/G48A. The lines represent fits to Eq. 6. Under these conditions (60 min of exchange, pH 6.7, room temperature), the midpoints of the SUPREX curves are shifted by 2.4 M [GdmCl] to the left relative to their corresponding CD denaturation curves (see Eq. 8).
It should be noted that the stability curves obtained from SUPREX analysis (Figs. 3B and 4) are not identical to conventional denaturation curves. The midpoint of transition in conventional denaturation curves is a function of the stability and the m value of the protein (see Materials and Methods for definition of m). In the SUPREX curves the midpoint is not only a function of these parameters but also depends on the time of exchange (t) and <kint>.
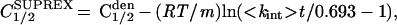 |
8 |
where C1/2SUPREX and C1/2den are the midpoints of transition in the SUPREX analysis and conventional denaturation curves, respectively and <kint> is itself a function of pH and temperature (13). As Eq. 8 demonstrates, a SUPREX curve is shifted to the left relative to a conventional denaturation curve, because <kint>t is always much greater than 0.693 (ln2). Under the conditions used for the experiments described here, this effect shifts the SUPREX curves of MBP and λ6–85 by 0.5 and 2.4 M, respectively. It is important to note that the C1/2SUPREX can purposely be altered by changing the pH, temperature, or exchange interval, t. High stabilities can be measured at higher pH and/or temperature, low stabilities at lower pH and/or temperature. Additionally, the exchange interval can conveniently range from a few minutes to hours. For example, according to Eq. 8, the midpoint of the SUPREX curve for λ6–85 can be shifted to the right by 1 M relative to the curves shown in Fig. 4 by changing the pH to 5.9 and the exchange time to 11 min. This sort of adjustment makes SUPREX very flexible for measurements over a wide range of stabilities.
As with all denaturation experiments, the major caveat of the quantitative analysis of SUPREX data is that the protein must unfold in a cooperative, two-state process (1). If one or more stable intermediates are present, the data will not fit Eq. 6. In such cases, a multiphasic titration curve is observed or the transition is broadened. Under these conditions, an extrapolated stability measurement is impossible. However, the midpoint of the curve still serves as a good qualitative gauge of stability. Multiphasic or broadened SUPREX curves would give poor fits with predicted m values; thus this method potentially can be used to identify proteins that have stable folding intermediates.
Another possible complication in the quantitative analysis of SUPREX data is the tendency of some proteins to enter the EX1 regime at high denaturant concentrations and/or pH (24–26). Under such conditions, the intrinsic exchange rate is greater than the folding rate and the exchange rate becomes a function of the unfolding rate rather the folding equilibrium constant. This situation will cause distortions in the SUPREX curves and it would not be possible to properly analyze the data without knowledge of the denaturant dependence of the unfolding rate. In practice, as is the case with MBP and λ6–85, SUPREX measurements most frequently will be obtained at lower denaturant concentrations and pH where EX1 conditions usually do not prevail.
Although the data described here were obtained by manual methods, the entire methodology of SUPREX can be easily automated and implemented for the analysis of a very large number of samples. For the above experiments, the E. coli cultures were grown and induced on 96-well plates. The exchange reaction and dilution into the matrix also were conducted in microtiter plates. The spotting onto a MALDI target from a microtiter well also can be done robotically, and commercially available MALDI instruments can record the mass spectra of all of the samples in quick succession. The analysis of the data also can be easily automated. We estimate that a single person, with access to a MALDI instrument, could use this technology to measure the stability of as many as 1,000 proteins per day. However, there are likely restrictions on the type of proteins amenable to this analysis. MBP and λ6–85 are both fairly soluble proteins with high expression levels in E. coli (≈50 mg protein per liter of culture). Poorly expressed proteins will be more difficult to visualize on a mass spectrum and might require an initial concentration procedure. Proteins that form aggregates in the lysate also will produce misleading results. Aggregation is likely to protect some hydrogens that are free to exchange in the monomeric soluble form, thus creating artificially high observed stability. Complications also will arise if the recombinant protein requires some manipulation such as reduction or renaturation for its folding. However, many of these complications have been minimized in the past by the addition of moderate concentrations of denaturant sufficient to disaggregate or encourage proper disulfide bond formation but not high enough to unfold the protein. Fortunately, these are just the sort of conditions under which SUPREX measurements are made. For this reason, we are optimistic that SUPREX may be useful even for proteins that are difficult to study by conventional methods.
We foresee several immediate applications of the SUPREX method. Recent genomic sequencing efforts have provided the DNA sequences of thousands of previously unknown genes (TIGR Microbial Genome Database, www.tigr.org). Assigning a function to all of these unknown sequences will be one of the major scientific tasks of the coming decades. Whether this process involves biochemical analysis or structure determination, the magnitude of the task is so great that it will be important to intelligently select out the most promising candidates for study. Many cellular proteins are not stable under typical in vitro conditions. Because both structural and functional analyses require stability, SUPREX can act as a fast screen for selecting promising stable proteins for further study. Similarly, large-scale screening of cloned and expressed polymorphic genes could efficiently aid in the identification of alleles that code for unstable proteins that cause a diseased phenotype.
Combinatorial and directed-evolution methods have proven to be promising techniques for designing proteins of novel structure and function (27, 28). These methods systematically generate a large number of sequences and it is imperative to be able to detect successful de novo designs from a large background of unfolded polypeptides. The ability to rapidly screen a large number of sequences for stable folding is crucial to the success of these methods. With its ease of use and high-throughput capability, SUPREX can perform this function and provide a fast convenient way to select for stable designs.
Finally, the results with MBP show that SUPREX can detect binding through a change in protein stability. The thermodynamic linkage between stability and binding is well established (3, 29) and recently has been implemented as a method to detect ligand binding (30). A large number of potential ligands can be rapidly screened for this effect. These ligands could be small molecules, proteins, or nucleic acids. SUPREX provides a convenient alternative in experiments where a binding assay is not available or difficult to use. This approach could be applied to a method analogous to the yeast two-hybrid screen (31), in which two proteins of interest (e.g., a target and a library) are coexpressed and the target protein is screened for stability by SUPREX. The method also could be modified to allow screening of large ligand libraries directly on MALDI plates with predeposited protein. Additional studies should help determine the applicability of the method to expression cell types other than bacteria to allow binding studies under nearly in vivo conditions.
SUPREX provides a simple, rapid, and economical way to measure the stability of a large number of cloned proteins and is useful in any application where stability provides important functional and thermodynamic information about a series of proteins. The method should prove to be a very important component in the arsenal of modern proteomic technology.
Acknowledgments
We thank Jennifer Hunt and Novartis, Inc. for pointing out the need for a high-throughput protein stability screen and Jonathon Marvin and Homme Hellinga for providing the MBP plasmid. This work was supported by National Institutes of Health Grant GM45322 (T.G.O.) and Duke University (M.C.F.).
Abbreviations
- MALDI
matrix-assisted laser desorption/ionization
- SUPREX
stability of unpurified proteins from rates of H/D exchange
- MBP
maltose binding protein
- λ6–85
monomeric λ repressor
- GdmCl
guanidinium monochloride
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.140111397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.140111397
References
- 1.Schellman J. Annu Rev Biophys Chem. 1987;16:115–137. doi: 10.1146/annurev.bb.16.060187.000555. [DOI] [PubMed] [Google Scholar]
- 2.Dobson C M. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 3.Schellman J. Biopolymers. 1975;14:999–1018. doi: 10.1002/bip.1975.360140113. [DOI] [PubMed] [Google Scholar]
- 4.Blackstock W P, Weir M P. Trends Biotechnol. 1999;17:121–127. doi: 10.1016/s0167-7799(98)01245-1. [DOI] [PubMed] [Google Scholar]
- 5.Hvidt A, Nielson S O. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 6.Englander S W, Sosnick T R, Englander J J, Mayne L. Curr Opin Struct Biol. 1996;6:18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huyghues-Despointes B M, Scholtz J M, Pace C N. Nat Struct Biol. 1999;6:910–912. doi: 10.1038/13273. [DOI] [PubMed] [Google Scholar]
- 8.Miranker A, Robinson C V, Radford S E, Dobson C M. FASEB J. 1996;10:93–101. doi: 10.1096/fasebj.10.1.8566553. [DOI] [PubMed] [Google Scholar]
- 9.Rosenbaum D M, Roy S, Hecht M H. J Am Chem Soc. 1999;121:9509–9513. [Google Scholar]
- 10.Deng Y, Smith D L. Anal Biochem. 1999;276:150–160. doi: 10.1006/abio.1999.4347. [DOI] [PubMed] [Google Scholar]
- 11.Mandell J G, Falick A M, Komives E A. Anal Chem. 1998;70:3987–3995. doi: 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- 12.Beavis R C, Chait B T. Methods Enzymol. 1996;270:519–551. doi: 10.1016/s0076-6879(96)70024-1. [DOI] [PubMed] [Google Scholar]
- 13.Bai Y, Milne J S, Englander S W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pace C N. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 15.Myers J K, Pace C N, Scholtz J M. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghaemmaghami S, Word J M, Burton R E, Richardson J S, Oas T G. Biochemistry. 1998;37:9179–9185. doi: 10.1021/bi980356b. [DOI] [PubMed] [Google Scholar]
- 17.Sheshadri S, Lingaraju G M, Varadarajan R. Protein Sci. 1999;8:1689–1695. doi: 10.1110/ps.8.8.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai Y W, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayo S L, Baldwin R L. Science. 1993;262:873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 20.Martineau P, Szmelcman S, Spurlino J C, Quiocho F A, Hofnung M. J Mol Biol. 1990;214:337–352. doi: 10.1016/0022-2836(90)90165-I. [DOI] [PubMed] [Google Scholar]
- 21.Huang G S, Oas T G. Proc Natl Acad Sci USA. 1995;92:6878–6882. doi: 10.1073/pnas.92.15.6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burton R E, Huang G S, Daugherty M A, Fullbright P W, Oas T G. J Mol Biol. 1996;263:311–322. doi: 10.1006/jmbi.1996.0577. [DOI] [PubMed] [Google Scholar]
- 23.Burton R E, Huang G S, Daugherty M A, Calderone T L, Oas T G. Nat Struct Biol. 1997;4:305–310. doi: 10.1038/nsb0497-305. [DOI] [PubMed] [Google Scholar]
- 24.Clarke J, Fersht A R. Fold Des. 1996;1:243–254. doi: 10.1016/S1359-0278(96)00038-7. [DOI] [PubMed] [Google Scholar]
- 25.Loh S N, Rohl C A, Kiefhaber T, Baldwin R L. Proc Natl Acad Sci USA. 1996;93:1982–1987. doi: 10.1073/pnas.93.5.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi Q, Scalley M L, Simons K T, Gladwin S T, Baker D. Fold Des. 1997;2:271–280. doi: 10.1016/S1359-0278(97)00038-2. [DOI] [PubMed] [Google Scholar]
- 27.Moore J C, Jin H M, Kuchner O, Arnold F H. J Mol Biol. 1997;272:336–347. doi: 10.1006/jmbi.1997.1252. [DOI] [PubMed] [Google Scholar]
- 28.Kamtekar S, Schiffer J M, Xiong H, Babik J M, Hecht M H. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 29.Pace C N, McGrath T. J Biol Chem. 1980;255:3862–3865. [PubMed] [Google Scholar]
- 30.Bowie J U, Pakula A. U.S. Patent. Vol. 5. 1997. p. 679,582. [Google Scholar]
- 31.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]