

Abstract
This study was designed to investigate the expression and molecular signaling of cyclooxygenase-1 (COX-1) in cervical carcinomas. Real-time quantitative reverse transcription-polymerase chain reaction and Western blot analysis confirmed enhanced expression of COX-1 RNA, and protein in squamous cell carcinomas and adenocarcinoma of the cervix. COX-1 expression in all carcinoma tissues was associated with enhanced expression of COX-2 RNA and protein. The site of COX-1 expression was localized by immunohistochemistry to the neoplastic epithelial cells in all squamous cell carcinomas and adenocarcinomas studied. Minimal COX-1 immunoreactivity was detected in normal cervix. To explore events associated with COX-1 up-regulation, we developed a doxycycline-regulated expression system in HeLa (cervical carcinoma) cells. Overexpression of COX-1 in HeLa cells resulted in induced expression of cyclooxygenase-2 (COX-2) and prostaglandin E synthase (PGES) concomitant with increased prostaglandin E2 (PGE2) synthesis. Treatment of HeLa cells overexpressing COX-1 with the dual COX enzyme inhibitor indomethacin or selective COX-2 inhibitor NS-398 significantly reduced PGE2 synthesis. Indomethacin, but not NS-398, treatment abolished the up-regulation of expression of COX-2 and PGES in HeLa cells, suggesting that the observed up-regulation of COX-2 and PGES was mediated by COX-1-enzyme products. To assess whether enhanced PGE2 synthesis after COX-1 induction would act in an autocrine/paracrine manner, we investigated the effect of COX-1 on the expression of the different isoforms of PGE2 receptors (EP1-EP4). We found that the cAMP-linked PGE2 receptors were significantly up-regulated by COX-1 overexpression coincident with enhanced cAMP responsiveness of these cells to exogenous PGE2 ligand. Finally, overexpression of COX-1 was associated with enhanced expression of the angiogenic factors basic fibroblast growth factor, vascular endothelial growth factor, angiopoietin-1, and angiopoietin-2. This up-regulation of angiogenic factor expression was abolished by indomethacin and partially reduced by NS-398. These data indicate that COX-1 up-regulation modulates the expression of factors that may act in an autocrine/paracrine manner to enhance and sustain tumorigenesis in neoplastic cervical epithelial cells. It is likely that similar mechanisms may act in vivo to modulate tumorigenesis of cervical carcinomas.
INTRODUCTION
Uterine cervical cancer is considered an important clinical problem in developing countries, with a high incidence of invasive disease reported for South African women (1). Three histological categories of epithelial tumors of the cervix are recognized by the World Health Organization (2): squamous cell carcinoma, adenocarcinoma, and other less common types of epithelial tumors. The most common histological type of cervical carcinoma is squamous cell carcinoma, which accounts for 60-80% of all cervical cancers. Adenocarcinoma accounts for <20% of invasive cervical carcinomas. Numerous studies have demonstrated that epithelial tumors may be regulated by COX-2-enzyme products (3-7). Two distinct isoforms of the COX enzyme, COX-1 and COX-2, have been reported (8-10). The relative contributions of COX-1- and/or COX-2-derived products in mediating events associated with cervical neoplasia remain to be elucidated. COX-1 expression is considered to be constitutive and generates prostaglandins for normal physiological functions (4, 11, 12). Transcription of COX-2 RNA and protein is up-regulated in several epithelial carcinomas (3, 12-15), including carcinomas of the cervix (16-18). This has prompted the suggestion that the increased level of prostaglandins and other eicosanoids present in cancer tissue is a consequence of induced COX-2. More recently, however, it has been demonstrated that both COX isoforms are inducible. In some cell types, including pulmonary artery endothelial cells, COX-1 levels are induced during differentiation (19, 20). COX-1 expression can be induced in vitro by VEGF (21), arachidonic acid, forskolin, dibutyryl-cAMP, and PGE2 (22). In addition, elevated COX-1 expression has been reported in mouse lung tumors (23), human breast cancer (24), and human prostate carcinoma (25). These data suggest that both COX enzymes and/or their products may function in promoting and maintaining the neoplastic state. COX catalyzes the double oxygenation and reduction of arachidonic acid after its release from membrane glycerophospholipids by phospholipase A2 to the intermediate form prostaglandin H2. This intermediate serves as the substrate for terminal prostanoid synthases, which produce their specific prostaglandins such as PGE2, being synthesized by PGES (26-28). PGE2 has been shown to stimulate gene transcription (29), influence mitogenesis of normal human bone cells (30), and promote growth and metastasis of tumors (31). More recently, enhanced synthesis of PGE2 resulting from up-regulated COX-2 has been shown to induce malignant change in epithelial cells through immunosuppression (32), inhibiting apoptosis (13), increasing metastatic potential of epithelial cells (6), and promoting angiogenesis (33, 34). Two segregated biosynthetic pathways have been described for PGE2 biosynthesis. These pathways synthesize PGE2 via PGES functionally and preferentially coupled with either COX-1 or COX-2 (27). The biological actions of PGE2 have been attributed to its interaction with G-protein-coupled seven-transmembrane-domain receptors, which belong to the rhodopsin superfamily of serpentine receptors (35). Four main subtypes of PGE2 receptors have been identified (EP1, EP2, EP3, and EP4), which use alternate and, in some cases, opposing intracellular pathways (36). Most studies have focused on neoplastic events associated with COX enzyme products as a consequence of COX-2 overexpression. In this study, we investigated (a) COX-1 expression and localization in cervical squamous cell carcinomas and adenocarcinomas compared with normal cervical tissue, and (b) a possible autocrine/paracrine role for COX-1 enzyme products in regulating the expression of COX-2, PGES, PGE2 receptors, and angiogenic factors in cervical epithelial carcinoma cells using an inducible expression system.
MATERIALS AND METHODS
Materials
The following antibodies used for Western blotting were purchased from Santa Cruz Biotechnology (Autogenbioclear, Wiltshire, United Kingdom): COX-1 goat polyclonal (sc-1752), COX-2 goat polyclonal (sc-1745), bFGF goat polyclonal (sc-1360), VEGF rabbit polyclonal (sc-152), Ang-1 goat polyclonal (sc-6319), Ang-2 goat polyclonal (sc-7016), and β-actin goat polyclonal (sc-1616), as well as the COX-1 and COX-2 blocking peptides (sc-1752p and sc-1745p). The PGES antibody raised against the microsomal glutathione-dependent inducible PGES (27) was purchased from Caymen Chemical Co. (Cheshire, United Kingdom); antigoat-alkaline phosphatase, antirabbit-alkaline phosphatase, cloning cylinders, G418, Hyg, DOX, and indomethacin were purchased from Sigma Chemical Co. (Dorset, United Kingdom). Samples and synthetic standards for the PGE2 ELISA were purchased from Applied Therapeutics (Paisley, United Kingdom), and NS-398 was purchased from Calbiochem (Nottingham, United Kingdom). HeLa Tet-Off cells and Tet system-approved fetal bovine serum were purchased from Clontech (Hampshire, United Kingdom). DMEM nutrient mixture F-12 was purchased from Life Technologies, Inc. (Paisley, United Kingdom), and penicillin-streptomycin was purchased from PAA (PAA Laboratories Ltd., Middlesex, United Kingdom). ECF chemiluminescence system was purchased from Amersham Biosciences (Little Chalfont, Buckinghamshire, United Kingdom).
Tissue Collection and Processing
Cervical specimens were obtained at the time of surgery/biopsy from patients who were attending the Gynaecological Oncology Clinic at Groote Schuur Hospital, Cape Town and who had been diagnosed previously with invasive carcinoma of the cervix. Punch biopsies were taken from the lesion by an experienced gynecologist with a special interest in oncology. A portion of the biopsy was excised and fixed in formalin, followed by paraffin wax embedding for histopathological typing. The remaining portion was snapfrozen in either dry ice or liquid nitrogen and stored at −70°C for RT-PCR and Western blot analysis. The extent of invasiveness of carcinoma biopsies (C1–C58) is represented in Table 1. Histologically normal cervical samples (N1–N21) were obtained from patients undergoing Wertheims hysterectomy for nonmalignant conditions. Pathological typing was defined according to the International Federation of Obstetricians and Gynaecologists (37) staging upon physical examination. The ages of the patients ranged from 29 to 80 years with a median age of 50 years. The study was approved by the University of Cape Town Research Ethics Committee, and informed consent was obtained from all patients before tissue collection.
Table 1. Extent of invasiveness of cervical carcinoma biopsy samples of South African women.
| Sample no. | Histological typing | FIGO stagea |
|---|---|---|
| C10, C14; C28–C32 | Squamous carcinoma | 1B; well differentiated |
| C5–C9; C24–C27; C37–C47 | Squamous carcinoma | 2B; well differentiated |
| C1–C4; C19–C23 | Squamous carcinoma | 3B; well differentiated |
| C36 | Adenocarcinoma | 1B; moderately differentiated |
| C15–C18; C33–C35; C48–C58 | Adenocarcinoma | 2B; well differentiated |
FIGO, Fédération Internationales des Gynaecologistes et Obstetristes.
Cell Culture
HeLa Tet-Off cells containing the regulatory plasmid (pTet-Off) were routinely maintained in DMEM nutrient mixture F-12 with Glutamax-1 and pyridoxine, supplemented with 10% fetal bovine serum, 100 μg/ml G418, and 1% antibiotics (stock, 500 IU/ml penicillin and 500 μg/ml streptomycin) at 37°C and 5% CO2 (v/v).
Cell Transfections
The Tet-Off expression system we used was developed by Gossen et al. (38) to deliver doxycycline-regulated expression based on the high specificity of the Escherichia coli tet repressor-operator-doxycycline interaction. In the Tet-Off expression system each clonal cell line is used as its own control (cells cultured in the presence of DOX), and the overexpression of the integrated target gene is modulated solely by removing DOX from the culture medium. This eliminates the need for a control clonal cell line transfected with vector alone (as used with constitutive stable expression systems), thereby overcoming the inherent variation that arises from different sites of integration of DNA between different clones. HeLa Tet-Off cells containing the pTet-Off vector stably transfected and constitutively expressing the tetracycline-controlled transactivator tTA (composed of a fusion of the TetR and VP16 activation domain) were purchased from Clontech. The pBS(SK-)/PSHI cDNA containing the full-length COX-1 gene (kindly supplied by Dr. Stephen Prescott, University of Utah, Salt Lake City, UT) was used as the template plasmid. The response plasmid pTRE2 (containing the minimal cytomegalovirus promoter containing Tet-operator sequences cloned upstream of the cDNA to be expressed) and the plasmid for antibiotic selection (pTK-Hyg) for use with the Tet-Off system were purchased from Clontech. The COX-1 gene was excised from the template plasmid and ligated at the BamHI site of the pTRE2 vector. The orientation of the insert was verified by dideoxy DNA sequencing using the sequence- specific primers 5′-CGCCTGGAGACGCCATCC-3′ and 5′-CCACACCTCCCCCTGAAC-3′ (Clontech). Cells were plated in 12-well dishes in complete medium containing 100 μg/ml G418 per well and were allowed to attach and grow overnight. The pTRE2 vector containing the COX-1 gene (2 μg) was cotransfected with pTK-Hyg (0.1 μg, which contains the Hyg gene under control of the minimal TK promoter) into the HeLa Tet-Off cell line at about 80% confluency using pfx-5 (Invitrogen, De Schelp, Netherlands) diluted in Optimem (Life Technologies, Inc.). Cells were incubated for 4 h at 37°C in 5% humidified CO2. Thereafter, the medium was replaced with fresh complete medium containing no G418. Cells were allowed to grow for 72 h. Transfected cells were then seeded together with wild-type cells. Clones were selected against 200 μg/ml Hyg in the presence of 1 μg/ml DOX. At least 50 Hyg-resistant clones were picked using cloning cylinders. Clones were allowed to grow under continuous selection with Hyg in the presence of DOX and then screened for the ability to express COX-1 in the presence and absence of DOX by immunoblot analysis. Three clones with the greatest inducible overexpression of COX-1 (clones 1.2, 2.2, and 3.1) were selected for additional experiments. All clones were characterized and exhibited identical phenotypic and biochemical alterations. The results of our studies using the COX-1 clone 1.2 are presented here. Similar reproducible results were obtained using clones 2.2 and 3.1. Unless otherwise stated, all clones were maintained uninduced in 1 μg/ml DOX, 200 μg/ml Hyg, and 100 μg/ml G418. COX inhibition studies were conducted by growing cells in medium containing 3 μg/ml indomethacin or 10 μm NS-398.
Real-time Quantitative RT-PCR
Real-time quantitative RT-PCR was performed to determine COX-1 and COX-2 expression in cervical carcinoma biopsies and normal cervical tissue as well as to assess the effect of COX-1 overexpression on expression of the different isoforms of PGE2 receptors (EP1, EP2, EP3, and EP4) in HeLa Tet-Off cells. RNA samples were extracted from cervical tissue (squamous cell carcinomas, C1–C14; adenocarcinomas, C15–C18; and normal cervix, N1–N8) using Tri-Reagent (Sigma Chemical Co.) as per the manufacturer’s instruction. To determine the effect of COX-1 overexpression on expression of EP receptors, cells (2 × 105) were seeded in six-well plates, and allowed to attach and grow overnight in the presence of DOX. The following day, the cells were synchronized by incubating with serum-free medium for 24 h. Thereafter, the medium was replaced with fresh complete medium, and COX-1 overexpression was induced by growing cells in medium containing no DOX. Control cells were maintained in DOX. Cells were harvested after 24, 48, and 72 h with 1 ml/well Tri-Reagent (Sigma Chemical Co.) as per the manufacturer’s protocol. RNA samples were reverse transcribed using MgCl2 (5.5 mm), dNTPs (0.5 mm each), random hexamers (1.25 μm), oligodeoxythymidylic acid (1.25 μm), RNase inhibitor (0.4 unit/μl), and multiscribe reverse transcriptase (1.25 units/μl), all from PE Biosystems (Warrington, United Kingdom). The mix was aliquoted into individual tubes (16 μl/tube), and template RNA was added (4 μl/tube of 250 ng/μl RNA). Samples were incubated for 60 min at 25°C, 45 min at 48°C, and then 5 min at 95°C. A reaction mix was made containing Taqman buffer (5.5 mm MgCl2, 200 μm dATP, 200 μm dCTP, 200 μm dGTP, 400 μm dUTP); ribosomal 18S forward and reverse primers and probe (all at 50 nm); forward and reverse primers for COX-1, COX-2, EP1, EP2, EP3, or EP4 receptor (300 nm); COX-1, COX-2, EP1, EP2, EP3, or EP4 receptor probe (200 nm); AmpErase UNG (0.01 unit/μl); and AmpliTaq Gold DNA Polymerase (0.025 unit/μl), all from PE Biosystems. A volume of 48 μl of reaction mix was aliquoted into separate tubes for each cDNA sample and 2 μl/replicate of cDNA were added. After mixing, 23 μl of sample were added to the wells on a PCR plate. Each sample was added in duplicate. A no-template control (containing water) was included in triplicate. Wells were sealed with optical caps, and the PCR reaction was run on an ABI Prism 7700 using standard conditions. COX-1, COX-2, and EP receptor primers and probe for quantitative PCR were designed using the PRIMER express program (PE Biosystems). The sequences of the COX-1 primers and probe were as follows. Forward: 5′-TGT TCG GTG TCC AGT TCC AAT A-3′; reverse: 5′-ACC TTG AAG GAG TCA GGC ATG AG-3′; probe (FAM labeled): 5′-CGC AAC CGC ATT GCC ATG GAG T-3′. The sequences of the COX-2 primers and probe were as follows. Forward: 5′-CCT TCC TCC TGT GCC TGA TG-3′; reverse: 5′-ACA ATC TCA TTT GAA TCA GGA AGC T-3′; probe (FAM labeled): 5′-TGC CCG ACT CCC TTG GGT GTC A-3′. The sequences of the EP1 receptor primers and probe were as follows. Forward: 5′-AGA TGG TGG GCC AGC TTG T-3′; reverse: 5′-GCC ACC AAC ACC AGC ATT G-3′; probe (FAM labeled): 5′-CAG CAG ATG CAC GAC ACC ACC ATG-3′. The sequences of the EP2 receptor primers and probe were as follows. Forward: 5′-GAC CGC TTA CCT GCA GCT GTA C-3′; reverse: 5′-TGA AGT TGC AGG CGA GCA-3′; Probe (FAM labeled): 5′-CCA CCC TGC TGC TGC TTC TCA TTG TCT-3′. The sequences of the EP3 receptor primers and probe were as follows. Forward: 5′-GAC GGC CAT TCA GCT TAT GG-3′; reverse: 5′-TTG AAG ATC ATT TTC AAC ATC ATT ATC A-3′; probe (FAM labeled): 5′-CTG TCG GTC TGC TGG TCT CCG CTC-3′. The sequences of the EP4 receptor primers and probe were as follows. Forward: 5′-ACG CCG CCT ACT CCT ACA TG-3′; reverse: 5′-AGA GGA CGG TGG CGA GAA T-3′; probe (FAM labeled): 5′-ACG CGG GCT TCA GCT CCT TCC T-3′. The ribosomal 18S primers and probe sequences were as follows. Forward: 5′-CGG CTA CCA CAT CCA AGG AA-3′; reverse: 5′-GCT GGA ATT ACC GCG GCT-3′; probe (VIC labeled): 5′-TGC TGG CAC CAG ACT TGC CCT C-3′. Expression of COX-1 and EP receptors was normalized to RNA loading for each sample using the 18S rRNA as an internal standard. Relative COX-1 and COX-2 expression in carcinoma tissue was calculated by dividing the expression in carcinoma tissue by the expression in normal cervix. Relative expression of EP receptors was calculated, from three independent experiments, by dividing the expression in induced cells by the expression in uninduced cells. The data are presented as mean ± SE.
Protein Extraction
Tissue
COX-1 and COX-2 protein expression in cervical carcinomas and normal cervix was assessed by Western blotting. Proteins were extracted from cervical tissue (squamous cell carcinomas, C19-C32; adenocarcinomas, C33–C36; and normal cervix, N9-N16) by homogenization in protein lysis buffer (1% Triton X-100, 150 mm NaCl, 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 0.1% SDS containing 2 mm phenylmethylsulfonyl fluoride). Thereafter, insoluble material was pelleted by centrifugation at 14,000 × g for 20 min at 4°C. The clarified lysate was removed to a new tube for protein quantification and SDS-PAGE. The protein content in the supernatant fraction was determined using protein assay kits (Bio-Rad, Hemel Hempstead, United Kingdom). A total of 50 μg of protein was resuspended in 20 μl of sample buffer (125 mm Tris-HCl, pH 6.8, 4% SDS, 5% 2-mercaptoethanol, 20% glycerol, and 0.05% bromphenol blue), boiled for 5 min at 95°C, and run on a 10% SDS-polyacrylamide gel before Western blotting.
Cells
Cells were seeded in 5-cm dishes and allowed to attach overnight. The following day, the cells were synchronized by incubating with serum-free medium for 24 h. Thereafter, the medium was replaced with fresh complete medium, and the cells were grown in the presence or absence of DOX for 24, 48, and 72 h, respectively. In parallel, cells were cotreated with indomethacin or NS-398. Cells were harvested by lysing in protein lysis buffer (150 mm NaCl, 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1% Triton X-100, 0.1% SDS). The protein content in the supernatant fraction was determined as described above. The clarified cell lysates (20 μg) were denatured and electrophoresed on 4-20% Tris-glycine gels (NOVEX, Invitrogen).
Western Blotting
Immunoblot analysis was performed on supernatant fractions of cervical tissues and HeLa COX-1 Tet-Off cells. The proteins were transferred onto a PVDF membrane (Millipore, Watford, United Kingdom) and subjected to immunoblot analysis. Membranes were blocked for 1 h at 25°C in 5% skimmed milk powder diluted in TBS-Tween [50 mm Tris-HCl, 150 mm NaCl, and 0.05% (v/v) Tween 20]. Thereafter, membranes were incubated overnight with either COX-1 (1:500)-, COX-2 (1:500)-, β-actin (1:500)-, PGES (1:250), bFGF (1:500)-, VEGF (1:500)-, Ang-1 (1:250)-, or Ang-2 (1:250)-specific antibodies. After transfer, membranes were subsequently incubated for 1 h with rabbit antigoat secondary antibody (for COX-1/2, β-actin, Ang-1/2, and bFGF) at a dilution of 1:30,000 or goat antirabbit secondary antibody (PGES or VEGF) at a dilution of 1:30,000. Thereafter, membranes were washed in TBS-Tween and developed by the ECF chemiluminescence system following the manufacturer’s instructions. Proteins were revealed and quantified by PhosphorImager analysis using the STORM 860 system (Molecular Dynamics, Amersham Biosciences, Buckinghamshire, United Kingdom). Fold induction in induced cells was determined relative to uninduced cells, after normalizing to β-actin, by dividing the expression in induced cells by the expression in uninduced cells. The molecular weights of the respective proteins were determined from the relative mobility on SDS-PAGE compared with molecular weight standards. COX-1 and COX-2 negative controls for determination of antibody specificity were performed by incubating membranes with goat anti-COX-1/2 antibody preadsorbed to blocking peptide as per the manufacturer’s protocol. Data are presented as mean ± SE from four independent experiments.
Immunohistochemistry
The site of COX-1 expression was localized in cervical tissues by immunohistochemistry using archival cervical blocks (squamous cell carcinomas, C37–C47; adenocarcinomas, C48–C58; and normal cervix, N19–N23) obtained from the Department of Anatomical Pathology, University of Cape Town, South Africa. Five-micrometer paraffin wax-embedded tissue sections were cut and mounted onto coated slides (TESPA, Sigma Chemical Co.). Sections were dewaxed in xylene, rehydrated in graded ethanol, and washed in water followed by TBS (50 mm Tris-HCl, 150 mm NaCl, pH 7.4), and blocked for endogenous endoperoxidase (1% H2O2 in methanol). Antigen retrieval was performed by pressure cooking for 2 min in 0.01 m sodium citrate pH 6. Sections were blocked using 5% normal rabbit serum diluted in TBS. Subsequently the tissue sections were incubated with polyclonal goat anti-COX-1 antibody (sc-1752; Autogenbioclear) at a dilution of 1:200 at 4°C for 18 h. Control tissue was incubated with goat anti-COX-1 antibody preadsorbed to blocking peptide (sc-1752p; Autogenbioclear) as per the manufacturer’s protocol. After thorough washing with TBS, the tissue sections probed with the goat antihuman COX-1 primary antibody were incubated with biotinylated rabbit antigoat secondary IgG antibody (DAKO, Buckinghamshire, United Kingdom) at a dilution of 1:500 at 25°C for 40 min. Thereafter, the tissue sections were incubated with streptavidin-peroxidase complex (DAKO) at 25°C for 20 min. Color reaction was developed by incubation with 3,3′-diaminobenzidine (DAKO). The tissue sections were counterstained in aqueous hematoxylin, followed by sequential dehydration using graded ethanol and xylene, before mounting and coverslipping.
PGE2 Assay
HeLa COX-1 Tet-Off cells were seeded in 5-cm dishes at a cell density of 5 × 105 cells/dish and were allowed to grow and attach overnight. The following day, the cells were synchronized by incubating with serum-free medium for 24 h. COX-1 expression was induced for 24, 48, and 72 h, respectively, by DOX withdrawal from the culture medium, in the presence or absence of indomethacin or NS-398. Arachidonic acid to a final concentration of 5 μg/ml was added to the culture medium after induction for 6 h. Thereafter, 1 ml of medium was removed and added to 1 ml of methyloximating solution. Control uninduced cells were treated similarly but maintained with DOX supplemented daily. PGE2 secretion into the culture medium was assayed by ELISA (39). The ELISA was performed using 96-well plates (amine-binding plates; Costar, High Wycombe, United Kingdom) coated with donkey antirabbit antibody. Plates were then coated with rabbit IgG (1 mg/ml diluted in PBS with 1% carbonate buffer, pH 9.6) at 200 μl/well for 16 h at 4°C. The solution was aspirated, and blocking solution (50 mm glycine, 10 mg/ml BSA) was added at 25 μl/well for 2 h at 23°C. The plates were then washed, and donkey antirabbit serum (Scottish Antibody Production Unit, Carluke, United Kingdom) was added to a final volume of 150 μl/well, before washing, air drying, and storage with desiccant at 4°C. The link was prepared by ether extraction and reverse-phase chromatography using 20 mg of synthetic PGE2, 320 μl of dry dimethylformamide, 3 μl butylchloroformate, and 0.05 mm biocytin. Samples and synthetic standards were diluted in ELISA buffer (150 mm NaCl, 100 mm Tris-HCl, 0.05% Tween 20, 50 mm phenol red, 1 mm 2-methylisothiazolone, 1 mm bromonitrodioxane, 2 mm EDTA, 2 mg/ml BSA to a final pH of 7.2), and 100 μl of each were added in duplicate to the plate. The link was diluted 1:1.5 × 106 in ELISA buffer, and 50 μl were added to each well. Antisera, diluted 1:50,000 in ELISA buffer, were added to a final volume of 50 μl to all wells except those used for measuring nonspecific binding. Plates were incubated at 4°C for 16 h and washed, and 100 μl/well of 0.2 unit/ml streptavidin-peroxidase were added. Plates were then incubated for 20 min at 23°C on an orbital shaker and washed, and substrate (0.3 g/liter urea-hydrogen peroxide, 0.1 g/liter tetramethylbenzene in 100 mm sodium acetate, pH 6.0) was added to a final volume of 200 μl/well for 10 min before quenching with 50 μl/well 1 m sulfuric acid. Color reaction was measured at 450 nm by spectrophotometry. The rabbit antiserum that was raised against PGE2-complexed keyhole limpet hemocyanin has been characterized previously (40). Data are presented as mean ± SE from three independent experiments.
PGE2 Stimulation and cAMP Measurement
Functionality of the up-regulated PGE2 receptors was assessed by measuring cAMP accumulation after COX-1 induction in the presence or absence of indomethacin. Cells (2 × 105) were plated in six-well dishes containing 4 ml/well of complete medium containing DOX. Cells were allowed to attach overnight. The following day, the cells were synchronized by incubating with fresh medium containing no fetal bovine serum for 24 h. COX-1 Tet-Off cells were induced by DOX withdrawal from the culture medium for 48 h at 37°C in humidified 5% CO2 in the presence or absence of indomethacin. In parallel, control uninduced cells were supplemented daily with DOX. Thereafter the culture medium was removed and replaced with serum-free medium containing 1-methyl-3-isobutylxanthine (Sigma Chemical Co.) to a final concentration of 1 mm for 40 min at 37°C. Cells were then stimulated with 0 or 300 nm PGE2 for 5, 10, 20, or 30 min, respectively. After stimulation, the medium was removed and the cells were lysed in 0.1 m HCl. cAMP concentration was quantified by ELISA using a cAMP kit (Biomol; Affiniti, Exeter, United Kingdom) as per the manufacturer’s protocol and normalized to the protein concentration of the lysate. Protein concentrations were determined using protein assay kits (Bio-Rad). The data are presented as mean ± SE from three independent experiments.
Statistical Analysis
The data in this study were analyzed by ANOVA using StatView 5.0 (Abacus Concepts, Berkeley, CA).
RESULTS
Expression of COX-1 and COX-2 in Cervical Carcinomas and Normal Cervix
Expression of COX-1 and COX-2 in cervical carcinomas was investigated using real-time quantitative RT-PCR (Fig. 1A) and Western blot analysis (Fig. 1B). Expression of COX-1 and COX-2 RNA was significantly up-regulated in 78 and 100% of cases, respectively, of squamous cell carcinoma and 100% of cases of adenocarcinoma investigated. By contrast, minimal COX-1 and COX-2 transcript was detected in normal cervical tissue by quantitative RT-PCR. COX-1 and COX-2 expression, as assessed by quantitative RT-PCR, was 19.9 ± 5.9- and 118 ± 32-fold greater in cervical carcinoma tissues than that observed in normal cervical tissue (P < 0.01). Western blot analysis confirmed enhanced expression of COX-1 and COX-2 in cervical squamous cell carcinoma (85 and 100% of cases, respectively; Fig. 1B, panel I) and adenocarcinoma (100% of cases; Fig. 1B, panel II). Basal expression of COX-1 protein was detected in 87% of cases of normal cervix. No COX-2 expression was detected in normal cervical tissue by Western blot analysis (Fig. 1B, panel III). Specificity of detection of the 72-kDa COX-1 and COX-2 protein was performed by competition studies using a specific immunogen (blocking) peptide (data not shown).
Fig. 1.
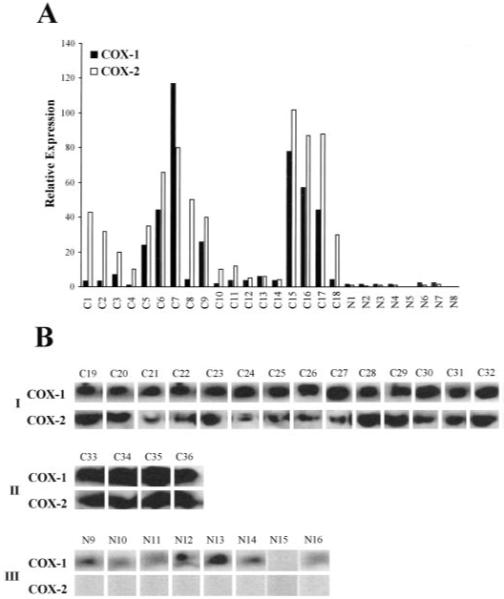
A, relative expression of COX-1 and COX-2 RNA in cervical squamous cell carcinoma (C1–C14), adenocarcinoma (C15–C18), and normal cervix (N1–N8) as determined by real-time quantitative RT-PCR. B, Western blot analysis of 50 μg of total protein isolated from human cervical carcinoma tissue. The proteins were loaded onto a 10% SDS-polyacrylamide gel, electrophoresed, and subsequently transferred to a PVDF membrane. The immunoblot was probed with antibody raised against the COOH terminus of human COX-1 or COX-2. A specific band of approximately 72 kDa was detected in all squamous cell carcinomas (panel I; C19–C32) and adenocarcinomas (panel II; C33–C36). Basal COX-1 expression was detected in seven of eight normal cervices. No COX-2 expression was detected in normal cervical tissue (panel III, N9–N16).
Localization of the Site of COX-1 Expression in Cervical Carcinomas and Normal Cervix
The site of COX-1 expression in the carcinoma tissue was investigated by immunohistochemistry. COX-1 expression was up-regulated in all carcinoma samples. COX-1 was localized to the neoplastically transformed squamous epithelium in squamous cell carcinoma (Fig. 2A), and to the neoplastically transformed columnar epithelium lining the endocervical canal and the glandular epithelium of the endocervical glands in adenocarcinomas (Fig. 2C). Little or no immunoreactivity for COX-1 was observed in the normal cervical tissues (Fig. 2E). Preadsorbing the antibody with the blocking peptide (COX-1 negative control) abolished the COX-1 immunoreactivity in all carcinoma samples. Representative sections incubated with the blocking peptide are shown in Fig. 2, B, D, and F for squamous cell carcinoma, adenocarcinoma, and normal cervical tissues, respectively.
Fig. 2.
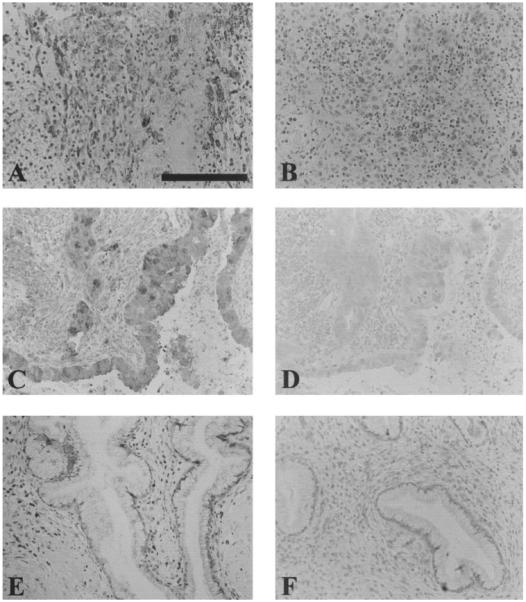
Localization of COX-1 expression in epithelial cells of squamous cell carcinomas and columnar and glandular epithelium of adenocarcinomas (A and C, respectively). Minimal COX-1 signal was detected in normal cervical tissue (E). Sections that were stained with preadsorbed COX-1 sera are shown in B, D, and F for squamous cell carcinoma, adenocarcinoma, and normal cervix, respectively (negative controls). Scale bar, 100 μm.
Inducible COX-1 Expression in HeLa Cells
To investigate the effect of COX-1 overexpression in HeLa neoplastic cervical epithelial cells, we established a DOX-regulated expression system. As shown in Fig. 3A, a 72-kDa immunoreactive COX-1 band was observed to increase in intensity 48 h after DOX withdrawal from the culture medium. Maximal sustained induction was achieved after 72 h. The fold induction for COX-1 overexpression above basal for 24, 48, and 72 h was determined to be 1.5 ± 0.34-, 3.7 ± 0.45-, and 4.7 ± 0.56-fold, respectively. COX-1 expression was normalized against β-actin on the same blot. Cells maintained in DOX for 72 h showed no elevation of COX-1 expression above basal. Preadsorbing the COX-1 antibody with the blocking peptide abolished the COX-1 immunore-activity, indicating specificity of the COX-1 antibody. These data indicate that high levels of inducible overexpression of COX-1 were achieved in HeLa cells. To determine whether COX-1 expression was altered by cell confluency or the addition of DOX to the culture medium, wild-type HeLa Tet-Off cells were grown for 72 h in the presence or absence of DOX. No increase in COX-1 expression above basal was observed, suggesting that neither DOX nor cell density affected the expression of COX-1 in wild-type HeLa Tet-Off cells (Fig. 3A).
Fig. 3.
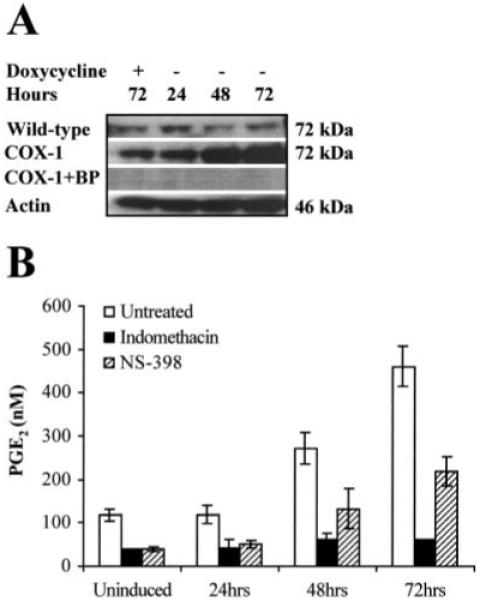
A, Western blot analysis of 20 μg of total protein isolated from wild-type HeLa Tet-Off and HeLa COX-1 Tet-Off cells grown for 24, 48, and 72 h, respectively, in the absence of DOX. In parallel, control uninduced HeLa COX-1 Tet-Off and wild-type HeLa Tet-Off cells were maintained for 72 h under the same conditions supplemented daily with DOX to a final concentration of 1 μg/ml. The proteins were loaded onto a 4-20% SDS-polyacrylamide gel, electrophoresed, and subsequently transferred to a PVDF membrane. The immunoblot was probed with antibody raised against the COOH terminus of human COX-1. A specific band of approximately 72 kDa was detected. No immunoreactivity was detected by preadsorbing the antibody with the blocking peptide (BP). COX-1 was normalized for protein loading against β-actin on the same blot. B, The functionality of the transfected COX-1 cDNA was assessed by ELISA, by measuring PGE2 secretion into the culture medium after COX-1 induction in the presence or absence of the COX enzyme inhibitor indomethacin, and treatment of HeLa cells with 5 μg/ml arachidonic acid.
The functionality of the transfected COX-1 cDNA was assessed by measuring PGE2 secretion into the culture medium after COX-1 induction for 24, 48, and 72 h, respectively. A time-dependent increase in PGE2 secretion into the culture medium accompanied the induction of COX-1 expression. PGE2 production was significantly elevated after 48 h (272.2 ± 18.8 nm; P < 0.05) and 72 h (537 ± 22.5 nm; P < 0.01) when compared with PGE2 levels in uninduced cells (118 ± 6.75 nm; Fig. 3B). The addition of indomethacin reduced the PGE2 levels to 62 ± 7 nm and 76 ± 0.7 nm after 48 and 72 h, respectively (P < 0.01). Cotreatment of cells with NS-398 (selective COX-2 inhibitor) partially reduced PGE2 levels to 132 ± 26.2 and 268 ± 17 nm after 48 and 72 h, respectively (P < 0.05).
COX-1 Overexpression Induces COX-2 and PGES
COX enzyme products including PGE2 are known to induce COX-2 expression (22). To investigate the effect of COX-1 enzyme products on expression of COX-2 and the microsomal glutathione-dependent inducible PGES, COX-1 Tet-Off HeLa cells were grown in the presence or absence of the dual COX enzyme inhibitor indomethacin or the highly selective COX-2 inhibitor NS-398 for 24, 48, and 72 h. After DOX withdrawal from the culture medium, a time-dependent increase in COX-1 overexpression was observed with maximal sustained overexpression after 72 h (Fig. 4A). Concomitant with this increase in COX-1 expression was a 3.2 ± 8.9-fold increase in COX-2 expression after 72 h and a 2.5 ± 0.45- and 1.3 ± 0.78-fold increase in PGES after 24 and 48 h, respectively (Fig. 4A). After 72 h, PGES levels had returned to basal. Cotreatment of the HeLa cells, induced for 24, 48, and 72 h, respectively, with indomethacin or NS-398 showed no alteration in COX-1 overexpression (Fig. 4, B and C). However indomethacin treatment inhibited COX-2 as well as PGES induction (Fig. 4B). No significant change in COX-2 expression was observed after treatment of HeLa cells with NS-398 (Fig. 4C). Induction of PGES by COX-1 overexpression was delayed by 24 h after treatment of HeLa cells with NS-398 (Fig. 4C).
Fig. 4.
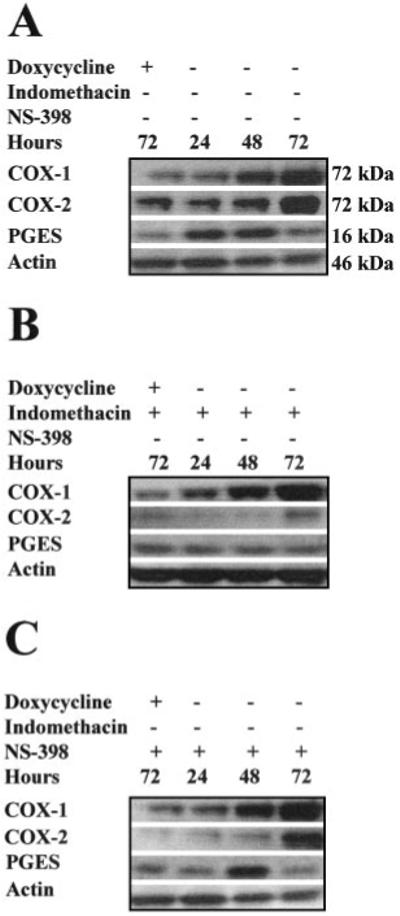
Western blot analysis of 20 μg of total clarified cell lysate isolated from HeLa COX-1 Tet-Off cells grown for 72 h in the presence of DOX (uninduced) or 24, 48, and 72 h, respectively, in the absence of DOX to induce COX-1 expression. A, expression of COX-2 and PGES was induced coincident with COX-1 overexpression in HeLa cells. B, cotreatment of HeLa cells with indomethacin abolished the COX-1-mediated up-regulation of COX-2 and PGES. C, partial inhibition of the COX-1-mediated up-regulation of COX-2 and PGES expression was observed after cotreatment with the selective COX-2 inhibitor NS-398. Proteins were normalized for loading against β-actin on the same blot.
COX-1 Overexpression in HeLa Cells Induces PGE2 Receptor Expression
The effect of COX-1 overexpression on the four subtypes of PGE2 receptors, namely EP1-EP4, was investigated by real-time quantitative RT-PCR, after DOX withdrawal from the culture medium and subsequent induction of COX-1. Induced overexpression of COX-1 for 24, 48, and 72 h had no significant effect on EP1 receptor expression when compared with cells grown in the presence of indomethacin. COX-1 overexpression for 48 and 72 h significantly induced expression of EP2 receptor transcript when compared with indomethacin-treated cells (Fig. 5; P < 0.01). Levels of EP3 receptor transcript were significantly induced after 48 h of COX-1 overexpression (P < 0.05) compared with cells grown in the presence of indomethacin. EP4 receptor transcript was significantly up-regulated after COX-1 overexpression for 24, 48, and 72 h compared with cells cotreated with the COX enzyme inhibitor (P < 0.05).
Fig. 5.
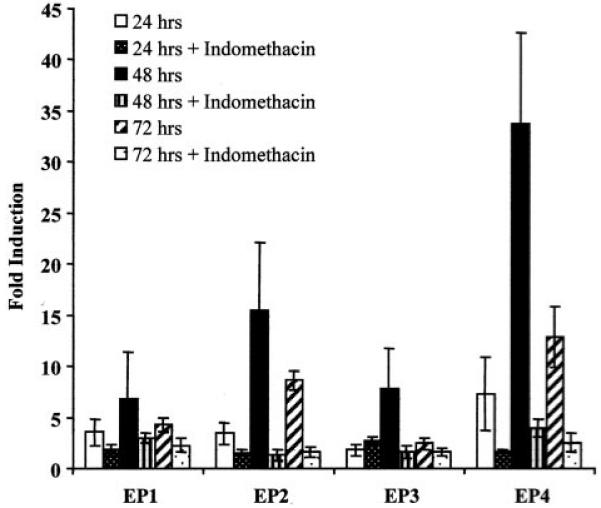
Fold induction of expression of PGE2 receptors (EP1-EP4) in HeLa COX-1 Tet-Off cells as determined by real-time quantitative RT-PCR. COX-1 expression was induced for 24, 48, and 72 h in the presence or absence of the COX enzyme inhibitor indomethacin. Fold induction was determined by dividing the relative expression in induced cells by the relative expression in uninduced cells.
cAMP Production in COX-1-overexpressing Cells in Response to PGE2
The effect of COX-1-induced up-regulation of the cAMP-linked PGE2 receptors on cAMP production was determined after overexpression of COX-1 and stimulation with exogenous PGE2. No significant difference in basal cAMP production was detected in uninduced and induced cells (Fig. 6). Treatment of uninduced cells with 300 nm PGE2 resulted in a 2.43 ± 1.07-fold increase in cAMP production (P < 0.05). Cells in which COX-1 was induced for 48 h before stimulation with exogenous PGE2 showed a rapid, transient 12.67 ± 3.7-fold cAMP response (P < 0.01). This rapid, elevated cAMP production in COX-1-overexpressing cells in response to PGE2 was abolished when cells were grown in medium containing the COX enzyme inhibitor indomethacin. The activity of EP1 receptor was investigated by measuring inositol phosphate accumulation (41). No inositol phosphate accumulation above basal level was observed in COX-1 Tet-Off HeLa cells after induced expression of COX-1 and PGE2 stimulation (data not shown).
Fig. 6.
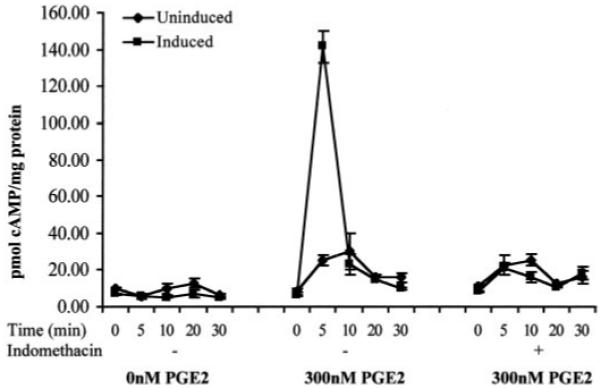
cAMP levels in HeLa COX-1 Tet-Off after treatment with 0 or 300 nm PGE2. Cells were either maintained with 1 μg/ml DOX (uninduced) or induced by incubation in culture medium without DOX for 48 h in the presence or absence of the COX enzyme inhibitor indomethacin.
Induction of Angiogenic Factors in Response to COX-1 Overexpression
The effect of COX-1 on expression of the angiogenic factors bFGF, VEGF, Ang-1, and Ang-2 was assessed by Western blot analysis. Overexpression of COX-1 for 72 h resulted in a 2.3 ± 0.45-fold increase in bFGF (Fig. 7A), a 4.5 ± 1.2-fold increase in VEGF (Fig. 7B), a 2.3 ± 0.78-fold increase in Ang-1 (Fig. 7C), and a 2.1 ± 0.98-fold increase in Ang-2 expression (Fig. 7D), respectively. Indomethacin treatment inhibited the COX-1-associated up-regulation of bFGF, VEGF, Ang-1, and Ang-2 (Fig. 7). Treatment of cells with NS-398 partially reduced the up-regulation of bFGF, VEGF, Ang-1, and Ang-2 expression (Fig. 7), suggesting that products from both COX enzymes were modulating expression of these factors.
Fig. 7.
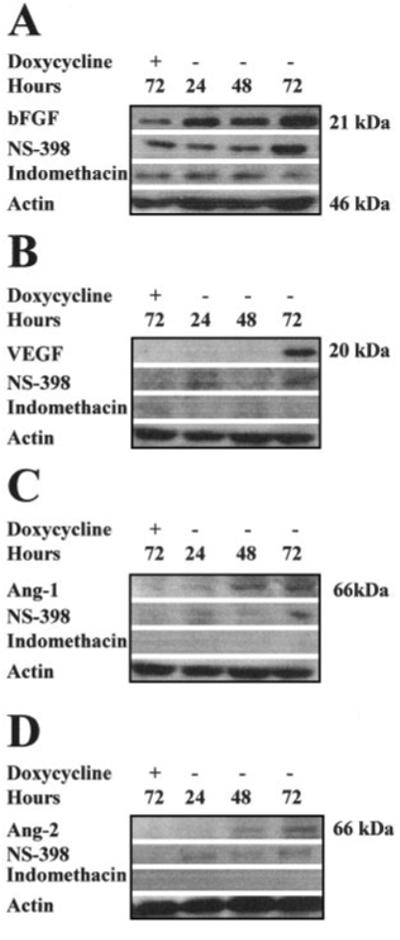
Western blot analysis of 20 μg of total clarified cell lysate isolated from HeLa COX-1 Tet-Off cells grown for 72 h in the presence of DOX or for 24, 48, and 72 h in the absence of DOX to induce COX-1 expression. A, immunoblot of bFGF expression after DOX withdrawal from the culture medium. bFGF expression was induced coincident with COX-1 overexpression. Up-regulated bFGF expression was abolished by indomethacin and partially inhibited by NS-398. B, immunoblot of VEGF expression after DOX withdrawal from the culture medium. VEGF was induced after 72 h of COX-1 overexpression. Up-regulated VEGF expression was abolished by indomethacin and partially inhibited by NS-398. C, immunoblot of Ang-1 expression after DOX withdrawal from the culture medium. Ang-1 was induced coincident with COX-1 overexpression after 48 h. Up-regulated Ang-1 expression was abolished by indomethacin and partially inhibited by NS-398. D, immunoblot of Ang-2 expression after DOX withdrawal from the culture medium. Ang-1 was induced after 48 h of COX-1 overexpression. Up-regulated Ang-2 expression was abolished by indomethacin and partially inhibited by NS-398. Proteins were normalized for loading against β-actin.
DISCUSSION
Recent studies have demonstrated up-regulated and inducible expression of COX-1 in different biological models. COX-1 expression is up-regulated in human breast cancer (24), human prostate cancer (25), and murine models of lung tumorigenesis (23). In addition, COX-1 expression can be induced in vitro by tobacco carcinogen (42), VEGF (21), arachidonic acid, forskolin, dibutyryl-cAMP, and PGE2 (22). In an in vitro model, COX-1 overexpression in endothelial cells implanted in mice was associated with enhanced tumorigenicity (5). This study confirms up-regulation of COX-1 expression in squamous cell carcinoma and adenocarcinoma of the human cervix as demonstrated by real-time quantitative RT-PCR, Western blot analysis, and immunohistochemistry. The up-regulation of expression of COX-1 was associated with enhanced expression of COX-2. Moreover, the site of COX-1 expression localized to the neoplastic epithelial cells of all squamous cell carcinomas and adenocarcinomas investigated, demonstrating a pattern of expression for COX-1 in cancer of the cervix that is similar to that demonstrated for COX-2 (16, 18) and PGE2 (18). These data suggest that both COX-enzymes and/or their products may contribute toward the development of cervical cell neoplasias.
To investigate the effect of overexpression of COX-1, we have established a DOX-regulated expression system in HeLa cells. Initial studies performed on wild-type HeLa Tet-Off cells showed no elevation of COX-1 expression above basal levels when wild-type cells were grown for 72 h in the presence or absence of DOX. These data demonstrate that neither cell growth nor DOX affected the basal expression of COX-1. Overexpression of COX-1 in HeLa cells up-regulates expression of COX-2 and PGES concomitant with increased PGE2 production. These data suggest that COX-2 and inducible PGES are co-regulated. In an in vitro model system, administration of interleukin 1β to A549 cells rapidly induced the expression of COX-2 and PGES (43). Similarly, inducible PGES activity has been described in lipopolysaccharide-stimulated rat peritoneal macrophages, coincident with COX-2 expression and PGE2 biosynthesis (44, 45). Indomethacin, but not NS-398, treatment abolished the up-regulation of expression of COX-2 and PGES and synthesis of PGE2. Up-regulation of COX-2 and PGES in HeLa cells may thus be mediated by prostanoids produced following overexpression of COX-1. NS-398 treatment significantly reduced PGE2 synthesis at 72 h but not 48 h. This is not surprising, because COX-2 expression in HeLa cells was only maximally induced at 72 h. This suggests that PGE2 production detected at 72 h after COX-1 overexpression is enhanced by the activity of both COX enzymes. In other model systems, COX-2 expression is up-regulated by PGE2 via the cAMP-dependent PGE2 receptors (22). In vitro studies have shown that cAMP activity accompanies a concomitant increase in COX-2 synthesis, suggesting that cAMP is the primary secondary messenger in regulating COX-2, presumably via the upstream cAMP response element located on the COX-2 gene (46). The biological actions of PGE2 have been attributed to its interaction with G-protein-coupled receptors, of which four subtypes (EP1-EP4) have been identified (35). COX-1 overexpression in HeLa cells resulted in significant up-regulation of the cAMP-dependent PGE2 receptors after 48 h of COX-1 overexpression. This up-regulation was inhibited by growing cells in medium containing indomethacin, suggesting that the up-regulation was mediated by COX enzyme products. Previous studies have demonstrated enhanced PGE2 synthesis in cervical carcinomas together with up-regulated expression of EP2 and EP4 receptors and enhanced cAMP-responsiveness of cervical tumor tissue to PGE2 (18). Because COX-1 overexpression in HeLa cells induces COX-2 and EP receptor expression, it is feasible that PGE2 may facilitate the process of cervical tumorigenesis in an autocrine/paracrine manner after enhanced EP receptor expression and ligand-receptor interaction. A direct role for EP receptors in tumorigenesis has been reported recently in colon cancer cells. In this model, enhanced proliferative and tumorigenic effects were mediated by PGE2 after interaction with the EP4 receptor (47). It is likely that similar mechanisms may exist in cervical carcinomas to enhance growth and proliferation via EP receptors in a cAMP-dependent manner. Because both COX enzymes catalyze the same reaction, enzyme products such as PGE2 from both COX enzymes may regulate EP receptor expression. The choice of COX enzyme for biosynthesis of prostaglandins may depend on the relative expression of each COX isoform in the cell because, in many cells, COX-2 levels are typically only 20-30% of COX-1 levels (46).
Functionality of the induced EP receptors in our model system was assessed by measuring cAMP in response to stimulation with exogenous PGE2. cAMP activity was measured in HeLa cells after overexpression of COX-1 for 48 h and stimulation with exogenous PGE2. A significant fold increase in cAMP production was observed after 5 min of PGE2 stimulation in COX-1-induced compared with uninduced cells. This augmented cAMP response was abolished by growing cells in medium containing indomethacin. These data suggest that PGE2 produced by COX-1 overexpression may be acting in an autocrine/paracrine manner via the cAMP-linked PGE2 receptors to mediate its effect on target genes, such as COX-2, via the cAMP-dependent protein kinase pathway by activating adenylate cyclase and increasing cAMP. Because COX-1 overexpression had no significant effect on EP1 expression, and stimulation of HeLa cells with PGE2 resulted in no increase in inositol phosphate accumulation above basal levels, this suggested that although PGE2 may be functioning via EP1 receptors coupled to inositol phosphate production and release of intracellular calcium in these cells, its contribution to events associated with COX-1 up-regulation was minimal.
Cancer cells produce a wide variety of factors that contribute to angiogenesis, including bFGF, VEGF, bFGF-binding protein, and platelet-derived growth factor (34). Our data demonstrate that COX-1 overexpression in HeLa cells results in the up-regulation of expression of proangiogenic factors. Induced overexpression of COX-1 resulted in an increase in bFGF, VEGF, Ang-1, and Ang-2 expression. Cotreatment of these cells with indomethacin abolished the up-regulation of these angiogenic factors. This suggests that the up-regulation of these factors is mediated by prostanoids produced by COX-1 overexpression. Moreover, because the effects of COX-1 overexpression can be reversed by COX inhibition with indomethacin, this confirms that these effects are not an artifact of forced overproduction of the enzyme. Partial reduction in expression of these factors by treatment with NS-398 suggests that both enzymes (COX-1 and COX-2) converge to regulate expression of target genes, possibly through common prostanoid synthetic pathways. In another model system, COX-2 overexpression and increase in PGE2 synthesis in colon carcinoma cells results in the up-regulation of bFGF and VEGF and this is associated with arrangement of endothelial cells into tubular structures (34). The up-regulation of angiogenic factors by COX enzymes is important in regulating angiogenesis and maintenance of the neoplastic tissue. As the demand for nutrients and oxygen increases for tissue development, an increased vascularization is necessary to supsply nutrients to the tumor (48). In this study, we also observe the regulation of the angiogenic factors Ang-1 and Ang-2 by COX enzymes. Ang-1 is a Tie-2 receptor agonist, which is required for recruitment of perivascular cells leading to the formation and stabilization of capillaries, vessel maturation, and endothelial cell survival (49, 50). Ang-1 and other angiogenic factors such as VEGF may act synergistically to increase vascular sprouting and branching (51, 52). In addition, Ang-1/Tie-2 interaction enhances the mitogenic effect of VEGF on endothelial cell growth (53). By contrast, Ang-2 is a natural Tie-2 receptor antagonist, destabilizing cell contacts and thus allowing access to angiogenic factors such as VEGF (54). In our model system, enhanced synthesis of prostanoids as a consequence of up-regulated COX-1 may thus act in an autocrine/paracrine manner to up-regulate the expression of COX-2 and target receptors as well as the intracellular signaling to a host of angiogenic factors, which could act on endothelial cells and lead to the recruitment of new blood vessels to enhance tumor mass.
The abbreviations used are
- COX
cyclooxygenase
- VEGF
vascular endothelial growth factor
- PGE2
prostaglandin E2
- PGES
prostaglandin E synthase
- bFGF
basic fibroblast growth factor
- Ang
angiopoietin
- Hyg
hygromycin
- DOX
doxycycline
- RT-PCR
reverse transcription-PCR
- TK
thymidine kinase
- PVDF
polyvinylidene difluoride
- TBS
Tris-buffered saline.
REFERENCES
- 1.Sitas F, Madhoo J, Wessie J. Incidence of Histologically Diagnosed Cancer in South Africa, 1993-1995. National Cancer Registry of South Africa, South African Institute for Medical Research; Johannesburg: 1998. [Google Scholar]
- 2.Scully RE, Bonfiglio TA, Kurman RJ, Silverberg SG, Wilkinson EJ. Histological Typing of Female Genital Tract Tumors. Springer-Verlag; Berlin: 1994. [Google Scholar]
- 3.DuBois RN, Radhika A, Reddy BS, Entingh AJ. Increased cyclooxygenase-2 levels in carcinogen-induced rat colonic tumors. Gastroenterology. 1996;110:1259–1262. doi: 10.1053/gast.1996.v110.pm8613017. [DOI] [PubMed] [Google Scholar]
- 4.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 5.Narko K, Ristimaki A, MacPhee M, Smith E, Haudenschild CC, Hla T. Tumorigenic transformation of immortalized ECV endothelial cells by cyclooxygenase-1 overexpression. J. Biol. Chem. 1997;272:21455–21460. doi: 10.1074/jbc.272.34.21455. [DOI] [PubMed] [Google Scholar]
- 6.Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc. Natl. Acad. Sci. USA. 1997;94:3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J. Clin. Investig. 2000;105:1589–1594. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. USA. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herschman HR. Regulation of prostaglandin synthase-1 and prostaglandin synthase-2. Cancer Metastasis Rev. 1994;13:241–256. doi: 10.1007/BF00666095. [DOI] [PubMed] [Google Scholar]
- 10.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 11.Kargman SL, O’Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S. Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res. 1995;55:2556–2559. [PubMed] [Google Scholar]
- 12.Herschman HR. Prostaglandin synthase 2. Biochim. Biophys. Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 13.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 14.Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT, Fahey TJ., III Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999;59:987–990. [PubMed] [Google Scholar]
- 15.Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- 16.Kulkarni S, Rader JS, Zhang F, Liapis H, Koki AT, Masferrer JL, Subbaramaiah K, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in human cervical cancer. Clin. Cancer Res. 2001;7:429–434. [PubMed] [Google Scholar]
- 17.Ryu HS, Chang KH, Yang HW, Kim MS, Kwon HC, Oh KS. High cyclooxygenase-2 expression in stage IB cervical cancer with lymph node metastasis or parametrial invasion. Gynecol. Oncol. 2000;76:320–325. doi: 10.1006/gyno.1999.5690. [DOI] [PubMed] [Google Scholar]
- 18.Sales KJ, Katz AA, Davis M, Hinz S, Soeters RP, Hofmeyr MD, Millar RP, Jabbour HN. Cyclooxygenase-2 expression and prostaglandin E2 synthesis are up-regulated in carcinomas of the cervix: a possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. J. Clin. Endocrinol. Metab. 2001;86:2243–2249. doi: 10.1210/jcem.86.5.7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brannon TS, North AJ, Wells LB, Shaul PW. Prostacyclin synthesis in ovine pulmonary artery is developmentally regulated by changes in cyclooxygenase-1 gene expression. J. Clin. Investig. 1994;93:2230–2235. doi: 10.1172/JCI117220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith CJ, Morrow JD, Roberts L. J. d., Marnett LJ. Differentiation of monocytoid THP-1 cells with phorbol ester induces expression of prostaglandin endoperoxide synthase-1 (COX-1) Biochem. Biophys. Res. Commun. 1993;192:787–793. doi: 10.1006/bbrc.1993.1483. [DOI] [PubMed] [Google Scholar]
- 21.Bryant CE, Appleton I, Mitchell JA. Vascular endothelial growth factor up-regulates constitutive cyclooxygenase 1 in primary bovine and human endothelial cells. Life Sci. 1998;62:2195–2201. doi: 10.1016/s0024-3205(98)00197-0. [DOI] [PubMed] [Google Scholar]
- 22.Maldve RE, Kim Y, Muga SJ, Fischer SM. Prostaglandin E2 regulation of cyclooxygenase expression in keratinocytes is mediated via cyclic nucleotide-linked prostaglandin receptors. J. Lipid Res. 2000;41:873–881. [PubMed] [Google Scholar]
- 23.Bauer AK, Dwyer-Nield LD, Malkinson AM. High cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2) contents in mouse lung tumors. Carcinogenesis (Lond.) 2000;21:543–550. doi: 10.1093/carcin/21.4.543. [DOI] [PubMed] [Google Scholar]
- 24.Hwang D, Scollard D, Byrne J, Levine E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J. Natl. Cancer Inst. (Bethesda) 1998;90:455–460. doi: 10.1093/jnci/90.6.455. [DOI] [PubMed] [Google Scholar]
- 25.Kirschenbaum A, Klausner AP, Lee R, Unger P, Yao S, Liu X, Levine AC. Expression of cyclooxygenase-1 and cyclooxygenase-2 in the human prostate. Urology. 2000;56:671–676. doi: 10.1016/s0090-4295(00)00674-9. [DOI] [PubMed] [Google Scholar]
- 26.Marnett LJ. Aspirin and the potential role of prostaglandins in colon cancer. Cancer Res. 1992;52:5575–5589. [PubMed] [Google Scholar]
- 27.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 28.Forsberg L, Leeb L, Thoren S, Morgenstern R, Jakobsson P. Human glutathione dependent prostaglandin E synthase: gene structure and regulation. FEBS Lett. 2000;471:78–82. doi: 10.1016/s0014-5793(00)01367-3. [DOI] [PubMed] [Google Scholar]
- 29.Simonson MS, Herman WH, Dunn MJ. PGE2 induces c-fos expression by a cAMP-independent mechanism in glomerular mesangial cells. Exp. Cell Res. 1994;215:137–144. doi: 10.1006/excr.1994.1325. [DOI] [PubMed] [Google Scholar]
- 30.Baylink TM, Mohan S, Fitzsimmons RJ, Baylink DJ. Evaluation of signal transduction mechanisms for the mitogenic effects of prostaglandin E2 in normal human bone cells in vitro. J. Bone Miner. Res. 1996;11:1413–1418. doi: 10.1002/jbmr.5650111007. [DOI] [PubMed] [Google Scholar]
- 31.Yano T, Yano Y, Uchida M, Murakami A, Hagiwara K, Otani S, Ichikawa T. The modulation effect of vitamin E on prostaglandin E2 level and ornithine decarboxylase activity at the promotion phase of lung tumorigenesis in mice. Biochem. Pharmacol. 1997;53:1757–1759. doi: 10.1016/s0006-2952(96)00869-6. [DOI] [PubMed] [Google Scholar]
- 32.DeWitt DL. Prostaglandin endoperoxide synthase: regulation of enzyme expression. Biochim. Biophys. Acta. 1991;1083:121–134. doi: 10.1016/0005-2760(91)90032-d. [DOI] [PubMed] [Google Scholar]
- 33.Jones MK, Wang H, Peskar BM, Levin E, Itani RM, Sarfeh IJ, Tarnawski AS. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat. Med. 1999;5:1418–1423. doi: 10.1038/70995. [DOI] [PubMed] [Google Scholar]
- 34.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 35.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- 36.Ashby B. Co-expression of prostaglandin receptors with opposite effects: a model for homeostatic control of autocrine and paracrine signaling. Biochem. Pharmacol. 1998;55:239–246. doi: 10.1016/s0006-2952(97)00241-4. [DOI] [PubMed] [Google Scholar]
- 37.International Federation of Obstetricians and Gynaecologists . TNM Atlas. Ed. 3 Springer-Verlag; Heidelberg: 1992. p. 196. [Google Scholar]
- 38.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denison FC, Calder AA, Kelly RW. The action of prostaglandin E2 on the human cervix: stimulation of interleukin 8 and inhibition of secretory leukocyte protease inhibitor. Am. J. Obstet. Gynecol. 1999;180:614–620. doi: 10.1016/s0002-9378(99)70263-2. [DOI] [PubMed] [Google Scholar]
- 40.Kelly RW, Graham BJ, O’Sullivan MJ. Measurement of PGE2 as the methyl oxime by radioimmunoassay using a novel iodinated label. Prostaglandins Leukotrienes Essent. Fatty Acids. 1989;37:187–191. doi: 10.1016/0952-3278(89)90084-7. [DOI] [PubMed] [Google Scholar]
- 41.Berg KA, Clarke WP, Sailstad C, Saltzman A, Maayani S. Signal transduction differences between 5-hydroxytryptamine type 2A and type 2C receptor systems. Mol. Pharmacol. 1994;46:477–484. [PubMed] [Google Scholar]
- 42.Rioux N, Castonguay A. The induction of cyclooxygenase-1 by a tobacco carcinogen in U937 human macrophages is correlated to the activation of NF-κB. Carcinogenesis (Lond.) 2000;21:1745–1751. doi: 10.1093/carcin/21.9.1745. [DOI] [PubMed] [Google Scholar]
- 43.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naraba H, Murakami M, Matsumoto H, Shimbara S, Ueno A, Kudo I, Oh-ishi S. Segregated coupling of phospholipases A2, cyclooxygenases, and terminal prostanoid synthases in different phases of prostanoid biosynthesis in rat peritoneal macrophages. J. Immunol. 1998;160:2974–2982. [PubMed] [Google Scholar]
- 45.Matsumoto H, Naraba H, Murakami M, Kudo I, Yamaki K, Ueno A, Oh-ishi S. Concordant induction of prostaglandin E2 synthase with cyclooxygenase-2 leads to preferred production of prostaglandin E2 over thromboxane and prostaglandin D2 in lipopolysaccharide-stimulated rat peritoneal macrophages. Biochem. Biophys. Res. Commun. 1997;230:110–114. doi: 10.1006/bbrc.1996.5894. [DOI] [PubMed] [Google Scholar]
- 46.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 47.Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J. Biol. Chem. 2001;276:18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 48.Seed MP, Brown JR, Freemantle CN, Papworth JL, Colville-Nash PR, Willis D, Somerville KW, Asculai S, Willoughby DA. The inhibition of colon-26 adenocarcinoma development and angiogenesis by topical diclofenac in 2.5% hyaluronan. Cancer Res. 1997;57:1625–1629. [PubMed] [Google Scholar]
- 49.Papapetropoulos A, Garcia-Cardena G, Dengler TJ, Maisonpierre PC, Yancopoulos GD, Sessa WC. Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab. Investig. 1999;79:213–223. [PubMed] [Google Scholar]
- 50.Asahara T, Chen D, Takahashi T, Fujikawa K, Kearney M, Magner M, Yancopoulos GD, Isner JM. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ. Res. 1998;83:233–240. doi: 10.1161/01.res.83.3.233. [DOI] [PubMed] [Google Scholar]
- 51.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature (Lond.) 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 52.Koblizek TI, Weiss C, Yancopoulos GD, Deutsch U, Risau W. Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr. Biol. 1998;8:529–532. doi: 10.1016/s0960-9822(98)70205-2. [DOI] [PubMed] [Google Scholar]
- 53.Huang XL, Takakura N, Suda T. In vitro effects of angiopoietins and VEGF on hematopoietic and endothelial cells. Biochem. Biophys. Res. Commun. 1999;264:133–138. doi: 10.1006/bbrc.1999.1472. [DOI] [PubMed] [Google Scholar]
- 54.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science (Wash. DC) 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]