

Abstract
Hermansky-Pudlak syndrome (HPS) develops from defects in the biogenesis and/or function of lysosome-related organelles essential to membrane and protein trafficking. Of the eight known human subtypes, only HPS-1 and HPS-4 develop pulmonary fibrosis in addition to the general clinical manifestations of oculocutaneous albinism and bleeding diathesis. We identified HPS-1 in three unrelated patients from different regions of India, who presented with iris transillumination, pale fundi, hypopigmentation, nystagmus, decreased visual acuity, and a bleeding diathesis. Two of these patients carried the homozygous mutation c.398+5G>A (IVS5+5G>A) in HPS1, resulting in skipping of exon 5 in HPS1 mRNA. The third patient carried a novel homozygous c.988-1G>T mutation that resulted in in-frame skipping of HPS1 exon 12 and removes 56 amino acids from the HPS1 protein. Given the discovery of HPS-1 in an ethnic group where oculocutaneous albinism (OCA) is highly prevalent, it is possible that HPS in India is under-diagnosed. We recommend that unconfirmed OCA patients in this ethic group be considered for mutational screening of known HPS genes, in particular c.398+5G>A and c.980-1G>T, to ensure that patients can be monitored and treated for clinical complications unique to HPS.
Keywords: lysosome-related organelle, albinism, Indian descent, splice site mutation, bleeding diathesis
Introduction
Hermansky-Pudlak syndrome (HPS; MIM #203300) is an autosomal recessive disorder characterized by oculocutaneous albinism and a bleeding diathesis; some patients exhibit lysosomal accumulation of ceroid lipofuscin, granulomatous colitis, or pulmonary fibrosis that is fatal in the fourth or fifth decade [1-5]. Genetic heterogeneity is supported by the existence of at least fifteen HPS mouse models [6], and mutations in eight different genes have been identified in humans with HPS [7, 8]. The HPS genes with known function have roles in the biogenesis and/or function of intracellular organelles required in membrane and protein trafficking [8]. The genetic heterogeneity of HPS gives rise to clinical heterogeneity. For example, only patients with mutations in HPS1 or HPS4 develop pulmonary fibrosis [8-10].
HPS is a rare disorder that has been identified in patients worldwide, including Puerto Rico, Japan, Northern Europe, and Israel [3, 7, 8, 11-15]. In addition, HPS-1, the most common HPS subtype, was recently reported in two African American siblings [16]. HPS-1 results from mutations in the HPS1 gene located on 10q23.1-q23.3 [17] and occurs largely as a genetic isolate in individuals from northwest Puerto Rico [11]. Among non-Puerto Ricans, HPS-1 accounts for ∼50% of all HPS cases [7, 18]. Here we report the first diagnosis of HPS in patients of Indian descent arising from two different mutations in HPS1.
Materials and Methods
Patients and cells
Written, informed consent was obtained for enrollment in a protocol approved by the National Human Genome Research Institute (NHGRI) institutional review board to evaluate the clinical and molecular aspects of HPS. Patient numbers conformed to a master list of NIH subjects with HPS. The diagnosis was based upon the absence of platelet dense bodies on whole-mount electron microscopy and the presence of hypopigmentation, decreased visual acuity, and nystagmus. Primary skin fibroblast and epidermal melanocyte cultures were obtained from 4-mm punch forearm skin biopsies and cultured as previously described [14, 19].
Electron microscopy of platelet dense bodies
Platelet-rich plasma prepared from fresh citrated blood was placed on copper grids and treated as described [2, 11]. The grids were air-dried and examined using a Philips model 301 electron microscope.
Molecular analysis
Genomic DNA was extracted from whole blood using the Gentra® Puregene® Blood Kit (Qiagen, Valencia, CA) and screened for mutations in known candidate genes HPS1 (NM_000195), HPS3 (NM_032383), HPS4 (NM_022081), HPS5 (NM_181507), and HPS6 (NM_024747) by sequencing each genes' exons and intronic boundaries (Agencourt Biosciences, Beverly, MA). Primer sequences are available upon request. Sequence analysis was performed using Sequencher™ 4.7 (Gene Codes, Ann Arbor, MI).
RNA was extracted from cultured normal and patient fibroblasts (for patient HPS159) or melanocytes (for patient HPS193) using the RNeasy® Mini Kit (Qiagen) and transcribed into cDNA using the SuperScript III system per the manufacturer's instructions (Invitrogen, Carlsbad, CA). cDNA was subjected to standard PCR amplification using HotStar Taq Master Mix (Qiagen) at an annealing temperature of 60°C.
To demonstrate skipping of HPS1 exon 5 in patient HPS159 (Fig. 2C), patient and normal cDNA was amplified using the primers 5′-AAGTTCGGGCAGTCAGAGAA-3′ and 5′-AGCTGGCACTGTGGCTAGA-3′, amplifying a normal 598-bp fragment spanning the exon-exon boundaries between exons 3 and 7 of the primary transcript of HPS1 (NM_000195) (Fig. 2A). To investigate HPS1 mRNA splicing effects of exon 12 in patient HPS193 (Fig. 3B), primers 5′-TCAGAGCTCAGGTAGCACCA-3′ and 5′-CACCAAGCCTGGGAAGTCT-3′, were used to amplify a normal 700-bp fragment spanning the exon-exon boundaries between exons 11 and 17 (Fig. 3A). Resultant PCR fragments were cloned into the TOPO TA Cloning® Kit per the manufacturer's instructions (Invitrogen). Clones were purified using the QIAprep® Spin Miniprep Kit (Qiagen) and subjected to automated sequence analysis on a Beckman CEQ™ 8000 using the CEQ™ Dye Terminator Cycle sequencing kit, according to the manufacturer's instructions (Beckman Coulter, Fullerton, CA).
Fig. 2. Molecular analysis of patients HPS152 and HPS159.
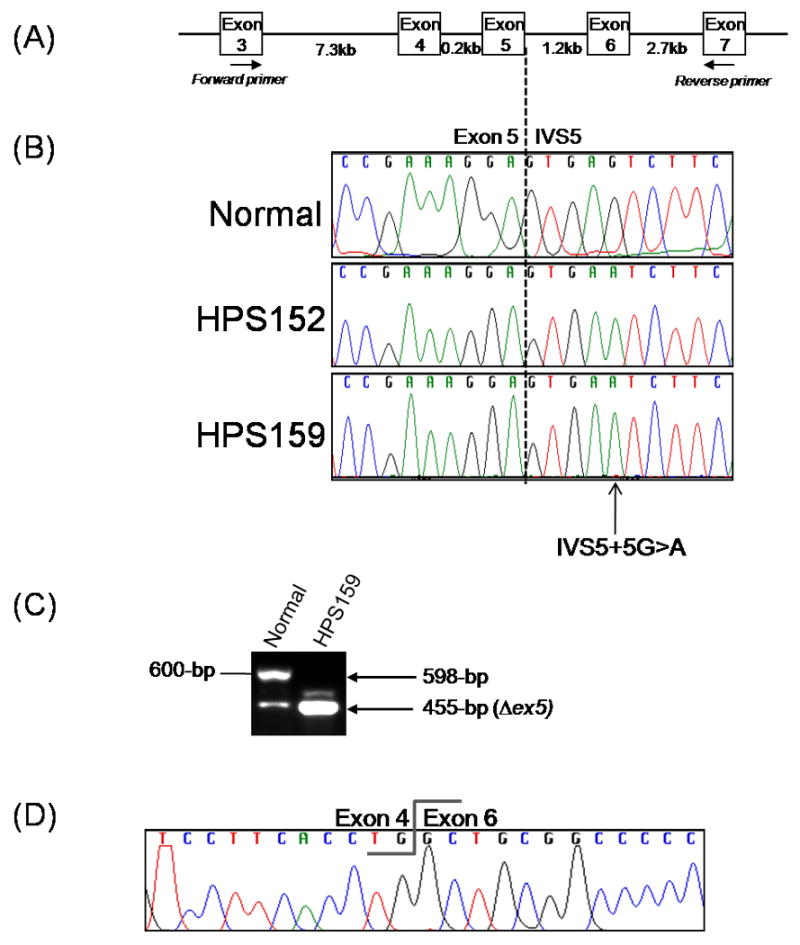
(A) Schematic representation of a part of the HPS1 gene and the primer positions used for RT-PCR analysis shown in (c).
(B) Genomic sequence analysis in HPS152 and HPS159 compared with the normal sequence indicating a homozygous IVS5+5 G>A mutation (c.398+5G>A) in both patients.
(C) RT-PCR analysis of the HPS1 mRNA transcript in fibroblasts of patient HPS159 showing homozygous skipping of exon 5 resulting in a 455-bp PCR fragment as compared to a 598-bp fragment in normal cDNA.
(D) Sequence analysis of the 455-bp RT-PCR fragment of patient HPS159, shown in (c), demonstrating the lack of HPS1 exon 5.
Fig. 3. Molecular and cellular analysis of patient HPS193.
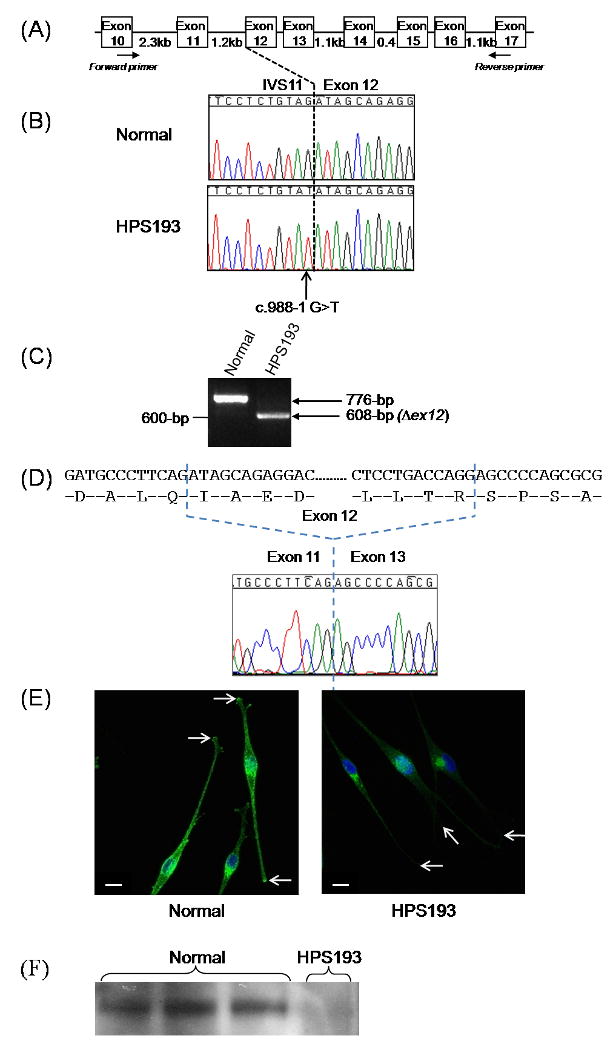
(A) Schematic representation of the HPS1 gene and the primer positions used for RT-PCR analysis shown in (c).
(B) Genomic sequencing results of the 3′ splice junction of HPS1 exon 12 in HPS193, compared with the normal sequence, indicating a homozygous IVS11-1G>T mutation in intron 11 (c.988-1G>T).
(C) RT-PCR analysis of the HPS1 mRNA transcript in melanocytes of patient HPS193 showing homozygous skipping of exon 12 resulting in a 608-bp PCR fragment as compared to a 776-bp fragment in normal cDNA.
(D) Sequence analysis of the 608-bp RT-PCR fragment of patient HPS193, shown in (c), demonstrating the lack of HPS1 exon 12.
(E) TYRP1 (green) and nuclear (blue) staining of melanosomes in normal (left panel) and HPS193 (right panel) melanocytes. There is decreased intensity and aberrant perinuclear distribution of TYRP1 in HPS193 melanocytes compared to the normal distribution. TYRP1 staining in the dendritic tips was greatly reduced in HPS193 melanocytes, compared to normal (arrows). Images are representative of three independent experiments and are 1D projections of confocal Z-sections. Size bar, 10 μm.
(F) Immunoblot analysis of HPS4 in melanocyte cell lysates from 3 normal controls (lanes 1-3) and HPS193 (lane 4). HPS4 is absent in HPS193 melanocytes indicating that HPS1 is not functional in this patient with respect to BLOC-3 and is likely subject to degradation. HPS4 was detected in 25μg of total protein lysate from skin-biopsied melanocytes using an in-house affinity purified polyclonal rabbit antibody and a corresponding anti-rabbit horseradish-peroxidase conjugated secondary antibody. The blot was developed using ECL™ Western Blotting Detection Reagent.
Immunocytochemistry
Normal or HPS193 melanocytes were grown for 48 hours on glass slides. Cells were fixed in 3% paraformaldehyde, blocked in PBS containing 0.1% saponin, 100 μM glycine and 2% donkey serum followed by incubation with melanosome-specific mouse monoclonal antibody MEL-5, recognizing tyrosinase-related protein 1 (TYRP1, 1:100; Covance, Princeton, NJ). The cells were washed and incubated with secondary donkey anti-mouse antibodies conjugated to Alexa Fluor®-488 (1:200; Invitrogen). Cells were washed again and subjected to nuclear staining with TO-PRO®-3 iodide (642/661) (1:600; Invitrogen) for 5 minutes. After two additional washes, slides were mounted with VECTASHIELD® (Vector Laboratories, Burlingame, CA). The slides were imaged with a Zeiss LSM510 META confocal laser-scanning microscope (Carl Zeiss, Microimaging Inc., Thornwood, NY). Images were acquired using a Plan-Apochromat® 63X/1.4 oil DIC objective. All images are 1D projections of 5 confocal Z-sections.
Immunoblot Analysis
Cell extract (25μg) from HPS193 melanocytes was fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a pre-cast 4-12% gradient gel (Invitrogen), transferred to polyvinylidene disulfluoride membranes, and probed using an affinity-purified rabbit polyclonal antibody against HPS4 (1:200) raised by our laboratory, essentially as previously described [20]. After incubation with the corresponding anti-rabbit horseradish-peroxidase conjugated secondary antibody (1:5000), the blot was developed using ECL™ Western Blotting Detection Reagent (GE Healthcare, Piscataway, NJ).
Results
Case Reports
Patient HPS152 was a female adopted from Hyderbad, India at 14 months of age, and was 8 years old at the time of our initial evaluation. At the time of adoption, an ophthalmologist diagnosed oculocutaneous albinism (Fig. 1A-C). Frequent bruising and epistaxis occurred, with bleeding for up to one hour. These findings and the absence of platelet dense bodies on electron microscopy (Fig. 1D) led to the diagnosis of HPS at age 7. No transfusions, DDAVP (1-deamino-8-D-arginine vasopressin), or cautery were required for bleeding episodes, and there were no episodes of hematochezia or respiratory complaints. Isoniazid was administered for 6 months for a positive tuberculosis skin test at the time of adoption. On initial evaluation at the NIH Clinical Center, her weight was 24.7 kg (35th centile) and her height was 127.1 cm (38th centile). Visual acuity was 20/125 OD and 20/160+2 OS. Pendular horizontal nystagmus, retinal pallor (Fig. 1B), and significant iris transillumination (Fig. 1C), retinal hypopigmentation and absence of foveal reflex (Fig. 1C) were noted.
Fig. 1. Clinical features of HPS-1 patients of Indian descent.
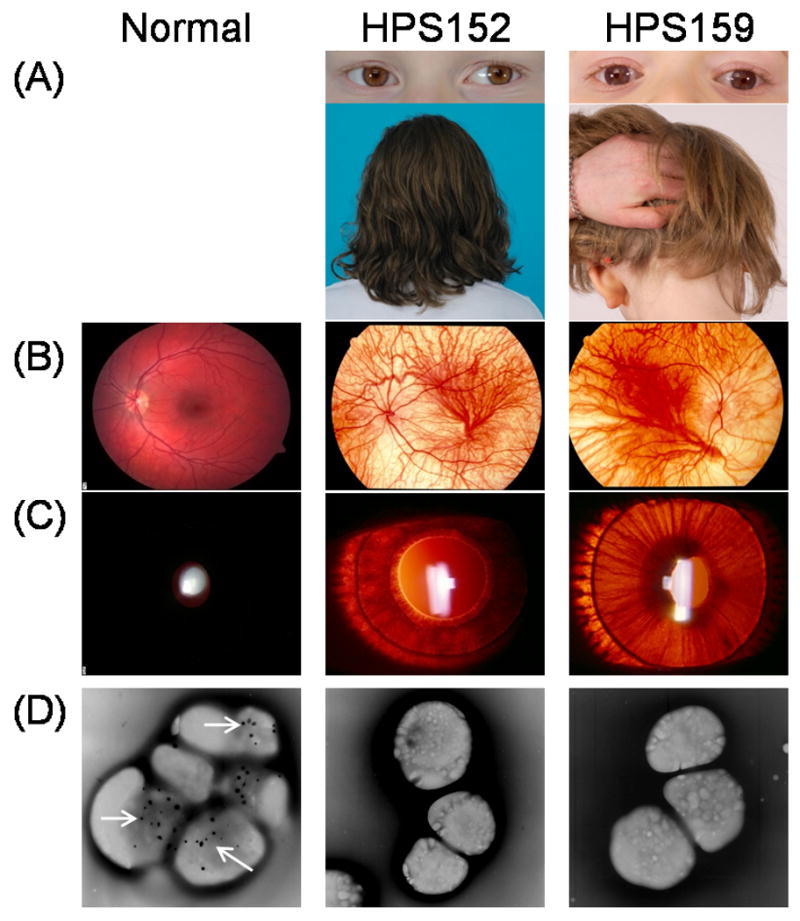
(A) Reduced skin and hair pigmentation in HPS patients contrary to patients' ethnic background. Note that patients' iris colors are light brown, which is darker than seen in most HPS patients.
(B) Retinal photographs demonstrating patchy, reduced pigmentation in HPS patients compared to normal.
(C) Transillumination of patients' irises. Normally, light reflected from the retina is not transmitted through the iris due to pigmentation.
(D) Absence of platelet dense bodies compared to normal platelets with multiple dense bodies, which appear as discrete dark spots (arrows in normal platelets).
Patient HPS159 was 8 years old at the time of her initial evaluation. She was adopted from Bangalore, India at 6 years of age. She had a clinically significant ventricular-septal defect (VSD), repaired at age 6 and oculocutaneous albinism (Fig. 1A) with characteristic horizontal nystagmus and decreased visual acuity. After her arrival in the United States, her adoptive family noted easy bruising and intermittent mucosal bleeding. She was noted to have prolonged bleeding from minor injuries. She received two scheduled platelet transfusions and a factor VIIA infusion at the time of open-heart surgery for VSD repair. An additional factor VIIA transfusion was given emergently after a traumatic facial contusion. DDAVP non-responsiveness was designated by an outside hematologist based on an assay of platelet aggregation before and after DDAVP administration. No episodes of hematochezia or respiratory complaints were reported. HPS was diagnosed based upon the absence of platelet dense bodies (Fig. 1D). At the time of our evaluation, her weight was 19.6 kg and her height was 120.53 cm. Best corrected visual acuity was 20/250 in both eyes. Bilateral iris transillumination (Fig. 1C), retinal hypopigmentation and foveal hypoplasia (Fig. 1B) were noted during eye exam.
Patient HPS193 was a boy from the Punjab region of northern India. At birth he had nystagmus and reduced pigmentation compared to his family members. An ophthalmologic evaluation revealed bilateral iris transillumination and retinal hypopigmentation. Molecular analysis of the tyrosinase gene detected one known pathogenic mutation, p.R402Q. A screen for the common OCA2 2.7-kb mutation was negative. Over time, the parents noticed easy bruisability and frequent, self-limited episodes of epistaxis. In pursuit of the possible diagnosis of HPS, they obtained whole-mount platelet electron microscopy, which showed an absence of dense bodies. At his initial evaluation at the NIH at age 4 years, 10/12 months, he weighed 15.2 kg (5th centile). His height was 109.9 cm (50th centile). He was hypopigmented relative to his parents. Visual acuity was 20/400 OU with pendular nystagmus and bilateral iris transillumination and foveal hypoplasia was noted.
Molecular analysis
Mutation analysis on genomic DNA of patients HPS152, HPS159, and HPS193 revealed mutations in the HPS1 gene. No mutations were found in the HPS3, HPS4, HPS5, or HPS6 genes.
Both patients HPS152 and HPS159 carried the homozygous mutation c.398+5G>A (IVS5+5) in intron 5 of HPS1 (Fig. 2B). This splicing mutation was previously reported in HPS patients of Japanese descent [18, 21, 22]. Using RNA extracted from HPS159 cultured fibroblasts, we confirmed that this mutation results in skipping exon 5 in the HPS1 mRNA transcript (Fig. 2C and 2D) as previously reported [22]. The resultant mutant mRNA is predicted to shift the reading frame, leading to 47 novel amino acids followed by a premature stop codon in exon 7. In addition, exon 5 appears to be alternatively spliced in normal individuals (lower band, Fig. 2C). This phenomenon is supported by at least one predicted transcript (NM_182638.1).
Patient HPS193 was homozygous for a novel c.988-1G>T (IVS11-1G>T) mutation in intron 11 of HPS1 (Fig. 3A). Given the location of this mutation on the 3′splice site of exon 12 (Fig. 3B), we predicted that splicing of the HPS1 transcript would be altered. RT-PCR analysis of cultured HPS193 melanocytes' mRNA indicated removal of exon 12 from the HPS1 transcript (Fig. 3C and 3D). Since exon 12 has 168 bases, this exon skipping event is in-frame and removes 56 amino acids from the HPS1 protein.
Cellular studies
Melanocytes were not available for HPS152 and HPS159. Using HPS193 cultured melanocytes, we examined the intracellular localization of melanosomes with immunofluorescence confocal microscopy. Staining for the melanogenic protein TYRP1 (Fig. 3E) showed a classic HPS-1 phenotype in HPS193 melanocytes, i.e. localization of melanosomes to the perinuclear region of the cell [8]. This indicated improper trafficking to the cell's dendritic tips compared to normal control melanocytes. Immunoblot analysis indicated the lack of HPS4, which is directly correlated to the lack of functional HPS1, in HPS193 melanocytes (Fig. 3F).
Discussion
Our mutation analysis of unclassified patients with Hermansky-Pudlak syndrome revealed three apparently unrelated patients from different regions of India with mutations in the HPS1 gene. All three patients displayed iris transillumination, pale fundi, hypopigmentation, nystagmus, decreased visual acuity, and absence of platelet dense bodies. None of the patients had developed pulmonary fibrosis or a decrease of pulmonary function at the time of evaluation, likely because all three patients were under age 10 and lung fibrosis typically develops in the second or third decade in HPS-1 patients [2, 5]. These patients should continue to be followed for the development of restrictive lung disease in order to determine whether pulmonary involvement occurs due to these HPS1 mutations and to anticipate therapy [2, 23].
Two patients (HPS152 and HPS159) were homozygous for a previously reported c.398+5G>A (IVS5+5G>A) splice-site mutation in HPS1 [18, 21, 22], while the third (HPS193) had a novel homozygous c.988-1G>T (IVS11-1G>T) splice-site mutation in HPS1. Our identification of the IVS5+5G>A mutation in these Indian patients indicates that this mutation is not unique to individuals of Japanese descent, as previously suggested [22], and may be an HPS1 allele that has spread in the Asian population.
The novel homozygous c.988-1G>T mutation, present in HPS193, is the fifth mutation identified that alters splicing of the pre-mRNA of HPS1. The previous four are c.1744-2 (IVS17-2A>C)[9], c.507G>A (E169E at exon 6 splice boundary)[16], c.398+5G>A (IVS5+5G>A)[18, 21, 22], and c.507+1G>A (IVS6+1G>A)[24]. The Neural Network Splice Site Prediction Tool (NNSPLICE, version 0.9; available online at http://www.fruitfly.org/seq_tools/splice.html) predicts that the c.988-1G>T mutation abolishes the consensus of the wild type 3′ splice site. RT-PCR analysis of HPS193 melanocytes supports that this alteration results in the in-frame skipping of exon 12 in HPS1 transcripts (Fig. 3B), causing loss of ∼6.3-kDa from the major endogenous 79.3 kDa protein.
Melanocytes are an ideal cell type in which to examine the biogenesis of melanosomes, i.e., lysosome-related organelles whose formation is affected in all HPS subtypes [7, 8, 25, 26]. TYRP1 is a resident melanosomal protein involved in pigment formation; in normal melanocytes, TYRP1 is dispersed with accumulation in the dendritic tips (Fig. 3D)[8]. TYRP1 distribution in HPS193 melanocytes remained perinuclear, with decreased staining in the cell periphery and no accumulation in the dendritic tips (Fig. 3D). This aberrant pattern resembles previously reported distribution of TYRP1 in HPS-1 patients [8, 25, 27], thus confirming an HPS-1 phenotype at the cellular level in patient HPS193.
Currently, HPS1 protein function is unknown. It has no homology to any known protein, and no functional domains have been identified. HPS1 is known to interact with HPS4 in the Biogenesis of Lysosome-related Organelles Complex (BLOC)-3 [28]. From the analysis of some cases of HPS-1 and HPS-4, it has been discovered that mutations in either of these genes results in destabilization and degradation of the BLOC-3 complex [20, 29]. Figure 3F represents the lack of HPS4 in HPS193 melanocytes indicating that, if produced, the mutant HPS1 protein in this patient is not functional with respect to BLOC-3 and is likely subject to degradation. In addition, these results insinuate that the amino acids encoded by exon 12 contain a critical domain essential for HPS1 function, structure, and/or stability, especially within the BLOC-3 complex.
Among the ethnic groups of eastern and southern India, OCA1 (MIM #606933) occurs with a frequency of 1 in 40,000 individuals [30, 31] and accounts for ∼50-60% of all oculocutaneous albinsim (OCA) because of a few founder mutations in the causative gene tyrosinase [31-33]. OCA4, due to mutations in SLC45A2, accounts for another approximately 10% of OCA in India [33]. Of all of the known HPS genes, only mutations in HPS4 have been previously identified in two sisters of Indian descent [10]. Our patient HPS193 carries one known pathogenic mutation (p.R402Q) in the tyrosinase gene, raising the possible diagnosis of classic OCA; however, the absence of platelet dense bodies, the presence of two pathogenic HPS1 mutations, and an HPS-1 cellular phenotype clearly suggest HPS-1 disease in this patient.
The characteristic features of OCA, a heterogeneous group of autosomal recessive disorders, include iris transillumination, nystagmus, strabismus, photophobia, and foveal hypoplasia, but not the fatal pulmonary fibrosis and granulomatous colitis associated with HPS-1 and HPS-4 [2, 10, 32]. Given our identification of HPS1 mutations and the previously reported mutations in HPS4 [10] in albinism patients of Indian descent, we suggest that other unconfirmed OCA patients in this ethic group be considered for testing for mutations in known HPS genes, in particular c.398+5G>A and c.988-1G>T in HPS1. An accurate diagnosis of the HPS subtype has important prognostic and treatment implications since the pulmonary function in HPS-1 and HPS-4 patients can be addressed with studies of the small molecule pirfenidone [23].
Acknowledgments
Isa Bernardini, Roxanne Fischer, Heidi Dorward, and Wendy Westbroek provided excellent technical assistance. This work was supported by the Intramural Research programs of the National Human Genome Research Institute and National Eye Institute, National Institutes of Health, Bethesda, MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood. 1959;14:162–169. [PubMed] [Google Scholar]
- 2.Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, Duffy LF, Kuehl EM, Troendle J, Bernardini I. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 3.Brantly M, Avila NA, Shotelersuk V, Lucero C, Huizing M, Gahl WA. Pulmonary function and high-resolution CT findings in patients with an inherited form of pulmonary fibrosis, Hermansky-Pudlak syndrome, due to mutations in HPS-1. Chest. 2000;117:129–136. doi: 10.1378/chest.117.1.129. [DOI] [PubMed] [Google Scholar]
- 4.Schinella RA, Greco MA, Cobert BL, Denmark LW, Cox RP. Hermansky-Pudlak syndrome with granulomatous colitis. Ann Intern Med. 1980;92:20–23. doi: 10.7326/0003-4819-92-1-20. [DOI] [PubMed] [Google Scholar]
- 5.Gahl WA. GeneReviews at Genetests: Medical Genetics Information Resource (database online) University of Washington; Seattle: 2007. Hermansky-Pudlak Syndrome. [Google Scholar]
- 6.Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky-Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26:616–628. doi: 10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- 7.Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function Pigment. Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 8.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermos CR, Huizing M, Kaiser-Kupfer MI, Gahl WA. Hermansky-Pudlak syndrome type 1: gene organization, novel mutations, and clinical-molecular review of non-Puerto Rican cases. Hum Mutat. 2002;20:482. doi: 10.1002/humu.9097. [DOI] [PubMed] [Google Scholar]
- 10.Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113:10–17. doi: 10.1007/s00439-003-0933-5. [DOI] [PubMed] [Google Scholar]
- 11.Witkop CJ, Nunez BM, Rao GH, Gaudier F, Summers CG, Shanahan F, Harmon KR, Townsend D, Sedano HO, King RA. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R. 1990;82:333–339. [PubMed] [Google Scholar]
- 12.Schallreuter KU, Frenk E, Wolfe LS, Witkop CJ, Wood JM. Hermansky-Pudlak syndrome in a Swiss population. Dermatology. 1993;187:248–256. doi: 10.1159/000247258. [DOI] [PubMed] [Google Scholar]
- 13.Schreyer-Shafir N, Huizing M, Anikster Y, Nusinker Z, Bejarano-Achache I, Maftzir G, Resnik L, Helip-Wooley A, Westbroek W, Gradstein L, Rosenmann A, Blumenfeld A. A new genetic isolate with a unique phenotype of syndromic oculocutaneous albinism: clinical, molecular, and cellular characteristics. Hum Mutat. 2006;27:1158. doi: 10.1002/humu.9463. [DOI] [PubMed] [Google Scholar]
- 14.Huizing M, Anikster Y, Fitzpatrick DL, Jeong AB, D'Souza M, Rausche M, Toro JR, Kaiser-Kupfer MI, White JG, Gahl WA. Hermansky-Pudlak syndrome type 3 in Ashkenazi Jews and other non-Puerto Rican patients with hypopigmentation and platelet storage-pool deficiency. Am J Hum Genet. 2001;69:1022–1032. doi: 10.1086/324168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito S, Suzuki T, Inagaki K, Suzuki N, Takamori K, Yamada T, Nakazawa M, Hatano M, Takiwaki H, Kakuta Y, Spritz RA, Tomita Y. High frequency of Hermansky-Pudlak syndrome type 1 (HPS1) among Japanese albinism patients and functional analysis of HPS1 mutant protein. J Invest Dermatol. 2005;125:715–720. doi: 10.1111/j.0022-202X.2005.23884.x. [DOI] [PubMed] [Google Scholar]
- 16.Merideth MA, Vincent LM, Sparks SE, Hess RA, Manoli I, O'Brien KJ, Tsilou E, White JG, Huizing M, Gahl WA. Hermansky-Pudlak Syndrome in Two African-American Brothers. Am J Hum Genet A. 2009 doi: 10.1002/ajmg.a.32757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailin T, Oh J, Feng GH, Fukai K, Spritz RA. Organization and nucleotide sequence of the human Hermansky-Pudlak syndrome (HPS) gene. J Invest Dermatol. 1997;108:923–927. doi: 10.1111/1523-1747.ep12294634. [DOI] [PubMed] [Google Scholar]
- 18.Oh J, Ho L, Ala-Mello S, Amato D, Armstrong L, Bellucci S, Carakushansky G, Ellis JP, Fong CT, Green JS, Heon E, Legius E, Levin AV, Nieuwenhuis HK, Pinckers A, Tamura N, Whiteford ML, Yamasaki H, Spritz RA. Mutation analysis of patients with Hermansky-Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. Am J Hum Genet. 1998;62:593–598. doi: 10.1086/301757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naeyaert JM, Eller M, Gordon PR, Park HY, Gilchrest BA. Pigment content of cultured human melanocytes does not correlate with tyrosinase message level. Br J Dermatol. 1991;125:297–303. doi: 10.1111/j.1365-2133.1991.tb14161.x. [DOI] [PubMed] [Google Scholar]
- 20.Nazarian R, Huizing M, Helip-Wooley A, Starcevic M, Gahl WA, Dell'Angelica EC. An immunoblotting assay to facilitate the molecular diagnosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2008;93:134–144. doi: 10.1016/j.ymgme.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horikawa T, Araki K, Fukai K, Ueda M, Ueda T, Ito S, Ichihashi M. Heterozygous HPS1 mutations in a case of Hermansky-Pudlak syndrome with giant melanosomes. Br J Dermatol. 2000;143:635–640. doi: 10.1111/j.1365-2133.2000.03725.x. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T, Ito S, Inagaki K, Suzuki N, Tomita Y, Yoshino M, Hashimoto T. Investigation on the IVS5 +5G --> a splice site mutation of HPS1 gene found in Japanese patients with Hermansky-Pudlak syndrome. J Dermatol Sci. 2004;36:106–108. doi: 10.1016/j.jdermsci.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Gahl WA, Brantly M, Troendle J, Avila NA, Padua A, Montalvo C, Cardona H, Calis KA, Gochuico B. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2002;76:234–242. doi: 10.1016/s1096-7192(02)00044-6. [DOI] [PubMed] [Google Scholar]
- 24.Natsuga K, Akiyama M, Shimizu T, Suzuki T, Ito S, Tomita Y, Tanaka J, Shimizu H. Ultrastructural features of trafficking defects are pronounced in melanocytic nevus in Hermansky-Pudlak syndrome type 1. J Invest Dermatol. 2005;125:154–158. doi: 10.1111/j.0022-202X.2005.23743.x. [DOI] [PubMed] [Google Scholar]
- 25.Huizing M, Gahl WA. Disorders of vesicles of lysosomal lineage: the Hermansky-Pudlak syndromes. Curr Mol Med. 2002;2:451–467. doi: 10.2174/1566524023362357. [DOI] [PubMed] [Google Scholar]
- 26.Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. 2001;152:809–824. doi: 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarangarajan R, Budev A, Zhao Y, Gahl WA, Boissy RE. Abnormal translocation of tyrosinase and tyrosinase-related protein 1 in cutaneous melanocytes of Hermansky-Pudlak Syndrome and in melanoma cells transfected with anti-sense HPS1 cDNA. J Invest Dermatol. 2001;117:641–646. doi: 10.1046/j.0022-202x.2001.01435.x. [DOI] [PubMed] [Google Scholar]
- 28.Martina JA, Moriyama K, Bonifacino JS. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem. 2003;278:29376–29384. doi: 10.1074/jbc.M301294200. [DOI] [PubMed] [Google Scholar]
- 29.Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- 30.King RA, Hearing VJ, Creel DJ, Oettin WS, Scriver CR, Beaudet AL, Sly WS, Valle D Albinism. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 5587–5627. [Google Scholar]
- 31.Chaki M, Sengupta M, Mukhopadhyay A, Subba RI, Majumder PP, Das M, Samanta S, Ray K. OCA1 in different ethnic groups of india is primarily due to founder mutations in the tyrosinase gene. Ann Hum Genet. 2006;70:623–630. doi: 10.1111/j.1469-1809.2006.00247.x. [DOI] [PubMed] [Google Scholar]
- 32.Chaki M, Mukhopadhyay A, Chatterjee S, Das M, Samanta S, Ray K. Higher prevalence of OCA1 in an ethnic group of eastern India is due to a founder mutation in the tyrosinase gene. Mol Vis. 2005;11:531–534. [PubMed] [Google Scholar]
- 33.Sengupta M, Chaki M, Arti N, Ray K. SLC45A2 variations in Indian oculocutaneous albinism patients. Mol Vis. 2007;13:1406–1411. [PubMed] [Google Scholar]