

Abstract
Coxsackievirus B3 (CVB3) is a causative agent of viral myocarditis, meningitis, pancreatitis, and encephalitis. Much of what is known about the coxsackievirus intracellular replication cycle is based on the information already known from a well-studied and closely related virus, poliovirus. Like that of poliovirus, the 5′ noncoding region (5′ NCR) of CVB3 genomic RNA contains secondary structures that function in both viral RNA replication and cap-independent translation initiation. For poliovirus IRES-mediated translation, the interaction of the cellular protein PCBP2 with a major secondary structure element (stem-loop IV) is required for poliovirus gene expression. Previously, the complete secondary structure of the coxsackievirus 5′ NCR was determined by chemical structure probing and overall, many of the RNA secondary structures bear significant similarity to those of poliovirus; however, the functions of the coxsackievirus IRES stem-loop structures have not been determined. Here we report that a CVB3 RNA secondary structure, stem-loop IV, folds similarly to poliovirus stem-loop IV and like its enterovirus counterpart, coxsackievirus stem-loop IV interacts with PCBP2. We used RNase foot-printing to identify RNA sequences protected following PCBP2 binding to coxsackievirus stem-loop IV. When nucleotide substitutions were separately engineered at two sites in coxsackievirus stem-loop IV to reduce PCBP2 binding, inhibition of IRES-mediated translation was observed. Both of these nucleotide substitutions were engineered into full-length CVB3 RNA and upon transfection into HeLa cells, the specific infectivities of both constructs were reduced and the recovered viruses displayed small plaque phenotypes and slower growth kinetics compared to wild type virus.
Keywords: coxsackievirus, cap-independent translation, internal ribosome entry site (IRES), RNA secondary structure, poly(rC)-binding protein (PCBP)
Introduction
Coxsackievirus B3 (CVB3) is a member of the enterovirus genus in the family Picornaviridae. CVB3 infection of heart cells leads to cardiomyopathies (Beck et al., 1990; Esfandiarei and McManus, 2008). In a mouse model, cardiovirulence of coxsackievirus was demonstrated by extensive inflammatory lesions and necrosis of heart tissue (Tracy et al., 1992; for review see Kim et al., 2001). Like other picornaviruses, coxsackievirus has a single-stranded, positive-sense RNA genome encapsidated by proteins that form an icosahedron of approximately 28 nm (Crowell and Landau, 1997). Two integral membrane receptors have been identified for viral entry, coxsackievirus and adenovirus receptor (CAR) (Bergelson et al., 1997) and CD55, a decay-accelerating factor (DAF) (Shafren et al., 1995). Once inside the cell, coxsackievirus completes its replication cycle in the cytoplasm.
Like poliovirus, the prototypic picornavirus, the coxsackievirus genome has a long stretch of noncoding sequence (5′ NCR) upstream of the authentic start codon. The ~ 740-nucleotide 5′ NCR (Lindberg et al., 1987) is highly structured, containing multiple stem-loop elements (Yang et al., 1997; Bailey and Tapprich, 2007). Unlike cellular messenger RNAs, translation of picornavirus genomes occurs via a cap-independent pathway. In the canonical mechanism of cap-dependent translation initiation, the 5′ cap on cellular mRNAs is recognized by the eIF4F cap-binding complex, which results in the recruitment of ribosomes (Merrick, 1990); however, picornaviruses lack the 7-methyl G cap structure and instead have a viral protein (VPg) that is covalently linked to the 5′ terminus of the genome (Lee et al., 1977; Flanegan et al., 1977). Also, during an enterovirus infection, the virus-encoded proteinase 2A cleaves eIF4G to disrupt the cap-binding complex (Etchison et al., 1982; Krausslich et al., 1987). Thus, the cleavage of eIF4G acts to inhibit eIF4F-mediated ribosome scanning in cap-dependent translation. Without a 5′ cap to initiate cap-recognition and ribosome scanning, picornaviruses utilize an alternative, internal pathway for translation initiation. The RNA secondary structures that form within the 5′ NCR serve as an internal ribosome entry site (IRES) for translation initiation (Jang et al., 1988; Pelletier and Sonenberg, 1988). The exact mechanism of IRES-mediated translation initiation has not been elucidated; however, it has been postulated that the interaction of trans-acting host factors with cis-acting stem-loop structures and helices acts to recruit canonical and non-canonical translation factors and/or stabilize the RNA for translation. The IRES elements of picornaviruses are divided into three types based on sequence and structural homology. Viruses containing type I IRES elements include poliovirus, coxsackievirus, and rhinovirus; those of type II include foot and mouth disease virus (FMDV) and encephalomyocarditis (EMCV). Hepatitis A virus (HAV) has a type III IRES (Wimmer et al., 1993; Jackson and Kaminski, 1995; Borman et al., 1997).
The RNA secondary structures of poliovirus and coxsackievirus 5′ NCRs have been solved through both enzymatic and chemical structure probing (Skinner et al., 1989; Bailey and Tapprich, 2007; Stewart and Semler, 1998). Overall, the RNA secondary structures that form in the 5′ NCR of these two enteroviruses are well conserved. The secondary structures of the 5′ NCR of enteroviruses have two distinct functions: cap-independent translation and RNA replication (Ehrenfeld and Teterina, 2002). At the very 5′ end of RNA is a highly conserved cloverleaf-like secondary structure termed stem-loop I. Enterovirus stem-loop I has been shown to be important for negative-strand RNA synthesis (Andino et al., 1990; Parsley et al., 1997; Bell et al., 1999). Stem-loops II though VI make up the IRES and are required for cap-independent translation (Pelletier and Sonenberg, 1988; Trono et al., 1988; Murray et al., 2004). Poliovirus stem-loop IV, a large 205 nucleotide-long cruciform-like structure in the middle of the IRES, has been identified as a major cis-acting element for translation initiation (Blyn et al., 1995). Poliovirus stem-loop IV presents two poly(C) loops and a GNRA tetra-loop, identified as a very stable motif, at the apex (Antao et al., 1991). A single nucleotide mutation at position 325 of stem-loop IV reduced translation of poliovirus in HeLa cell extract (Blyn et al., 1995). Nucleotide substitutions in the poly(C) region at position 332 completely abolished poliovirus IRES-mediated translation (Gamarnik and Andino, 2000).
To date, the only cellular protein identified to have a definitive role in both enterovirus RNA replication and IRES-mediated translation is the cellular poly(rC) binding protein (PCBP) (Parsley et al., 1997; Gamarnik and Andino, 1997; Walter et al., 2002). PCBPs are RNA binding proteins that preferentially bind to single-stranded stretches of cytidines (for review see, Makeyev and Liebhaber, 2002). There are four isoforms of PCBPs, each having three K-homology (KH) domains, which are consensus RNA binding domains that fold according to a β1α1α2β2β3α3 motif (Siomi et al., 1993; Dejgaard and Leffers, 1996). In the cell, PCBPs function by interacting with poly(C) stretches in the 3′ NCR of specific cellular mRNAs and stabilizing these messenger RNAs, as in the case of α-globin, or by modulating translation, as in the case of lipoxygenase (Weiss and Liebhaber, 1994; Ostareck et al., 1997). Co-crystal structures of the KH1 domain of PCBP2 with human telomeric DNA shows that the poly(C) nucleotides interact with a groove that is generated between α1α2 and β2β3 (Du et al., 2005). Similar to cellular mRNAs, it has also been shown that the interaction of PCBPs with poliovirus stem-loop I contributes to the overall stability of the viral RNA in vitro (Murray et al., 2001).
In addition to forming ribonucleoprotein (RNP) complexes with RNA, PCBPs can also participate in protein-protein interactions. In yeast-two hybrid screens, it was shown that PCBP1, PCBP2, and hnRNP K can homo- and hetero-dimerize in various combinations (Kim et al., 2000; Bedard et al., 2004). PCBP2 has also been shown to interact with multiple RNA-binding proteins, including poly(A) binding protein (PABP) and two proteins involved in splicing, SRp20 and 9G8 (Funke et al., 1996; Wang et al., 1999; Bedard et al., 2007).
Of the four PCBP isoforms, only PCBP1 and PCBP2 have been experimentally shown to have roles in the replication cycles of enteroviruses (i.e., poliovirus and coxsackievirus). The interaction of PCBP1/2 and the viral polymerase precursor 3CD with poliovirus stem-loop I RNA forms a ternary complex, a required step in negative-strand RNA synthesis (Andino et al., 1990; Parsley et al., 1997; Gamarnik and Andino, 1997; Perera et al., 2007). Ternary complex formation might facilitate circularization of the viral RNA template, through an interaction with PABP on the 3′ poly(A) tract, for negative-strand RNA synthesis (Barton et al., 2001; Herold and Andino, 2001). In HeLa cell cytoplasmic extracts depleted of PCBPs, poliovirus RNA replication was inhibited; however, addition of recombinant PCBPs rescued RNA replication to near mock-depleted levels (Walter et al., 2002).
Beyond its role in viral RNA replication, PCBP2 is an IRES trans-acting factor (ITAF). In HeLa cell cytoplasmic extracts depleted of PCBPs, poliovirus IRES-mediated translation is greatly reduced (Blyn et al., 1997). This translation defect can be rescued by addition of recombinant PCBP2, but not PCBP1 (Blyn et al., 1997). The ability of PCBP2 to rescue poliovirus IRES-mediated translation results from it’s ability to bind poliovirus stem-loop IV RNA, whereas PCBP1 can not (Walter et al., 2002; Sean et al., 2008). PCBP2 interaction with poliovirus stem-loop IV requires a stretch of single-stranded poly(C) residues (Blyn et al., 1996; Gamarnik and Andino, 2000). Specific nucleotide insertions and substitutions in poliovirus stem-loop IV RNA that inhibit PCBP2 binding also inhibit poliovirus IRES-mediated translation (Blyn et al., 1996; Gamarnik and Andino, 2000). In fact, PCBP2 has been shown to be required for cap-independent translation initiation of several type I IRES-containing picornaviruses (Blyn et al., 1997; Walter et al., 1999). The exact mechanism by which PCBP2 mediates translation has not been elucidated, but it is possible that the interaction of PCBP2 with poliovirus stem-loop IV RNA recruits ribosomes via protein-protein interactions or by stabilizing the RNA structure for internal ribosome entry (Bedard et al., 2007).
Genetic evidence suggests that a conserved mechanism of virus-host interaction is required for the intracellular replication cycles of poliovirus and coxsackievirus. Semler and colleagues showed that a chimeric poliovirus containing the 5′ NCR of coxsackievirus displayed a near wild type growth phenotype (Johnson and Semler, 1988) even though at the nucleotide level, the 5′ NCRs of these two viruses share only ~70% identity (Semler et al., 1986). We reasoned that the coxsackievirus IRES, like that of poliovirus, contains RNA secondary structures which form RNP complexes with host cell proteins to mediate cap-independent translation initiation. In this report, we describe enzymatic RNA structure probing and RNase foot-printing experiments to identify the CVB3 stem-loop IV sequences required for interaction with PCBP2. Our data confirm previous structure probing studies (Gamarnik and Andino, 2000; Bailey and Tapprich, 2007), showing that coxsackievirus stem-loop IV RNA folds into a secondary structure similar to that of poliovirus stem-loop IV RNA, including a GNRA tetra-loop at the apex, a poly(C) loop at a 5′ proximal hairpin, and a single-stranded polypyrimidine (Py)-bulge that is adjacent to the GNRA tetra-loop. Using mobility shift assays, we show that coxsackievirus stem-loop IV RNA interacts with PCBP2 at a reduced binding affinity compared to poliovirus stem-loop IV. Foot-printing experiments revealed three major regions of stem-loop IV RNA that are protected by PCBP2 from RNase digestion, two of which included or were proximal to the poly(C)-loop and (Py)-bulge sequences. When the poly(C) sequences and (Py)-bulge sequences in coxsackievirus stem-loop IV were separately mutated to guanosines, the interaction of the RNAs with PCBP2 was greatly reduced [especially for the (Py)-bulge substitution to poly(G)]. Using in vitro translation assays of luciferase reporter constructs, we found that substitution of poly(G) for either the poly(C)-loop or the (Py)-bulge in coxsackievirus stem-loop IV produced four- or ten-fold reductions, respectively, in IRES-mediated translation. Surprisingly, when these same poly(G) nucleotide substitutions were engineered into full-length CVB3 RNA transcripts, both constructs produced infectious virus. However, the specific infectivities of the mutated RNAs were reduced and the recovered viruses displayed smaller plaque sizes and slower replication kinetics compared to wild type CVB3. We discuss the implications of these findings in the context of additional host factors that may play a role in coxsackievirus translation initiation.
Results
Enzymatic structure probing and m-fold prediction of coxsackievirus stem-loop IV RNA
To determine if the coxsackievirus 5′ NCR contains an RNA secondary structure that folds and functions like poliovirus stem-loop IV RNA, we employed both enzymatic structure probing experiments and computer-based RNA structure modeling. A coxsackievirus stem-loop IV RNA was generated by sub-cloning sequences corresponding to poliovirus stem-loop IV from the 5′ NCR of a full-length coxsackievirus genomic cDNA (Chapman et al., 1994). In vitro transcribed 32P end-labeled coxsackievirus stem-loop IV RNAs were subjected to partial digestion by single-strand, base-specific RNases (A and T1) and a double-strand specific RNase, V1. The reactions were then resolved on sequencing gels in parallel with untreated coxsackievirus stem-loop IV RNA. An example of the results of one of several structure-probing experiments carried out on CVB3 stem-loop IV RNA is shown in Fig. 1A. The nucleotides identified from enzymatic structure probing were then used as constraints for m-fold (Mathews et al., 1999; Zuker, 2003), a computer algorithm that predicts secondary structure based on the thermodynamics of base pair interactions. The resulting output reveals a cruciform-like RNA secondary structure (Fig. 1B). Nucleotides in bold were identified by our enzymatic structure experiments. Overall, the secondary structure of coxsackievirus stem-loop IV RNA bears a strong similarity to poliovirus stem-loop IV RNA, having a GNRA tetra-loop at the apex, a poly(C)-loop at the 5′ proximal hairpin, and a polypyrimidine-bulge [poly(Py)-bulge] that is adjacent to the GNRA tetra-loop. Slight differences between the two stem-loop structures were also observed. Coxsackievirus stem-loop IV RNA has an internal bulge that is 18 nucleotides in length, and the poly(C) stem is shorter than poliovirus stem-loop IV by 7 paired nucleotides. Our structure of coxsackievirus stem-loop IV confirms, for the most part, a previously-published secondary structure that was determined by chemical probing methods (Bailey and Tapprich, 2007). However, there are predicted differences in the sizes of several internal bulge sequences as well as in the length of base-paired stem regions.
Figure 1. Secondary structure determination of coxsackievirus stem-loop IV RNA.


A. Enzymatic structure probing of coxsackievirus stem-loop IV RNA. Radio-labeled coxsackievirus stem-loop IV RNA (20–40 ng) was digested with RNase A (lanes 2–5), T1 (lanes 6–13) and V1 (lanes 14–17). The RNA was digested for 1–3 minutes at 25°C with RNase dilutions of 1:1 and 1:10 from stock. Urea (1 mM) was added to the T1 digestion to obtain a G-ladder (lanes 8–9 and 12–13). The single-strand specific RNases used were RNase A, which cleaves after C and U residues, and RNase T1, which cleaves after G residues. RNase V1 was used to identify base-paired nucleotides. Undigested, full-length end-labeled coxsackievirus stem-loop IV RNAs are seen at the top of the gel. This shows “one-hit kinetics” of RNase digestion and ensures that the observed cleavage products are from completely formed RNA structures. The bands in the RNase-digested lanes correspond to specific nucleotides cleaved by the RNases. B. M-fold predicted secondary structure of coxsackievirus stem-loop IV RNA. Nucleotides in bold were identified through RNase structure probing. The structure shown represents nucleotides 235 to 430 for coxsackievirus B3. The poly(C)-loop, poly(Py)-bulge, and GNRA tetra-loop are indicated by dashed-line boxes.
Coxsackievirus stem-loop IV RNA binds to PCBP2
The secondary structure of coxsackievirus stem-loop IV presents two single-stranded polypyrimidine motifs (refer to Fig. 1A), which are potential PCBP binding sites (Leffers et al., 1995; Blyn et al., 1996; Parsley et al., 1997). Previously, it was shown that PCBP2 can interact with two sites on coxsackievirus stem-loop I RNA, at the b-loop and at the poly(C) region directly downstream (Bell et al., 1999; Zell et al., 2008). Also, the complete 5′ NCR of coxsackievirus RNA can compete with poliovirus stem-loop IV for PCBP2 binding in mobility shift assays (Walter et al., 1999). To determine if coxsackievirus stem-loop IV RNA interacts directly with PCBP2, we performed electrophoretic mobility shift assays with recombinant PCBP2. At low concentrations of PCBP2 (50–100 nM), distinct RNP complexes are readily detected with 32P-labeled poliovirus stem-loop IV RNA (Fig. 2A, lanes 2–3). The apparent affinity of PCBP2 appears to be weaker for coxsackievirus stem-loop IV RNA, as evidenced by probe smearing and the lack of discrete RNP complex formation (Fig. 2A, lanes 5–6). At higher concentrations of PCBP2 (250–750 nM), a discrete RNP complex forms in a dose-dependent manner, along with a concomitant disappearance of free probe (Fig. 2A, lanes 8–10).
Figure 2. Mobility shift assays of coxsackievirus stem-loop IV RNA with PCBP2 and PCBP1.


A. Electrophoretic mobility shift assays of poliovirus and coxsackievirus stem-loop IV RNA with recombinant PCBP2 and PCBP1. In vitro transcribed, 32P-labeled stem-loop IV RNA at a final concentration of 0.1 nM was incubated with increasing amounts of purified recombinant PCBP2 for 10 min at 30°C, and the reaction was then subjected to native polyacrylamide gel electrophoresis. Lanes 1, 4, 7, and 11 are the RNA alone. Poliovirus stem-loop IV RNA (lanes 2–3) and coxsackievirus stem-loop IV RNA (lanes 5–6 and 8–10) can form ribonucleoprotein (RNP) complexes with PCBP2. The RNP complex is denoted by the arrow. PCBP1 (lanes 12–13) is unable to interact with coxsackievirus stem-loop IV RNA. B. Competition electrophoretic mobility shift assay of poliovirus stem-loop IV RNA and PCBP2 using unlabeled coxsackievirus stem-loop IV RNA. In vitro transcribed 32P-labeled poliovirus stem-loop IV RNA (1 nM) was incubated with PCBP2 (50 nM) in the presence of increasing amounts of unlabeled poliovirus (lanes 3–6) or coxsackievirus stem-loop IV RNAs (lanes 7–10) for 10 min at 30°C. The RNP complexes were then resolved on a native polyacrylamide gel. RNP complexes are denoted by the arrow. C. Graph of relative binding affinities of PCBP2 for poliovirus and coxsackievirus stem-loop IV RNA. The relative affinities of PCBP2 for the stem-loop IV RNAs were determined by measuring the band intensity of the bound RNA divided by the sum of the free RNA and bound RNA. It was observed that 15 nM unlabeled poliovirus stem-loop IV RNA was enough to dissociate half of the labeled poliovirus stem-loop IV-PCBP2 RNP complex, while it required 35 nM unlabeled coxsackievirus stem-loop IV RNA to obtain the same level of competition.
Mobility shift assays of coxsackievirus stem-loop IV RNA and PCBP1, an isoform that does not interact with poliovirus stem-loop IV RNA, were also performed (Walter et al., 2002). In agreement with data reported for poliovirus, coxsackievirus stem-loop IV does not interact with PCBP1 (Fig. 2A, lanes 12–13). The selective interaction of coxsackievirus stem-loop IV with PCBP2 and not PCBP1 suggests the conservation of protein utilization in IRES-mediated translation between both poliovirus and coxsackievirus (Sean et al., 2008).
The mobility shift assays indicate that the apparent affinity of PCBP2 for coxsackievirus stem-loop IV RNA is lower compared to poliovirus stem-loop IV. To determine the relative affinity of PCBP2 for coxsackievirus stem-loop IV RNA, we performed competition electrophoretic mobility shift assays of radiolabeled poliovirus stem-loop IV RNA and PCBP2 (Fig. 2B). RNP complexes were formed with poliovirus stem-loop IV RNA and 50 nM PCBP2 (Fig. 2B, lane 2). Increasing molar excesses of either unlabeled poliovirus or coxsackievirus stem-loop IV RNAs were then added to compete for binding to PCBP2. At 10–25 nM unlabeled poliovirus stem-loop IV RNA, half of the radiolabeled poliovirus stem-loop IV/PCBP2 RNP complex disappears (Fig. 2B, lanes 3–4); however, 50 nM unlabeled coxsackievirus stem-loop IV RNA is required to compete for PCBP2 binding (Fig. 2B, lanes 8–9). We calculated the percentage of radiolabeled poliovirus stem-loop IV RNA bound against molar excess of unlabelled competitor RNA and determined that the apparent KD of PCBP2 for poliovirus stem-loop IV is ~15 nM (Fig. 2C), which is in agreement with previously published data (Gamarnik and Andino, 2000). The apparent KD of PCBP2 for coxsackievirus stem-loop IV RNA is ~35 nM. The lowered affinity of PCBP2 for coxsackievirus stem-loop IV RNA (compared to poliovirus stem-loop IV) might be due to differences in structural features, which include a shorter stem presenting the poly(C)-loop and only two predicted single-stranded cytidines in the poly(Py)-bulge, compared to three single-stranded cytidines on poliovirus stem-loop IV (Fig. 1B).
RNase foot-printing analysis of coxsackievirus stem-loop IV RNA and PCBP2
After determining that the apparent affinity of PCBP2 for coxsackievirus stem-loop IV is lower when compared to poliovirus stem-loop IV, we performed RNase foot-printing analysis to more carefully examine this interaction. The higher affinity observed for PCBP2 and poliovirus stem-loop IV (compared to coxsackievirus stem-loop IV) may be the result of PCBP2 interactions with three regions of the RNA that were protected from RNase T1 and T2 digestion (Gamarnik and Andino, 2000). Foot-printing data showed that PCBP2 interacted with two single-stranded loops and a bulge sequence in poliovirus stem-loop IV RNA (Gamarnik and Andino, 2000). We performed RNase foot-printing assays with 5 μM PCBP2 using the same experimental conditions employed in our enzymatic structure probing experiments (Fig. 3A). PCBP2 protected three major regions of coxsackievirus stem-loop IV RNA from RNase A and V1 digestion, denoted by A, B, and C (Fig. 3B). The foot-printing assay shows that three cytidines which form the poly(C)-loop and adjacent stem were protected. A double-stranded region along the helix of coxsackievirus stem-loop IV RNA was also protected by PCBP2, as well as three helical regions adjacent to the poly(Py)-bulge. The RNase foot-printing assay showed that PCBP2 protects three major regions within coxsackievirus stem-loop IV RNA from nuclease digestion. The RNase protection observed on the stem structure may result from the variable nucleotide sequence motifs that interact with PCBP2. Although there is a preference for binding poly(rC) motifs, it has been shown that the KH domains of PCBPs can interact with homopolymers of poly(rU), poly(rG), double-stranded RNA, or DNA (Leffers et al., 1995; Dejgaard and Leffers, 1996). Given that our RNase foot-printing experiments identified three regions on the RNA that are protected by PCBP2 and that each protein has three KH domains (RNA-binding domains), it is possible that one molecule of RNA interacts with one molecule of PCBP2; however, the exact stoichiometry of the interaction cannot be determined accurately due to the fact that a molar excess of protein was used, and due to the homo-dimerization capability of PCBP2.
Figure 3. RNase foot-printing assay of coxsackievirus stem-loop IV RNA and PCBP2.


A.PCBP2 protection of multiple regions of coxsackievirus stem-loop IV RNA. Radio-labeled coxsackievirus stem-loop IV RNA (20–40 ng) was incubated with 5 μM of PCBP2, denoted by +, at 30°C for 10 minutes and subjected to RNase A (lanes 4–7), T1 (lanes 8–13) and V1 (lanes 14–17) digestion. The RNAs were digested for 1–3 minutes at 25°C with RNase dilution of 1:1 and 1:10 from stock. Urea (1 mM) was added to the T1 digestion to obtain a G-ladder (lanes 10 and 13). Boxes indicate nucleotides within coxsackievirus stem-loop IV RNA that were protected by PCBP2 from RNase cleavage. B. Secondary structure showing regions of sequence in coxsackievirus stem-loop IV RNA protected by PCBP2 from RNase digestion (denoted by boxes). PCBP2 protects the poly(C)-loop, the stem leading to the internal bulge, and the helix region adjacent to the poly(Py)-bulge.
Poly(G) substitutions in coxsackievirus stem-loop IV RNA reduce PCBP2 binding
Our RNase foot-printing experiments identified sequences in coxsackievirus stem-loop IV RNA that are protected from nuclease digestion; however, protection does not necessarily demonstrate direct interaction with the protein. The secondary structure of coxsackievirus stem-loop IV presents two single-stranded polypyrimidine tracts, which may provide binding sites for PCBP2 (Fig. 1B). To examine the importance of the single-stranded polypyrimidine residues of coxsackievirus stem-loop IV RNA, the poly(C)-loop and the poly(Py)-bulge were separately mutated to poly(G) sequences (Fig. 4A). Mutated stem-loop IV RNAs were then assayed for binding to PCBP2 and, subsequently, for the ability to mediate IRES-dependent translation. The poly(G)-loop was constructed by mutating the four cytidines in the loop to four guanosines. The poly(G)-bulge was constructed by mutating the three cytidines in the bulge and one adjacent cytidine in the helical domain to guanosines.
Figure 4. Mobility shift assay of poly(G) substituted coxsackievirus stem-loop IV RNA with PCBP2.


A.M-fold predicted secondary structure of wild type, poly(G)-loop and poly(G)- bulge substituted coxsackievirus stem-loop IV RNAs. Wild type sequences corresponding to the poly(C)-loop or poly(C)-bulge are boxed with solid lines while mutated sequences are boxed with dashed lines. B. In vitro transcribed 32P-labeled wild type (lanes 1–5), poly(G)-bulge (lanes 6–10), and poly(G)-loop (lanes 11–15) coxsackievirus stem-loop IV RNAs (1 nM) were incubated with increasing amounts of purified recombinant PCBP2 (250–750 nM) for 10 min at 30°C. The reactions were then resolved by native polyacrylamide gel electrophoresis. Lanes 1, 6, and 11 are the RNA alone. RNP complexes and free probe RNAs are denoted by arrows. Competition mobility shift assays were carried out with 100 nM unlabeled poliovirus stem-loop IV RNA (lanes 5, 10, and 15).
The results of mobility shift assays of PCBP2 with the poly(G)-substituted coxsackievirus stem-loop IV RNAs are displayed in Fig. 4B. When increasing amounts of PCBP2 were added to wild type coxsackievirus stem-loop IV RNA, there was a concomitant dose-dependent increase in RNP complex formation and disappearance of free probe, indicating a stable interaction (Fig. 4B, lanes 2–4). As noted above, we used high concentrations of PCBP2 (250–750 nM) because 50 to 100 nM PCBP2 did not result in the formation of discrete RNP complexes (Fig. 2A). When the same concentrations of PCBP2 were incubated with the poly(G)-bulge coxsackievirus stem-loop IV RNA, no RNP complexes could be detected, even at the highest concentration (Fig. 4B, lanes 7–9). When increasing amounts of PCBP2 were added to the poly(G)-loop coxsackievirus stem-loop IV RNA, a dose-dependent RNA-protein interaction was observed. The apparent affinity is lower than that of wild type stem-loop IV, since the signal for the RNP complex was not as intense (compare Fig. 4B, lanes 2 and 12) and there was not a complete disappearance of free probe as observed for the wild type RNA (Fig. 4B, lanes 12–14). The mobility shift assay shows that the poly(G) nucleotide substitutions in coxsackievirus stem-loop IV RNA reduced binding of PCBP2 to different extents. The poly(G)-loop reduced PCBP2 binding only slightly, whereas the poly(G)-bulge mutations completely abrogated binding to PCBP2, even though the poly(C)-loop is still intact in this second mutated construct. It is possible that the poly(G) nucleotide substitutions generated near the GNRA tetra-loop (refer to Fig. 4A) cause destabilization of the RNA.
Poly(G) substitutions in coxsackievirus stem-loop IV RNA cause impaired translation in vitro
The mobility shift assays indicated that the poly(C) sequences in coxsackievirus stem-loop IV RNA are important for binding to PCBP2. Previously, it was demonstrated that for poliovirus, cap-independent translation was abrogated when poliovirus stem-loop IV RNA failed to interact with PCBP2 (Blyn et al., 1996; Gamarnik and Andino, 2000). To examine the effects of the poly(G) substitutions on coxsackievirus IRES-mediated translation, we carried out in vitro translation assays using a reporter RNA in HeLa cell cytoplasmic extracts. We engineered the poly(G) nucleotide substitutions into a coxsackievirus 5′ NCR-luciferase reporter RNA in which translation of the luciferase protein is driven by the coxsackievirus IRES. Both poliovirus and wild type coxsackievirus 5′ NCR-luciferase reporter RNAs were efficiently translated in HeLa cell cytoplasmic extract (Fig. 5). Translation of the reporter RNAs containing the poly(G) nucleotide substitutions was greatly reduced compared to wild type, at ~10% and ~23% for the poly(G)-bulge and poly(G)-loop, respectively. The in vitro translation data show that the poly(G)-bulge nucleotide substitutions have a more pronounced inhibition of coxsackievirus IRES-mediated translation than the poly(G)-loop substitutions, a trend that is consistent with our mobility shift assay data. Although the mobility shift assays showed a nearly complete lack of PCBP2 binding for the poly(G)-bulge stem-loop IV RNA, these substitutions still mediated translation at levels 10% of wild type RNA, suggesting that a lack of PCBP2 interaction at this site does not completely abrogate IRES-mediated translation initiation.
Figure 5. Luciferase assay of coxsackievirus IRES with poly(G) substitutions in stem-loop IV.
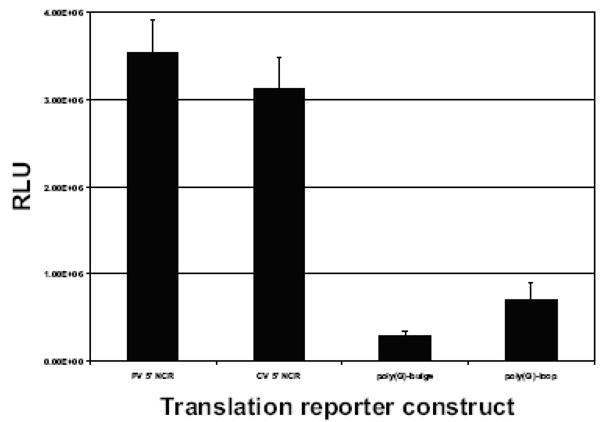
A.Luciferase reporter RNAs (50 fmol) containing the poliovirus 5′ NCR, coxsackievirus 5′ NCR, poly(G)-loop coxsackievirus 5′NCR, or poly(G)-bulge coxsackievirus 5′ NCR were incubated in HeLa S10 cytoplasmic extracts for 2.5 h at 30°C and then assayed for luciferase. The relative light unit (RLU) values are averages from three separate experiments.
Effects of stem-loop IV poly(G) substitutions on coxsackievirus infectivity and growth properties
As a functional complement to our in vitro analyses of PCBP2 interaction with coxsackievirus stem-loop IV RNAs, we used RNA transfections of HeLa cells to determine if full-length viral RNAs containing either of the two poly(G) substitutions in stem-loop IV were infectious. Wild type and mutated RNAs were transcribed in vitro, serially diluted, and then transfected into HeLa cell monolayers. Both mutated RNAs were capable of producing virus, and plaques were visible by 3 days post-transfection (Fig. 6A). Coxsackievirus RNAs containing the poly(G)-loop mutated stem-loop IV formed plaques with a specific infectivity approximately one-half of that observed for wild type RNAs (data not shown). In addition, the plaque sizes for this mutant were slightly smaller than those of wild type (refer to Fig. 6A). The poly(G)-bulge substituted stem-loop IV mutant formed plaques at a specific infectivity of less than one-tenth of that of wild type RNA (data not shown), and the plaque sizes were much smaller than those of wild type (Fig. 6A). Virus stocks generated from plaque isolates of these mutants (and wild type CVB3) were used to infect HeLa cells in single cycle growth experiments. As shown in Fig. 6B, the growth of both mutant viruses was delayed markedly compared to wild type virus, with titers reduced by more than one log10 unit at 4 h post-infection for the poly(G)-loop mutant and by nearly two log10 units for the poly(G)-bulge mutant. By eight h post-infection, both of the mutants had reached titers that were within one log10 unit of wild type coxsackievirus, suggesting that synthesis of viral proteins eventually achieved sufficient levels to produce near wild type titers of virus. Genomic RNAs recovered following infections of HeLa cells with these mutant viruses showed that the stem-loop IV targeted mutations were intact, and no additional nucleotide changes were detected in the 5′ NCRs of the mutant viruses (data not shown). The smaller plaque sizes and reduced virus growth kinetics observed for coxsackievirus RNAs containing the different poly(G) mutated stem-loop IV sequences correlated with the lower in vitro IRES-mediated translation efficiency and reduced PCBP2 binding that these mutations conferred.
Figure 6. Growth properties of coxsackievirus mutants with poly(G) substitutions in stem-loop IV RNA.

A.Plaque size morphology. In vitro transcribed full-length wild type, poly(G)-loop, or poly(G)-bulge mutated stem-loop IV coxsackievirus RNAs were transfected into HeLa cell monolayers. Readily visible plaques formed at 3 days post-transfection. Wild type coxsackievirus RNA produced large plaques, while the poly(G) mutated stem-loop coxsackievirus RNAs produced smaller plaques. B. Single cycle growth analysis of coxsackievirus mutants with poly(rG) nucleotide substitutions in stem-loop IV. HeLa cell monolayers were infected with wild type -◆-, poly(G)-loop -■-, or poly(G)-bulge -▲-mutated coxsackieviruses at a multiplicity of infection (MOI) of 10. The infected monolayers were harvested at the indicated times after infection and the virus titers were determined by plaque assay on HeLa cells as described in Material and Methods. The virus titers are reported as the log10 PFU (plaque forming units) per milliliter (ml).
Discussion
Immediately after uncoating in the cytoplasm, secondary structures that form in the 5′ NCR of coxsackievirus RNA recruit both canonical and non-canonical translation factors to mediate cap-independent translation. By hijacking cellular components for their own gene expression, picornaviruses exploit an alternative mechanism of internal initiation of translation that already exists in the cell. The interaction of IRES trans-acting factors (ITAFs) with RNA secondary structures has been shown to stimulate picornavirus IRES-mediated translation. Several RNA-binding proteins have been identified as ITAFs, including PCBP2, the La autoantigen, unr (upstream of N-Ras), and polypyrimidine tract binding protein (PTB) (Blyn et al., 1996; Meerovitch et al., 1993; Hunt et al., 1999; Jang and Wimmer, 1990; Hellen et al., 1993; Borman et al., 1993). The interaction of ITAFs with IRES elements may provide a functional equivalent to eIF4F binding to a 5′ cap structure, which picornaviruses lack, as well as an RNA chaperone-like function to stabilize IRES structures (Song et al., 2005).
Using dicistronic constructs in rabbit reticulocyte lysates supplemented with HeLa cell extract, Yang and colleagues determined that the 5′ NCR of coxsackievirus contains an internal ribosomal entry site for translation (Yang et al., 1997). In a subsequent study, the same group used coxsackievirus 5′ NCR deletion mutations to map the IRES to nucleotides ~430–640, encompassing several predicted stem-loop structures (Liu et al., 1999). Based on the work of Semler and colleagues, we predicted that the secondary structures of enterovirus 5′ NCRs are conserved in both form and function (Semler et al., 1986; Johnson and Semler, 1988). For poliovirus, the IRES sequence was reported to be between nucleotides ~130 and 600, encompassing all or part of stem-loops II-VI (Pelletier and Sonenberg, 1988; Skinner et al., 1989; Nicholson et al., 1991), indicating that the 5′ border is ~300 nucleotides upstream of that previously reported for CVB3. Stem-loop IV of CVB3 RNA has been shown to be contained within nucleotides ~240–445 (Bailey and Tapprich, 2007), suggesting that the coxsackievirus IRES encompasses sequences beyond those originally reported (Liu et al., 1999). In this report, we presented evidence that CVB3 stem-loop IV is a major cis-acting element important for coxsackievirus IRES-mediated translation.
Enzymatic structure probing with strand-specific RNases and m-fold computer modeling indicated that sequences in the coxsackievirus 5′ NCR, corresponding to poliovirus stem-loop IV, form a secondary structure similar to poliovirus stem-loop IV. Overall, the structure of coxsackievirus stem-loop IV forms a cruciform with three hairpin loops and a GNRA tetra-loop at the apex. Like poliovirus, coxsackievirus stem-loop IV RNA interacts with PCBP2, albeit with a lower affinity. Our RNase foot-printing data showed that PCBP2 protects three major regions of coxsackievirus stem-loop IV RNA from RNase digestion: the poly(C)-loop, the stem adjacent to the large internal bulge, and the region adjacent to the poly(Py)-bulge. Although there was not a clear PCBP2 protection due to multiple pyrimidine nucleotides that form the poly(Py)-bulge, we predict that the bulge motif is interacting with PCBP2 and thus protecting the adjacent regions from RNase digestion. The sequences of coxsackievirus stem-loop IV RNA that were protected by PCBP2 from RNase digestion were in the same general domains as those previously identified for poliovirus stem-loop IV (Gamarnik and Andino, 2000). Although our RNase footprinting data showed that multiple regions of the RNA are protected, several features of the PCBP2/stem-loop IV interaction make it difficult to determine the specific RNA:protein stoichiometry, including multiple RNA binding domains on the protein and multiple PCBP2 binding sites on the RNA. In mobility shift assays, when increasing amounts of PCBP2 (250–750 nM) were incubated with coxsackievirus stem-loop IV RNA, three complexes formed with slightly different electrophoretic mobilities (refer to Fig. 2A). We suspect that the formation of the slower migrating complex results from PCBP2 dimerization, rather than saturating RNA-protein contacts because at 250 nM PCBP2 there was an excess ratio of protein to RNA (2500:1). Ultimately, co-crystallization of stem-loop IV RNA and PCBP2 will be required to further illuminate the nature of this interaction.
When the poly(C)-loop was mutated to a poly(G)-loop in coxsackievirus stem-loop IV RNA, the interaction with PCBP2 was somewhat reduced; however, when the poly(Py)-bulge was mutated to poly(G), the interaction was almost completely abolished. The finding that the poly(G)-bulge dramatically inhibited interaction with PCBP2 is in agreement with previous studies on poliovirus stem-loop IV. A nucleotide mutation at position 325 or a five nucleotide substitution starting at position 332 in poliovirus stem-loop IV RNA inhibited both interaction with PCBP2 and IRES-mediated translation (Blyn et al., 1996; Gamarnik and Andino, 2000). NMR structure studies of the poly(C)-bulge of poliovirus stem-loop IV RNA show that when the cytidine at position 334 is changed to a uridine, the bulge becomes less flexible (when compared to wild type stem-loop IV) and adopts a U-shape instead of an L-shape (Du et al., 2004). We speculate that generating mutations in a region directly adjacent to the GNRA tetra-loop disrupts the overall RNA secondary structure, and not just the structure of the targeted motif.
When the two poly(G) nucleotide mutations were individually engineered into a coxsackievirus 5′ NCR-luciferase reporter RNA, IRES-mediated translation was greatly reduced, supporting previous evidence for poliovirus that interaction of stem-loop IV with PCBP2 stimulates IRES-mediated translation. Poliovirus stem-loop IV RNA contains two poly(C) sequences that interact with PCBP2 (Gamarnik and Andino, 2000). Similar to its poliovirus counterpart, coxsackievirus stem-loop IV RNA also presents two single-stranded sequences that may interact with PCBP2. The utilization of PCBPs to mediate important functions in the enterovirus life cycle results from the multi-functional properties of these proteins during interactions with both nucleic acids and proteins.
Previous studies have shown that sequence integrity of the picornavirus IRES is required for successful infection. Alterations to nucleotide sequence or secondary structure may have detrimental effects on protein binding and thus adversely affect translation and pathogenesis. A single point mutation in domain V of the poliovirus IRES was shown to attenuate the virus for poliomyelitis (Evans et al., 1985; Kawamura et al., 1989). It was shown that growth of the Sabin strain (which harbors this mutation) was restricted in neuronal cell lines (Agol et al., 1989; La Monica and Racaniello, 1989). For coxsackievirus, stem-loop II of the 5′ NCR is a major determinant for cardiovirulence (Dunn et al., 2003). In addition, a point mutation at position 234 of the 5′ NCR attenuates the virus for cardiovirulence, even though the virus harboring this mutation replicated to the same level as the cardiovirulent strain in cardiomyocytes (Tu et al., 1995). By exchanging the cardiovirulence domain of coxsackievirus (located in the 5′ NCR) with the corresponding region of poliovirus RNA, Chapman and colleagues showed that the chimeric virus displayed replication defects in tissue culture (Chapman et al., 2000). When mice were inoculated with this chimeric virus, it provided protection against heart and pancreatic lesions when challenged with a pathogenic strain.
We demonstrated that when the poly(C) motifs of coxsackievirus stem-loop IV were mutated to poly(G) in full-length cDNAs, the specific infectivities of the resulting RNA transcripts were reduced. The recovered mutant viruses had small-plaque phenotypes and significantly slower growth kinetics compared to wild type CVB3. Given the almost complete lack of binding of PCBP2 to mutated stem-loop IV RNA and the ~10-fold reduction in translation activity, the recovery of infectious virus harboring the poly(G)-bulge substitution was somewhat surprising. Since the engineered mutation was still present in recovered virus and no additional mutations were found in the 5′ NCR, we assume that other canonical or non-canonical factors in the cytoplasm of HeLa cells are able to substitute for a putative chaperone-like function of PCBP2 required for CVB3 translation, although at a much-reduced level of biological efficiency. Such factors may contribute to the cardiac- and pancreas-specific disease phenotype of CVB3. As such, it will be important to study any putative cell-specific defects of these mutant viruses in human coronary artery endothelial cells, primary murine heart fibroblasts, and human pancreas cells. These cells are natural targets of coxsackie B virus infections, and they may provide an important starting point in evaluating these mutant coxsackieviruses as potential vaccine candidates (Chapman et al., 2000).
Materials and Methods
Plasmid design and in vitro transcription
A plasmid harboring cDNA corresponding to the putative coxsackievirus stem-loop IV sequences was generated by PCR amplification of DNA sequences (235–430), corresponding to poliovirus stem-loop IV, from a coxsackievirus 5′ NCR-luciferase construct and cloning into a pGEM4Z vector (Walter et al., 1999). Linearization by EcoRI digestion followed by in vitro transcription of the plasmid generated a coxsackievirus stem-loop IV transcript that is 205 nucleotides in length. Radio-labeled coxsackievirus stem-loop IV RNA was generated by in vitro T7 polymerase (New England Biolabs) transcription using α-32P UTP (GE Health Sciences). After transcription the RNA was purified by Chromaspin chromatography (Clontech), phenol/chloroform extraction, and ethanol precipitation.
5′ end-labeling of coxsackievirus RNAs
Coxsackievirus stem-loop IV RNA was transcribed in vitro using T7 polymerase (Ambion). The 5′ phosphate of the RNA was removed using calf intestine phosphatase (New England Biolabs). Coxsackievirus stem-loop IV RNA was end-labeled with γ-32P-ATP using T4 polynucleotide kinase (New England Biolabs). The reaction products were then resolved on an 8% polyacrylamide gel in the presence of urea, and the RNA transcript was excised from the gel and eluted with elution buffer (500 mM NH4OAc, 1 mM EDTA and 0.1% SDS). The eluted RNA was subjected to phenol/chloroform extraction and ethanol precipitation.
PCBP2 purification
A pET22-PCBP2 plasmid was transformed into E. coli BL-21(DE3) cells. One liter of M9 media was inoculated with 1 ml of overnight culture. The cells were grown at 37°C until the culture reached an absorbance of OD600= 0.2. Protein expression was induced by addition of 1 mM IPTG. The bacteria were grown for an additional 3 h at 18°C and then collected by centrifugation. The bacterial pellet was resuspended in 20 ml of phosphate buffer (50 mM phosphate pH 7.0, 5% glycerol, and 300 mM NaCl) and lysed by sonication. The supernatant was adjusted to 20% ammonium sulfate and centrifuged to pellet the protein precipitate. The ammonium sulfate precipitate was resuspended in 10 ml of phosphate buffer and dialyzed overnight against 1 L of phosphate buffer. The protein was batch bound in 1 ml bed volume of Ni2+ Sepharose resin (Amersham). The resin was loaded onto a column and washed with 50 ml of 20 mM imidazole-phosphate buffer and eluted with 200 mM imidazole-phosphate buffer. The protein was dialyzed in a modified initiation factor buffer (5 mM Tris-Cl, 25 mM KCl, 0.5 mM DTT, 0.05 mM EDTA, and 5% glycerol) prior to use in mobility shift assays or RNase footprinting assays.
HeLa cell S10 preparation
HeLa S10 cytoplasmic extracts were prepared with slight modifications following a published protocol (Barton and Flanegan, 1993). Briefly, 6 liters of suspension HeLa cells were collected by centrifugation and resuspended in wash buffer (35 mM HEPES pH 7.4, 146 mM NaCl, and 11 mM glucose). After centrifugation, the HeLa cell pellet was then resuspended in hypotonic buffer (20 mM HEPES pH 7.4, 10 mM KCl, 1.5 mM MgOAc, and 1 mM DTT) and allowed to swell for 20 min on ice. Cells were lysed by Dounce homogenization, and the extract was adjusted by adding 1/10 of the extract volume of post-lysis buffer (20 mM HEPES pH 7.4, 120 mM KOAc, 4 mM MgOAc, and 5 mM DTT). The extract was then subjected to centrifugation at 10,000 × g for 20 min. The supernatant (S10) was then adjusted to 1 mM CaCl2 and 20 μg/ml micrococcal nuclease, and the mixture was incubated for 15 min at 14°C, followed by addition of EGTA to a final concentration of 4 mM. The extract was stored in liquid N2.
Electrophoretic mobility shift assays
PCBP2 proteins were pre-incubated (at a final concentration of 50 or 100 nM) with BSA (1mg/ml), yeast tRNA (0.8 mg/ml) and binding buffer (5 mM HEPES pH 7.5, 25 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, and 4% glycerol) in a volume of 9 μl for 10 min at 30°C. Then 1 μl of radio-labeled coxsackievirus stem-loop IV RNA (at a final concentration of 0.1 nM or 1.0 nM) was added, and the reaction was incubated for an additional 10 min at 30°C. The reaction mixture was adjusted to 10% glycerol and resolved by native polyacrylamide (4%) gel electrophoresis in the presence of glycerol.
In vitro translation and luciferase assays
Translation reactions were carried out as described (Walter et al., 1999). Briefly, the in vitro translation mixtures consisted of 60% HeLa S10 cytoplasmic extract, 1 μl of all-4 mix (1 mM ATP, 250 μM CTP, 250 μM GTP, and 250 μM UTP, 16 mM HEPES pH 7.4, 60 mM KOAc, 30 mM creatine-phosphate, and 400 μg creatine kinase), 50 fmol of PV 5′ NCR-luciferase RNA or CVB 5′ NCR-luciferase RNA in a total volume of 10 μl. The reactions were incubated for 2.5 h at 30°C. Translation was stopped by addition of 10 μl of passive lysis buffer (Promega). Translation efficiency was determined by measuring the light emission of 10 μl of the total in vitro translation reaction in 50 μl of luciferin substrate (Promega) in a luminometer (Berthold).
Enzymatic RNA structure probing and RNase foot-printing
The RNases used to carry out the enzymatic structure probing were from Ambion. The dilutions of RNase used were 1:1 and 1:10 from stock. A total of 25–50 ng of end-labeled coxsackievirus stem-loop IV RNA (5×105-1×106 cpm), 8 μg yeast tRNA, and 1X structure probing buffer (Ambion) in a final volume of 9 μl was heated to 68°C for 90 seconds to denature the RNA, and then cooled to 37°C for 2 minutes, and 25°C for 2 minutes to renature the RNA. Then 1 μl of RNase from stock (1:1 or a 1:10 dilution) was added. The 1:1 RNase dilution reactions were incubated for 1 min at 25°C while the reactions with the 1:10 dilutions of RNase were incubated for 3 minutes. For RNase foot-printing experiments, PCBP2 was added (at a final concentration of 5 μM) after the renaturation step and incubated for 10 min at 30°C. Then the reaction was subjected to the RNase digestion. For alkaline hydrolysis to generate an RNA ladder, a total of 25–50 ng of end-labeled coxsackievirus stem-loop IV RNA (5×105-1×106 cpm), 8 μg yeast tRNA, and 1X alkaline hydrolysis buffer (200 mM NaHCO3) was heated to 95°C for 4–6 minutes. The reactions were then cooled on ice to stop hydrolysis.
RNA transfections
In vitro transcribed wild type and poly(G)-substituted stem-loop IV coxsackievirus RNAs (Ambion) were transfected into HeLa cell monolayers in 60-mm plates using 1 mg/ml DEAE-dextran and TS buffer (137 mM NaCl, 4.4 mM KCl, 0.7 mM Na2HPO4-pH 7.45, 25 mM Tris, 0.5 mM MgCl2, and 0.68 mM CaCl2) for 30 minutes. The transfected cells were overlaid with a semi-solid mixture of MEM containing 8% newborn calf serum and 0.45% agarose. Transfected cells were incubated at 37°C for 3 days and then fixed with trichloroacetic acid (10%) and stained with crystal violet for plaque analysis.
Single cycle growth analysis of mutant coxsackieviruses
HeLa cell monolayers were infected with passage two stocks of wild type, poly(G)-loop, or poly(G)-bulge mutant coxsackieviruses at a multiplicity of infection (MOI) of 10. The virus stock was generated by RNA transfection experiments described above. At 2, 4, 6, or 8 hours post-infection, the infected monolayers and supernatants were harvested and the virus was released by five cycles of freeze-thaw. The virus preparation was subjected to centrifugation to pellet the cell debris. Virus growth was determined by plaque assay on HeLa (R19) cell monolayers. Briefly, the virus preparation was serially diluted 10-fold and adsorbed on cell monolayers for 30 minutes at room temperature. The cells were then overlaid with 0.45% agarose in MEM with 8% newborn calf serum and incubated at 37°C for 3 days. Cells were fixed with trichloroacetic acid (10%) and stained with crystal violet to visualize plaques.
Acknowledgments
We are grateful to Gwendolyn M. Jang for critical experimental discussions and Kerry D. Fitzgerald and Sarah Daijogo for helpful suggestions on the manuscript. We also thank Tara Crabb and Richard Virgen for their assistance in the structure probing experiments. This work was supported by Public Health Service grant AI 26765 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agol VI, Drozdov SG, Ivannikova TA, Kolesnikova MS, Korolev MB, Tolskaya EA. Restricted growth of attenuated poliovirus strains in cultured cells of a human neuroblastoma. J Virol. 1989;63:4034–4038. doi: 10.1128/jvi.63.9.4034-4038.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino R, Rieckhof GE, Baltimore D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell. 1990;63:369–380. doi: 10.1016/0092-8674(90)90170-j. [DOI] [PubMed] [Google Scholar]
- Antao VP, Lai SY, Tinoco I., Jr A thermodynamic study of unusually stable RNA and DNA hairpins. Nucleic Acids Res. 1991;19:5901–5905. doi: 10.1093/nar/19.21.5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JM, Tapprich WE. Structure of the 5′ nontranslated region of the coxsackievirus b3 genome: Chemical modification and comparative sequence analysis. J Virol. 2007;81:650–668. doi: 10.1128/JVI.01327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, Flanegan JB. Coupled translation and replication of poliovirus RNA in vitro: synthesis of functional 3D polymerase and infectious virus. J Virol. 1993;67:822–831. doi: 10.1128/jvi.67.2.822-831.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, O’Donnell BJ, Flanegan JB. 5′ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J. 2001;20:1439–1448. doi: 10.1093/emboj/20.6.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck MA, Chapman NM, McManus BM, Mullican JC, Tracy S. Secondary enterovirus infection in the murine model of myocarditis. Pathologic and immunologic aspects. Am J Pathol. 1990;136:669–681. [PMC free article] [PubMed] [Google Scholar]
- Bedard KM, Daijogo S, Semler BL. A nucleo–cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 2007;26:459–467. doi: 10.1038/sj.emboj.7601494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard KM, Walter BL, Semler BL. Multimerization of poly(rC) binding protein 2 is required for translation initiation mediated by a viral IRES. RNA. 2004;10:1266–1276. doi: 10.1261/rna.7070304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell YC, Semler BL, Ehrenfeld E. Requirements for RNA replication of a poliovirus replicon by coxsackievirus B3 RNA polymerase. J Virol. 1999;73:9413–9421. doi: 10.1128/jvi.73.11.9413-9421.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- Blyn LB, Chen R, Semler BL, Ehrenfeld E. Host cell proteins binding to domain IV of the 5′ noncoding region of poliovirus RNA. J Virol. 1995;69:4381–4389. doi: 10.1128/jvi.69.7.4381-4389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn LB, Swiderek KM, Richards O, Stahl DC, Semler BL, Ehrenfeld E. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5′ noncoding region: identification by automated liquid chromatography-tandem mass spectrometry. Proc Natl Acad Sci U S A. 1996;93:11115–11120. doi: 10.1073/pnas.93.20.11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn LB, Towner JS, Semler BL, Ehrenfeld E. Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J Virol. 1997;71:6243–6246. doi: 10.1128/jvi.71.8.6243-6246.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borman A, Howell MT, Patton JG, Jackson RJ. The involvement of a spliceosome component in internal initiation of human rhinovirus RNA translation. J Gen Virol. 1993;74:1775–1788. doi: 10.1099/0022-1317-74-9-1775. [DOI] [PubMed] [Google Scholar]
- Borman AM, Le MP, Girard M, Kean KM. Comparison of picornaviral IRES-driven internal initiation of translation in cultured cells of different origins. Nucleic Acids Res. 1997;25:925–932. doi: 10.1093/nar/25.5.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Ragland A, Leser JS, Hofling K, Willian S, Semler BL, Tracy S. A group B coxsackievirus/poliovirus 5′ nontranslated region chimera can act as an attenuated vaccine strain in mice. J Virol. 2000;74:4047–4056. doi: 10.1128/jvi.74.9.4047-4056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Tu Z, Tracy S, Gauntt CJ. An infectious cDNA copy of the genome of a non-cardiovirulent coxsackievirus B3 strain: its complete sequence analysis and comparison to the genomes of cardiovirulent coxsackieviruses. Arch Virol. 1994;135:115–130. doi: 10.1007/BF01309769. [DOI] [PubMed] [Google Scholar]
- Crowell RL, Landau BJ. A short history and introductory background on the coxsackieviruses of group B. Curr Top Microbiol Immunol. 1997;223:1–11. doi: 10.1007/978-3-642-60687-8_1. [DOI] [PubMed] [Google Scholar]
- Dejgaard K, Leffers H. Characterisation of the nucleic-acid-binding activity of KH domains. Different properties of different domains. Eur J Biochem. 1996;241:425–431. doi: 10.1111/j.1432-1033.1996.00425.x. [DOI] [PubMed] [Google Scholar]
- Du Z, Yu J, Chen Y, Andino R, James TL. Specific recognition of the C-rich strand of human telomeric DNA and the RNA template of human telomerase by the first KH domain of human poly(C)-binding protein-2. J Biol Chem. 2004;279:48126–48134. doi: 10.1074/jbc.M405371200. [DOI] [PubMed] [Google Scholar]
- Du ZH, Lee JK, Tjhen R, Li S, Pan H, Stroud RM, James TL. Crystal structure of the first KH domain of human poly(C)-binding protein-2 in complex with a C-rich strand of human telomeric DNA at 1.7 angstrom. J Biol Chem. 2005;280:38823–38830. doi: 10.1074/jbc.M508183200. [DOI] [PubMed] [Google Scholar]
- Dunn JJ, Bradrick SS, Chapman NM, Tracy SM, Romero JR. The stem loop II within the 5′ nontranslated region of clinical coxsackievirus B3 genomes determines cardiovirulence phenotype in a murine model. J Infect Dis. 2003;187:1552–1561. doi: 10.1086/374877. [DOI] [PubMed] [Google Scholar]
- Ehrenfeld E, Teterina NL. Initiation of translation of picornavirus RNAs: structure and function of the internal ribosome entry site. In: Semler BL, Wimmer E, editors. Molecular Biology of Picornaviruses. Washington, D.C.: ASM Press; 2002. pp. 159–169. [Google Scholar]
- Esfandiarei M, McManus BM. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol. 2008;3:127–155. doi: 10.1146/annurev.pathmechdis.3.121806.151534. [DOI] [PubMed] [Google Scholar]
- Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem. 1982;257:14806–14810. [PubMed] [Google Scholar]
- Evans DMA, Dunn G, Minor PD, Schild GC, Cann AJ, Stanway G, Almond JW, Currey K, Maizel JV. Increased Neurovirulence Associated with A Single Nucleotide Change in A Noncoding Region of the Sabin Type-3 Poliovaccine Genome. Nature. 1985;314:548–550. doi: 10.1038/314548a0. [DOI] [PubMed] [Google Scholar]
- Flanegan JB, Pettersson RF, Ambros V, Hewlett NJ, Baltimore D. Covalent linkage of a protein to a defined nucleotide sequence at the 5′-terminus of virion and replicative intermediate RNAs of poliovirus. Proc Natl Acad Sci U S A. 1977;74:961–965. doi: 10.1073/pnas.74.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke B, Zuleger B, Benavente R, Schuster T, Goller M, Stevenin J, Horak I. The mouse poly(C)-binding protein exists in multiple isoforms and interacts with several RNA-binding proteins. Nucleic Acids Res. 1996;24:3821–3828. doi: 10.1093/nar/24.19.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Two functional complexes formed by KH domain containing proteins with the 5′ noncoding region of poliovirus RNA. RNA. 1997;3:882–892. [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Interactions of viral protein 3CD and poly(rC) binding protein with the 5′ untranslated region of the poliovirus genome. J Virol. 2000;74:2219–2226. doi: 10.1128/jvi.74.5.2219-2226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CU, Witherell GW, Schmid M, Shin SH, Pestova TV, Gil A, Wimmer E. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc Natl Acad Sci U S A. 1993;90:7642–7646. doi: 10.1073/pnas.90.16.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J, Andino R. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol Cell. 2001;7:581–591. doi: 10.1016/S1097-2765(01)00205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SL, Hsuan JJ, Totty N, Jackson RJ. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 1999;13:437–448. doi: 10.1101/gad.13.4.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Kaminski A. Internal initiation of translation in eukaryotes: the picornavirus paradigm and beyond. RNA. 1995;1:985–1000. [PMC free article] [PubMed] [Google Scholar]
- Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol. 1988;62:2636–2643. doi: 10.1128/jvi.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SK, Wimmer E. Cap-independent translation of encephalomyocarditis virus RNA: structural elements of the internal ribosomal entry site and involvement of a cellular 57-kD RNA-binding protein. Genes Dev. 1990;4:1560–1572. doi: 10.1101/gad.4.9.1560. [DOI] [PubMed] [Google Scholar]
- Johnson VH, Semler BL. Defined recombinants of poliovirus and coxsackievirus: sequence-specific deletions and functional substitutions in the 5′-noncoding regions of viral RNAs. Virology. 1988;162:47–57. doi: 10.1016/0042-6822(88)90393-5. [DOI] [PubMed] [Google Scholar]
- Kawamura N, Kohara M, Abe S, Komatsu T, Tago K, Arita M, Nomoto A. Determinants in the 5′ noncoding region of poliovirus Sabin 1 RNA that influence the attenuation phenotype. J Virol. 1989;63:1302–1309. doi: 10.1128/jvi.63.3.1302-1309.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Hahm B, Kim YK, Choi M, Jang SK. Protein-protein interaction among hnRNPs shuttling between nucleus and cytoplasm. J Mol Biol. 2000;298:395–405. doi: 10.1006/jmbi.2000.3687. [DOI] [PubMed] [Google Scholar]
- Kim KS, Hufnagel G, Chapman NM, Tracy S. The group B coxsackieviruses and myocarditis. Rev Med Virol. 2001;11:355–368. doi: 10.1002/rmv.326. [DOI] [PubMed] [Google Scholar]
- Krausslich HG, Nicklin MJ, Toyoda H, Etchison D, Wimmer E. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J Virol. 1987;61:2711–2718. doi: 10.1128/jvi.61.9.2711-2718.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Monica N, Racaniello VR. Differences in replication of attenuated and neurovirulent polioviruses in human neuroblastoma cell line SH-SY5Y. J Virol. 1989;63:2357–2360. doi: 10.1128/jvi.63.5.2357-2360.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci U S A. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffers H, Dejgaard K, Celis JE. Characterisation of two major cellular poly(rC)-binding human proteins, each containing three K-homologous (KH) domains. Eur J Biochem. 1995;230:447–453. [PubMed] [Google Scholar]
- Lindberg AM, Stalhandske PO, Pettersson U. Genome of coxsackievirus B3. Virology. 1987;156:50–63. doi: 10.1016/0042-6822(87)90435-1. [DOI] [PubMed] [Google Scholar]
- Liu Z, Carthy CM, Cheung P, Bohunek L, Wilson JE, McManus BM, Yang D. Structural and functional analysis of the 5′ untranslated region of coxsackievirus B3 RNA: In vivo translational and infectivity studies of full-length mutants. Virology. 1999;265:206–217. doi: 10.1006/viro.1999.0048. [DOI] [PubMed] [Google Scholar]
- Makeyev AV, Liebhaber SA. The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA. 2002;8:265–278. doi: 10.1017/s1355838202024627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Meerovitch K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI, Keene JD, Sonenberg N. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J Virol. 1993;67:3798–3807. doi: 10.1128/jvi.67.7.3798-3807.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick WC. Overview: mechanism of translation initiation in eukaryotes. Enzyme. 1990;44:7–16. doi: 10.1159/000468743. [DOI] [PubMed] [Google Scholar]
- Murray KE, Roberts AW, Barton DJ. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA. 2001;7:1126–1141. doi: 10.1017/s1355838201010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KE, Steil BP, Roberts AW, Barton DJ. Replication of poliovirus RNA with complete internal ribosome entry site deletions. J Virol. 2004;78:1393–1402. doi: 10.1128/JVI.78.3.1393-1402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson R, Pelletier J, Le SY, Sonenberg N. Structural and functional analysis of the ribosome landing pad of poliovirus type 2: in vivo translation studies. J Virol. 1991;65:5886–5894. doi: 10.1128/jvi.65.11.5886-5894.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- Parsley TB, Towner JS, Blyn LB, Ehrenfeld E, Semler BL. Poly (rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA. 1997;3:1124–1134. [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- Perera R, Daijogo S, Walter BL, Nguyen JH, Semler BL. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J Virol. 2007;81:8919–8932. doi: 10.1128/JVI.01013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sean P, Nguyen JH, Semler BL. The linker domain of poly(rC) binding protein 2 is a major determinant in poliovirus cap-independent translation. Virology. 2008;378:243–253. doi: 10.1016/j.virol.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semler BL, Johnson VH, Tracy S. A chimeric plasmid from cDNA clones of poliovirus and coxsackievirus produces a recombinant virus that is temperature-sensitive. Proc Natl Acad Sci U S A. 1986;83:1777–1781. doi: 10.1073/pnas.83.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J Virol. 1995;69:3873–3877. doi: 10.1128/jvi.69.6.3873-3877.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993;21:1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MA, Racaniello VR, Dunn G, Cooper J, Minor PD, Almond JW. New model for the secondary structure of the 5′ non-coding RNA of poliovirus is supported by biochemical and genetic data that also show that RNA secondary structure is important in neurovirulence. J Mol Biol. 1989;207:379–392. doi: 10.1016/0022-2836(89)90261-1. [DOI] [PubMed] [Google Scholar]
- Song Y, Tzima E, Ochs K, Bassili G, Trusheim H, Linder M, Preissner KT, Niepmann M. Evidence for an RNA chaperone function of polypyrimidine tract-binding protein in picornavirus translation. RNA. 2005;11:1809–1824. doi: 10.1261/rna.7430405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SR, Semler BL. RNA structure adjacent to the attenuation determinant in the 5′-non- coding region influences poliovirus viability. Nucleic Acids Res. 1998;26:5318–5326. doi: 10.1093/nar/26.23.5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy S, Chapman NM, Tu Z. Coxsackievirus B3 from an infectious cDNA copy of the genome is cardiovirulent in mice. Arch Virol. 1992;122:399–409. doi: 10.1007/BF01317202. [DOI] [PubMed] [Google Scholar]
- Trono D, Andino R, Baltimore D. An RNA sequence of hundreds of nucleotides at the 5′ end of poliovirus RNA is involved in allowing viral protein synthesis. J Virol. 1988;62:2291–2299. doi: 10.1128/jvi.62.7.2291-2299.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z, Chapman NM, Hufnagel G, Tracy S, Romero JR, Barry WH, Zhao L, Currey K, Shapiro B. The cardiovirulent phenotype of coxsackievirus B3 is determined at a single site in the genomic 5′ nontranslated region. J Virol. 1995;69:4607–4618. doi: 10.1128/jvi.69.8.4607-4618.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter BL, Nguyen JH, Ehrenfeld E, Semler BL. Differential utilization of poly(rC) binding protein 2 in translation directed by picornavirus IRES elements. RNA. 1999;5:1570–1585. doi: 10.1017/s1355838299991483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter BL, Parsley TB, Ehrenfeld E, Semler BL. Distinct poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J Virol. 2002;76:12008–12022. doi: 10.1128/JVI.76.23.12008-12022.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Day N, Trifillis P, Kiledjian M. An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol Cell Biol. 1999;19:4552–4560. doi: 10.1128/mcb.19.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss IM, Liebhaber SA. Erythroid cell-specific determinants of alpha-globin mRNA stability. Mol Cell Biol. 1994;14:8123–8132. doi: 10.1128/mcb.14.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E, Hellen CU, Cao X. Genetics of poliovirus. Annu Rev Genet. 1993;27:353–436. doi: 10.1146/annurev.ge.27.120193.002033. [DOI] [PubMed] [Google Scholar]
- Yang D, Wilson JE, Anderson DR, Bohunek L, Cordeiro C, Kandolf R, McManus BM. In vitro mutational and inhibitory analysis of the cis-acting translational elements within the 5′ untranslated region of coxsackievirus B3: potential targets for antiviral action of antisense oligomers. Virology. 1997;228:63–73. doi: 10.1006/viro.1996.8366. [DOI] [PubMed] [Google Scholar]
- Zell R, Ihle Y, Seitz S, Gundel U, Wutzler P, Gorlach M. Poly(rC)-binding protein 2 interacts with the oligo(rC) tract of coxsackievirus B3. Biochem Biophys Res Commun. 2008;366:917–921. doi: 10.1016/j.bbrc.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]