

Abstract
Docosahexaenoic acid (DHA; 22:6n-3) is a critical constituent of the brain, but its metabolism has not been measured in the human brain in vivo. In monkeys, using positron emission tomography (PET), we first showed that intravenously injected [1-11C]DHA mostly entered nonbrain organs, with ∼0.5% entering the brain. Then, using PET and intravenous [1-11C]DHA in 14 healthy adult humans, we quantitatively imaged regional rates of incorporation (K*) of DHA. We also imaged regional cerebral blood flow (rCBF) using PET and intravenous [15O]water. Values of K* for DHA were higher in gray than white matter regions and correlated significantly with values of rCBF in 12 of 14 subjects despite evidence that rCBF does not directly influence K*. For the entire human brain, the net DHA incorporation rate Jin, the product of K*, and the unesterified plasma DHA concentration equaled 3.8 ± 1.7 mg/day. This net rate is equivalent to the net rate of DHA consumption by brain and, considering the reported amount of DHA in brain, indicates that the half-life of DHA in the human brain approximates 2.5 years. Thus, PET with [1-11C]DHA can be used to quantify regional and global human brain DHA metabolism in relation to health and disease.
Keywords: metabolism, blood flow, n-3 polyunsaturated fatty acid
The nutritionally essential n-3 PUFA, docosahexaenoic acid (DHA; 22:6n-3), is found in high concentrations in vertebrate brain and is reported to influence cell membrane fluidity, enzyme activity (1), ion channels (2), and neuroreceptors and their signaling (3–5). When released from membrane phospholipid by a phospholipase A2 (PLA2), unesterified DHA can act as a second messenger or can be metabolized to bioactive docosanoids and other products (6–9). Decreased dietary n-3 PUFA intake during early childhood is thought to be deleterious to brain function, whereas n-3 PUFA deprivation in later life may contribute to depression, bipolar disorder, and Alzheimer's disease (10–12).
To understand the role of PUFAs in brain function and structure, it would be useful to be able to quantitatively image brain PUFA metabolism in vivo. We have developed a method to do this in unanesthetized rodents for both DHA and arachidonic acid (ARA; 20:4n-6) in which regional incorporation coefficients K* of either PUFA are imaged using quantitative autoradiography following the intravenous injection of the respective radiolabeled PUFA (13–21). This method has demonstrated that intravenously injected radiolabeled DHA or ARA is rapidly incorporated mainly into the stereospecifically numbered (sn)-2 position of different brain phospholipids (22, 23). This method has also demonstrated that intravenously injected radiolabeled DHA or ARA incorporation is increased into synaptic membrane phospholipids following ligand activation of a neuroreceptor coupled to PLA2 and that this incorporation can be blocked by preadministration of a PLA2 inhibitor or by preadministration of an antagonist to the targeted receptor (18, 21, 24, 25). PUFA incorporation into brain phospholipid is unaffected by changes in regional cerebral blood flow (rCBF); thus, PUFA incorporation into brain phospholipid reflects only brain PUFA metabolism (14, 16, 22). The characteristics of brain DHA metabolism permit the use of an irreversible uptake model over the time course of a PET scan.
The product of the regional incorporation coefficient K* and the respective unesterified PUFA concentration in plasma (the net incorporation rate Jin), represents the regional rate of replacement of the quantity of PUFA that is metabolized in brain and lost from brain (18). This is because the other forms of plasma PUFA (i.e., esterified in lipoproteins) were shown not to contribute measurably to brain uptake (26, 27) and because circulating precursors of ARA (linoleic acid, 18:2n-6) and of DHA (α-linolenic acid, 18:3n-3) after entering the adult brain are largely lost by metabolism and are not elongated to ARA or DHA (28–31). Thus, Jin for DHA was shown to equal the rate of DHA loss from brain, calculated following intracerebral injection of radiolabeled DHA in unanesthetized rats (32). Consequently, measuring K* and Jin in vivo can provide an estimate of brain consumption of the PUFA in relation to aging and disease and under different experimental conditions. Furthermore, since PUFAs are nutritionally essential, these consumption rates might be used to estimate daily PUFA dietary requirements of the brain.
In view of the importance of PUFAs to brain structure and function, and of evidence that certain human brain disorders and human cognitive functions can be influenced by the dietary PUFA content, we thought it of interest to extend our in vivo animal method to quantitatively image regional PUFA metabolism in the human brain. To do this, we first developed a radiosynthetic method for positron-emitting [1-11C]polyhomoallylic acids, including ARA and DHA (33, 34). We then used intravenously injected [1-11C]ARA together with positron emission tomography (PET) to image regional brain incorporation coefficients K* and calculate regional incorporation rates Jin of ARA in monkey and humans in the resting state (35, 36) and following functional activation (37). In this study, after estimating radiation exposure parameters (organ distribution and radiation absorbed dose) from studies with [1-11C]DHA in rhesus monkeys (see Appendix), we performed PET to measure K* and Jin for DHA in 14 healthy human volunteers.
MATERIALS AND METHODS
Human subjects
The clinical protocol (No. 04-AA-0058) was approved by the Institutional Review Board of the National Institute on Alcohol Abuse and Alcoholism and by the National Institutes of Health Radiation Safety Committee. Written informed consent was obtained from participants, who were reimbursed for their participation. We studied eight female and six male healthy volunteers (mean age ± SD, 36 ± 15 years; range of 19 to 64 years) who were recruited from the Bethesda, MD area. Each participant was a nonsmoker and took no medication, drugs, or alcohol for at least 2 weeks prior to the PET scan. Subjects underwent an extensive history and physical examination with laboratory tests to insure that they were entirely healthy with no history of neurological or psychiatric disorders. Healthy subjects vary widely in dietary n-3 PUFA intake, resulting in wide individual variations in tissue n-3 PUFA concentrations. To avoid as much variation as possible, 3 days preceding the scan subjects avoided foods high in n-3 PUFAs (e.g., seafood) and were limited to one caffeinated beverage per day. Beginning 24 h before the PET scan, they consumed standardized meals; in addition, they did not eat for 12 h prior to the scan. On the morning of the PET scan, all subjects were confirmed to have fasting blood glucose levels in the normal range, with a mean ± SD of 91.2 ± 10.5 mg/100 ml.
Radiochemistry of [1-11C]DHA
[1-11C]DHA was synthesized by the Grignard reaction as previously described, with a specific activity >500 μCi/nmol (34). HPLC analysis of aliquots of the final product revealed >98.6 ± 0.9% (n = 8) radiochemical purity.
Human PET scanning
Subjects stayed in the hospital overnight and venous and arterial lines were inserted on the morning of the PET scan. The procedures followed those used in a PET study with [1-11C]ARA (36). Scans were performed with a General Electric Advance Tomograph (Waukesha, WI) that acquires 35 simultaneous slices separated by 4.25 mm. PET image counts were converted to μCi/cc by scanning a 16-cm cylindrical phantom containing known radioactivity. Each subject was scanned in a quiet room with his eyes open and ears unoccluded, while his head was held in place by a thermoplastic facemask. Scans were obtained parallel to the orbitomeatal line. A transmission scan was obtained first to correct for attenuation, followed by a bolus intravenous injection of 370 MBq (10 mCi) [15O]water to acquire a 60 s scan in three-dimensional mode. Automatic arterial sampling was used to produce PET images of rCBF (38). Fifteen minutes following the injection of [15O]water, 1118 ± 24 MBq (30.2 ± 0.7 mCi) [1-11C]DHA was infused intravenously for 3 min at a constant rate (Harvard Infusion Pump, South Natick, MA). Because of the highly specific activity of the infused [1-11C]DHA (see above), <0.06 μmol of unlabeled DHA was infused into a subject. Diluted in a plasma volume of ∼3000 ml, this amount would not have changed unesterified plasma DHA by >0.2% and thus was unlikely to have a significant metabolic or biological effect. Serial dynamic three-dimensional scans were acquired during 1 h following the start of infusion. Arterial blood samples (2–5 ml) were obtained at fixed times to determine radioactivity in whole blood and plasma. In addition, a subset of these samples (0, 3, 7, 10, 15, 20, 40, and 60 min) was used to measure blood [11C]CO2 and the parent fraction of [1-11C]DHA.
Correction for subject motion during the 60-min PET acquisition was performed with a mutual information registration of each time frame to a standard frame before attenuation correction. Based on calculated motion, transmission images were resliced and projected for final attenuation correction, reconstruction, and realignment. Final reconstructed images included corrections for attenuation, scatter, random coincidences, dead time, and decay. The final image resolution was 6 to 7 mm in all three dimensions.
Determination of plasma [1-11C]DHA input function (plasma curve)
Considering the short 20.4 min half-life of 11C, it was necessary to develop a method that would rapidly assay plasma [1-11C]DHA during a PET scan. To do this, we developed a solid phase extraction procedure to separate unesterified [1-11C]DHA from remaining plasma radioactivity. From plasma samples collected at 0, 3, 7, 10, 15, 20, 40, and 60 min postinfusion of [1-11C]DHA, total lipids were extracted into chloroform:methanol (1:1) by a method adapted from Bligh and Dyer (39). The original method was modified to allow for a rapid single-step extraction due to 11C decay. Briefly, 0.3 ml of plasma was placed into glass centrifuge screw-cup tubes containing a mixture of 1 ml methanol, 1 ml of chloroform, and 0.6 ml of water. The tubes were purged with nitrogen, sealed, vigorously vortexed for 30 s, and centrifuged at 4,000 rpm for 5 min. The bottom layer containing the total lipid extract was collected by aspiration. Using this single-pass extraction, recovery of available total lipids from the plasma sample was found to be in the range of 50–60%, with the remainder still found in the aqueous phase. Recovery of lipids from plasma for this extraction procedure was determined in a separate experiment using 14C- and 3H-labeled individual lipid probes.
Solid phase extraction analysis was carried out on 500 mg aminopropylsilane (NH2) cartridges (BAKERBOND spe™; JT Baker, Phillipsburg NJ) according to a method adapted from Agren, Julkunen, and Penttila (40). The total lipid extracts were evaporated to complete dryness under a stream of nitrogen gas in a heating block maintained at 45°C. The lipid extracts were dissolved in 0.25 ml of hexane-methyl tert-butyl ether-acetic acid (100:3:0.3) loading solvent.
A 0.1 ml aliquot of this solution was reserved for counting, and 0.1 ml was applied onto solid phase extraction cartridges for separation into lipid classes. The separations were performed on a vacuum manifold under 30–40 mm Hg vacuum. Four to ten samples were separated simultaneously. Prior to separation, the solid phase extraction cartridges were preconditioned with 10 ml of hexane. Combined cholesterol ester and triglyceride fractions were eluted with 10 ml of hexane-chloroform (2:1), nonesterified fatty acids with 10 ml of chloroform-methanol-acetic acid (100:2:2), and phospholipids with 8 ml of isopropanol-3N methanolic HCl (4:1).
All lipid fractions were collected and counted with a calibrated γ counter. This procedure also was applied to a reference blood sample taken before injection, to which ∼185 MBq (5 μCi) of [1-11C]DHA was added. Of the original plasma total lipid extract loaded on the solid phase extraction column, >98% of the counts were recovered in the nonesterified fatty acid fraction (second elution step), and very few counts, if any, were associated with the plasma triglycerides, cholesteryl ester, and phospholipids.
The final continuous function for the [1-11C]DHA fraction was determined as the product of two fitted curves, one for the time-varying recovery and one for the nonesterified fatty acid fraction determined by the ratio of the second fraction to the sum of the fractions. The continuous functions were chosen based on those described previously (36).
To verify the identity of the tracer determined from the solid phase extraction procedure, we conducted a separate procedure on a subset of samples. HPLC was performed on the unesterified plasma fatty acid fraction from the second elution step to determine percentage of radioactivity due to [1-11C]DHA. Measurements were made on 5-ml plasma samples at 10 min postinfusion of [1-11C]DHA as well as on plasma spiked with [1-11C]DHA. The fractions were dried under nitrogen gas, redissolved in methanol, and separated by HPLC (System Gold ® model 126; Beckman, Fullerton, CA) using a 25 cm × 4.6 mm i.d., C18 reverse phase column [Luna (I)™; Phenomenex, Torrence, CA]. Elution (2 ml/min) of unesterified fatty acids was by a linear gradient of acetonitrile/15 mM H3PO4 in water, initiated and held at 80: 20 (v/v) for 1 min, increased to 96: 4 (v/v) in 10 min, held at 96: 4 (v/v) for 10 min, and returned to 80:20 (v/v) in 1 min (41). Elution was monitored at 192 nm with a UV/visible light detector (Model 151; Gilson, Middleton, WI). Radioactivity profiles were obtained with an on-line flow scintillation counter (β-Ram, model 2B; IN/US Systems, Tampa, FL) using a 2:1 ratio of scintillation cocktail (IN-FLOW™ 2:1; IN/US Systems) to monitor HPLC column outflow. The radioactive signal was monitored using a Laura Lite 3 computer program (version 3.2; IN/US Systems, Lab Logic Systems). Following the manufacturer's instructions, the counting window was set at 80–1000 keV to capture both γ and β particle emissions from [11C] decay events. Peaks were identified against retention times of unlabeled standards of unesterified fatty acids as well as [1-14C]standards of unesterified palmitic (16:0), linoleic, α-linolenic, eicosapentaenoic (20:5n-3), arachidonic, and docosahexaenoic acids (Perkin-Elmer, New England Nuclear Life Science Products, Boston, MA). Under these conditions, recovery of unesterified fatty acids through the HPLC column was 95–98%.
Analysis of plasma unesterified fatty acid concentrations
To measure unesterified unlabeled fatty acid concentrations, total lipids were extracted from plasma samples taken at 0, 30, and 60 min after beginning [1-11C]DHA infusion, using a partition system of chloroform, methanol, and 0.5 M KCl (2: 1: 0.75, v/v/v) (42) at a ratio of 25:1 to plasma. Prior to extraction, unesterified unlabeled heptadecanoic acid (17:0) was added as an internal standard. Extracts were separated by TLC on silica gel 60 plates (EM Separation Technologies, Gibbstown, NJ) using a solvent system of heptane/diethyl ether/glacial acetic acid (60: 40: 3, v/v/v) (43). The TLC plates were sprayed with 0.03% (w/v) toluidine-2-naphthalene sulfonic acid in 50 mM Tris buffer (pH 7.4), and lipid bands were visualized under UV light. Positions for unesterified fatty acids were identified using standard 17:0 run separately on the plates. The lipid bands containing unesterified fatty acids were converted to fatty acid methyl esters using 1% H2SO4 in methanol (44), which then were separated on a 30 m × 0.25 mm i.d. capillary column (SP-2330; Supelco, Bellefonte, PA) using a gas chromatograph with flame ionization detector (Model 6890N; Agilent Technologies, Palo Alto, CA). Runs were initiated at 80°C, with a temperature gradient to 160°C (10°C/min) and 230°C (3°C/min) in 31 min, and held at 230°C for 10 min. Peaks were identified by retention times of fatty acid methyl ester standards. Unesterified fatty acid concentrations (nmol/ml) were calculated by proportional comparison, as a mol to mol ratio, of gas chromatograph peak areas for the fatty acids to the area of the added 17:0 internal standard. Data from the three time periods were averaged.
Magnetic resonance image acquisition
A 1.5 Tesla Horizon (General Electric, Milwaukee, Wisconsin) MR scanner was used to obtain T1-weighted volumetric spoiled gradient magnetic resonance (MR) images (echo time = 5.4 ms, flip angle = 20°, repetition time = 14 ms). Images were acquired in a coronal orientation (0.94 × 0.94 × 2 mm voxel size, 256 × 256 × 124 slices) and resliced to the transverse plane for analysis.
Modeling
For each subject, rCBF images from the [15O]water scan were registered to the MR brain volume using a six-parameter transformation and a mutual information cost function (45). The same transformation was applied to the [1-11C]DHA parametric images so that all analyses were performed in the subject's individual MR space.
The following equation was used to fit the dynamic PET data to estimate regional incorporation coefficients (K*) of [1-11C]DHA by nonlinear least squares:
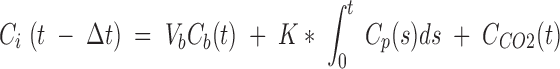 |
Eq. 1 |
Ci(t) is the whole-brain time activity curve determined from the dynamic PET data, Δt is the delay between the blood and brain curves, Cb(t) is the whole-blood time activity curve (nCi/ml), Vb is blood volume (ml blood/ml brain), Cp is the plasma input function (nCi/ml) corrected for metabolites, CCO2(t) is the predicted brain tissue concentration of [11C]CO2, and K* (μL/min/ml brain) is the unidirectional incorporation coefficient of [1-11C]DHA. This equation assumes that all [1-11C]DHA is irreversibly trapped in the brain and that no radioactive metabolite other than [11C]CO2 crosses the blood-brain barrier. Brain concentrations of [11C]CO2 were estimated from blood [11C]CO2 measurements (nCi/ml) as previously described (36). Equation 1 was used to estimate Δt, Vb, and K* from a whole-brain time activity curve. Then, on a pixel-by-pixel basis, images of K* and Vb were obtained by linearly fitting Equation 1 to the data with Δt fixed to the global value.
The incorporation coefficient, K*, when multiplied by the plasma concentration of unesterified unlabeled DHA, Cpl, gives the net influx of unesterified DHA from plasma into brain, Jin (18).
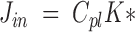 |
Eq. 2 |
As noted in the Introduction, Jin equals the rate of metabolic loss of DHA from brain (18, 29). Jin for the whole brain was obtained by summing Jin for all gray and white matter voxels.
Partial volume error correction
Because of the limited spatial resolution of a PET scan, underestimation of radioactivity can occur in high-activity gray matter regions of interest (ROI). To provide the most accurate measure of activity in specific regions of gray matter, we corrected for this partial volume error (PVE). This correction is not necessary when examining the brain as a whole. We used anatomic information from the MR images with an algorithm, as previously described, to determine values of K*, Vb, and rCBF for ROI (35). Gray matter pixels were corrected for spill-in of activity from white matter and for spill-out of activity. MR segmentation was performed after nonbrain regions were eliminated from the MR image using automatic and manual methods (46). An adaptive fuzzy C-means algorithm was used to perform segmentation (47).
ROI
ROI were drawn manually on individual MR images on six continuous axial MR slices. Non-PVE-corrected values of K*, Vb, and rCBF, as well as corrected values, were obtained for gray matter regions from PET images by limiting averaging to voxels identified as gray matter by the segmentation procedure. Global gray matter values were determined by averaging all voxels in the gray matter mask. K*, Vb, and rCBF for white matter were obtained from the PET images by limiting the averaging to voxels identified as 99% pure white matter from the smoothed white matter mask.
Statistics
Results are presented as means ± SD. Statistical analysis was performed using STATISTICA for Windows 6.0 (Statsoft, Tulsa, OK) with significance set at P ≤ 0.05.
RESULTS
[1-11C]DHA metabolism and plasma radioactivity
HPLC was used in four subjects to validate the solid phase extraction method of estimating the plasma [1-11C]DHA input function (Fig. 1). In the plasma sample that had been spiked with [1-11C]DHA, 98 ± 1% of the radioactivity was [1-11C]DHA (Fig. 1A). At 10 min postinfusion, by which time most of the brain incorporation of [1-11C]DHA had taken place, [1-11C]DHA represented 90 ± 4% of unesterified fatty acid radioactivity measured in human plasma (Fig. 1B). There was no measurable radioactivity in shorter-chain fatty acids at 10 min or evidence of recycled [11C] in saturated fatty acids, such as palmitate (16:0) or stearate (18:0). The remaining radioactivity was mainly found within a single unknown peak that eluted shortly after the [1-11C]DHA, at ∼10 min (Fig. 1B). This peak is unlikely to be an elongation, limited chain shortening (peroxisomal β-oxidation) or carbon recycling product of DHA; because DHA elongation does not biologically occur, carbon recycling of 11C into other fatty acids would be a slow/limited process, and the 11C-label would have been lost during any chain shortening event. Because traces of this peak also appear in the spike sample (Fig. 1A), the peak may have been derived from that sample.
Fig. 1.

HPLC radioactivity trace (relative response of detector in mV) versus elution time (min). A: Plasma unesterified fatty acids extracted from baseline (0 min), spiked with [1-11C]DHA. B: Plasma unesterified fatty acids collected 10 min postinfusion of [1-11C]DHA. Absorbance (192 nm) traces for A and B are not shown. The arrow represents an unknown peak (see text).
Plasma radioactivity as determined by solid phase extraction peaked sharply at the end of the 3-min infusion and declined to 10.9% of the peak value by 20 min (Fig. 2A), consistent with a plasma half-life of ∼2 min. The ratio of whole blood to plasma radioactivity equaled 0.62 ± 0.06 at 3 min and 0.64 ± 0.06 at 10 min, suggesting that red blood cell metabolism of DHA was not an important consideration for our calculations. Radioactivity due to [11C]CO2 tended to plateau at 4% of total after about 30 min (data not shown). Percentage of total plasma radioactivity present as unchanged [1-11C]DHA equaled 87 ± 4%, 79 ± 7%, 69 ± 7%, 58 ± 8%, and 35 ± 8% at 5, 10, 20, 30, and 60 min, respectively, following initiation of infusion (Fig. 2B). Aqueous radioactivity approximated 5% of total plasma radioactivity after 10 min. Approximately 1.3 ± 2.6% of the radioactivity was due to cholesteryl ester and triglycerides, and 0.5 ± 1.3% of the radioactivity was due to phospholipids and other polar lipids. In the HPLC analysis of the plasma-free fatty acid fraction, there were no peaks that had retention times corresponding to 11C-labeled saturated fatty acids, especially palmitic (16:0) and stearic (18:0) acids.
Fig. 2.


A: Plasma radioactivity normalized to injected dose, as a function of time after a 3-min intravenous infusion of [1-11C]DHA. Data represent mean of 14 subjects. B: Fraction of net plasma radioactivity due to the contribution of injected unesterified [1-11C]DHA (parent fraction) as a function of time after beginning injection. Data represent mean of 14 subjects.
Brain radioactivity and imaging
Figure 3 presents horizontal brain PVE-corrected rCBF and K* images from one subject compared with his respective anatomic MR image. The figure illustrates regional differences in rCBF and K* among gray matter regions and higher values of both parameters in gray than in white matter regions.
Fig. 3.

Transverse images from one subject at level of basal ganglia. In the color scheme, blue represents the minimum and red the maximum value. A: MR image. B: K* image after correction for PVE, color scaled to maximum (red) of 8 μL/min/ml. C: rCBF image corrected for PVE, color scaled to maximum (red) of 150 ml/min/100 g.
Non-PVE-corrected values for whole-brain gray matter K* equaled 2.64 ± 0.35 μL·min−1·ml−1 and for whole-brain white matter equaled 1.45 ± 0.41 μL·min−1·ml−1. For the entire brain (gray + white matter), uncorrected K* equaled 2.46 ± 0.35. The PVE correction of region gray matter increased values of K* by 12% in medial temporal cortex to 58% in orbitofrontal cortex region (data not shown), consistent with our prior study with [1-11C]ARA (35). As reported earlier (35), increases also were produced by PVE corrections in rCBF and Vb. Mean gray matter Vb equaled 0.057 ± 0.008 ml/ml before the PVE correction.
Table 1 lists mean PVE-corrected values of K*, rCBF, and Vb calculated by Equation 1, in each of 20 ROI. In gray matter regions, K* ranged from 2.32 μL·min−1·ml−1 in the medial temporal cortex to 4.88 μL·min−1·ml−1 in the superior parietal cortex and was on average 2.6-fold higher in gray than white matter. For comparison, non-PVE-corrected K* in gray matter regions ranged from 2.07 μL·min−1·ml−1 in the medial temporal cortex to 3.18 μL·min−1·ml−1 in the superior parietal cortex (data not shown), showing a greater PVE effect (+53%) on the latter region than on the former (+12%).
TABLE 1.
Partial volume-corrected values for regional DHA incorporation coefficients K*, brain blood volume Vb, and rCBF in 20 ROI
| Regions | K* (μL·min−1.ml−1) | Vb (ml/ml) | CBF (ml min−1 100 g−1) |
|---|---|---|---|
| Orbitofrontal | 4.41 ± 0.99 | 0.082 ± 0.025 | 68.86 ± 18.00 |
| Prefrontal | 3.71 ± 0.53 | 0.072 ± 0.014 | 65.17 ± 14.70 |
| Premotor | 4.20 ± 0.62 | 0.072 ± 0.011 | 77.29 ± 22.49 |
| Anterior cingulated | 3.03 ± 0.36 | 0.070 ± 0.017 | 70.78 ± 13.74 |
| Inferior temporal | 3.97 ± 0.53 | 0.080 ± 0.018 | 71.56 ± 9.14 |
| Middle temporal | 3.90 ± 0.47 | 0.073 ± 0.011 | 65.21 ± 10.43 |
| Superior temporal | 3.75 ± 0.87 | 0.101 ± 0.023 | 55.70 ± 12.50 |
| Medial temporal | 2.32 ± 0.33 | 0.070 ± 0.012 | 46.16 ± 4.99 |
| Sensorimotor | 4.20 ± 0.62 | 0.076 ± 0.010 | 70.48 ± 15.98 |
| Inferior parietal | 4.11 ± 0.66 | 0.072 ± 0.013 | 57.81 ± 19.53 |
| Superior parietal | 4.88 ± 1.34 | 0.142 ± 0.043 | 56.23 ± 25.53 |
| Medial parietal | 3.98 ± 0.57 | 0.074 ± 0.023 | 73.17 ± 11.34 |
| Posterior cingulated | 3.65 ± 0.56 | 0.082 ± 0.025 | 75.03 ± 11.48 |
| Occipital association | 4.32 ± 0.61 | 0.073 ± 0.011 | 65.34 ± 16.86 |
| Calcarine | 4.51 ± 0.63 | 0.076 ± 0.019 | 84.34 ± 17.41 |
| Thalamus | 3.69 ± 1.26 | 0.076 ± 0.017 | 75.44 ± 32.52 |
| Striatum | 3.17 ± 1.26 | 0.052 ± 0.013 | 67.93 ± 29.32 |
| Cerebellar hemispheres | 3.47 ± 0.50 | 0.083 ± 0.031 | 60.98 ± 9.80 |
| Cerebellar vermis | 3.31 ± 0.47 | 0.046 ± 0.013 | 52.59 ± 9.84 |
| White matter | 1.45 ± 0.41 | 0.028 ± 0.005 | 22.61 ± 5.00 |
| Mean gray matter (ROI) | 3.82 ± 0.45 | 0.078 ± 0.010 | 66.32 ± 12.42 |
| Global gray matter (whole brain) | 4.04 ± 0.53 | 0.096 ± 0.014 | 66.29 ± 11.28 |
| Gray to white matter ratio | 2.63 ± 0.63 | 2.81 ± 0.40 | 2.93 ± 0.94 |
PVE-corrected rCBF ranged from 46.2 ml·min−1·100 g−1 in the medial temporal cortex to 84.3 ml·min−1·100 g−1 in the calcarine cortex and on average was 2.9-fold higher than in white matter (Table 1). PVE-corrected gray matter Vb (ml/ml) ranged from 0.046 ± 0.013 in the cerebellar vermis to 0.142 ± .043 in the superior parietal cortex, whereas white matter Vb equaled 0.028 ± 0.005. High values of Vb may reflect close proximity of the ROI to vascular structures. There was no statistically significant gender difference in Vb, rCBF, or K*.
The relation of PVE-corrected K* to PVE-corrected rCBF for the 20 ROI (19 gray matter regions and one white matter region) identified in Table 1 was determined for each of the 14 subjects. Figure 4 presents data for one such subject, which had a statistically significant Pearson correlation coefficient between K* and rCBF of 0.83 (P < 0.0001). Twelve of the 14 correlation coefficients were statistically significant; the mean value for the 14 subjects was 0.63 ± 0.23. The mean coefficient derived from the original PVE uncorrected data (10 of 14 were statistically significant) equaled 0.56 ± 0.25.
Fig. 4.

Relation in one subject between PVE-corrected incorporation coefficients for docosahexaenoic acid (K*) and rCBF after partial volume correction for both, in 19 gray matter and one white matter ROI. The Pearson correlation coefficient of the data is 0.83 (P < 0.0001). The white matter region has the lowest value for K* and CBF; the next lowest pair of values is in the medial temporal lobe ROI, which includes the hippocampus and the amygdala.
DHA incorporation rate Jin
Table 2 presents mean plasma concentrations of certain unesterified fatty acids, which are consistent with a previous report (48). Using the mean concentration of unesterified DHA, 2.63 ± 1.17 nmol/ml, the incorporation rate Jin of DHA into whole brain was calculated by Equation 2 from the global value for K* before PVE correction. For the 14 subjects, Jin equaled 2.64 ± 0.35 μL·min−1·ml−1, equivalent to 0.0093 ± 0.0044 μmol·ml−1·day−1. Taking into account whole-brain volume as determined by MR, 1231 ml, this is equivalent to a DHA incorporation rate of 3.8 ± 1.7 mg/day for the brain as a whole.
TABLE 2.
Plasma unesterified fatty acid concentrations, averaged from samples at 0, 30, and 60 min after injection of [1-11C]DHA (n = 14)
| Fatty Acids | Mean (nmol/ml) ± SD |
|---|---|
| Palmitic, 16:0 | 112.4 ± 25.0 |
| Palmitoleic, 16:1n-7 | 11.9 ± 4.53 |
| Stearic, 18:0 | 67.8 ± 12.9 |
| Oleic, 18:1n-9 | 219.4 ± 51.3 |
| Linoleic, 18:2n-6 | 97.0 ± 24.75 |
| α-Linolenic, 18:3n-3 | 6.6 ± 3.1 |
| Arachidonic, 20:4n-6 | 4.3 ± 1.0 |
| Eicosapentaenoic, 20:5n-3 | 0.3 ± 0.14 |
| Docosapentaenoic, 22:5n-3 | 0.1 ± 0.03 |
| Docosahexaenoic, 22:6n-3 | 2.63 ± 1.17 |
DISCUSSION
We used PET with intravenously injected [1-11C]DHA and an irreversible uptake model to determine regional brain incorporation coefficients K* for DHA in 14 healthy adult volunteers, in whom potentially confounding diet or medication effects on brain fatty acid metabolism were minimized. As has been shown, the calculated net incorporation rate of DHA into the brain, Jin, represents the net rate of brain DHA consumption (18, 29, 32). The calculated rate in this article for the human brain, 0.0093 ± 0.0044 μmol·ml−1·day−1, is equivalent to a DHA consumption rate of 3.8 ± 1.7 mg/day.
Dividing Jin by 5 g, the reported amount of DHA in the human brain (49), gives an estimated whole-brain DHA turnover rate (daily incorporation rate/amount) of 0.076% per day, equivalent to a half-life (0.695/turnover rate) of 911 days (2.5 years). However, since the 5 g figure was derived from limited published data, these calculations should be considered preliminary.
While a half-life of 2.5 years may seem at first very long, using this value shows that brain DHA would fall by 5% within 49 days of its disappearance from plasma. We don't know how sensitive human brain function would be to a 5% drop in brain DHA content, but if it is very sensitive, dietary n-3 PUFA deprivation for only a few months might lead to functional brain changes. For comparison, 15 weeks (105 days) of dietary n-3 PUFA deprivation in the postweaning rat produced a 30% reduction in brain DHA content and altered behavior (32, 50).
A half-life measured in years leads us to consider that any potential benefit of increasing brain DHA concentration though dietary change may not be fully manifest in clinical trials whose duration is measured in weeks. A DHA half-life in years also leads us to question the physiological basis of reported benefits within weeks of increasing dietary DHA in patients with psychiatric disorders (12). Such relatively rapid improvements, if they occur, may not depend on alterations in the total overall DHA concentration of brain membranes. Instead, the mechanism for such effects might involve a rapid peripheral action of n-3 PUFAs on cytokines which could secondarily influence neuropsychiatric functioning (51). Alternatively, an acute dietary DHA change may selectively affect the composition of newly formed neuronal membranes in discrete regions of the adult brain or influence synaptic remodeling and neurogenesis (52). In the adult human, newly formed neurons (and perhaps synapses also) may help to shape existing neural circuitry and thereby permit rapid changes in neuropsychiatric function in response to dietary change (53, 54).
When injected, [1-11C]DHA was taken up to a greater extent in gray than white matter and varied 2-fold in gray matter regions, consistent with previous studies in rats (19, 20) (Table 1). High uptake into the neocortex is consistent with evidence that incorporation of DHA occurs into phospholipids of brain synaptic membranes (21), that DHA is enriched in aminophospholipids of gray matter (55, 56), and that high levels of DHA are found in gray matter (57). Such uptake can be increased during neurotransmission or reduced by visual denervation (19, 22, 24) and also may represent active DHA metabolism during synaptogenesis and docosanoid formation.
The much lower values of K* in the medial temporal cortex are consistent with previous reports for [1-11C]ARA and with values for rCBF (35, 36). The data likely reflect the unique architecture of this region, although there is some effect of the PVE correction (e.g., +12% for medial temporal cortex compared with +53% for superior parietal cortex; see Results).
A PVE correction of data is critical when comparing individuals and populations of different ages or when examining effects of disorders such as alcoholism or Alzheimer's disease, which are accompanied by cerebral atrophy (35, 58, 59). Our PVE-corrected (and uncorrected) values for rCBF were comparable to values reported in other studies; mean gray matter PVE-uncorrected Vb (0.057 ± 0.008 ml/ml) was similar to the mean reported in a prior PET ARA study (0.055 ± 0.006 ml/ml) (35, 36, 60).
Our PVE-corrected K* in global gray matter equaled 4.04 ± 0.53 μl·min−1·ml−1, and our PVE-corrected gray to white matter ratio equaled 2.63 ± 0.63 (Table 1). These data can be compared with a prior PET study measuring K* for ARA, which reported global PVE-corrected gray matter values of 7.03 and 7.34 μl·min−1·ml in young and old subjects, respectively. Considering that the unesterified plasma ARA concentration equaled 3.8 ± 1.7 nmol·ml−1, global gray matter Jin equaled 0.025–0.039 μmol·g−1·day−1, which is double the Jin for DHA (0.0093 ± 0.0044 μmol·ml−1·day−1) (35). This suggests that the rate of brain consumption of ARA is approximately twice that of DHA.
Regional values of K* for DHA were significantly correlated with rCBF in 12 of the 14 subjects. This correlation was not determined by a dependence of K* on rCBF but by the fact that both parameters represent common aspects of regional brain metabolism. K* for DHA is thought to reflect the rate of DHA consumption by brain, in relation to the role of DHA in signaling and to its metabolism to docosanoids and other products, including reactive oxygen species (21, 22, 61–63). rCBF, which is coupled to brain glucose consumption (64), is thought to reflect any of a number of energy-demanding metabolic processes, including synaptic activation and reincorporation of unesterified DHA into membrane phospholipid (65, 66).
K* for fatty acids has been shown to be independent of changes in rCBF during functional activation or hypercapnia in rats and in patients with Alzheimer's disease (16, 22, 67). The basis for this independence is the ability of bound unesterified fatty acids to rapidly and reversibly dissociate from plasma albumin as blood passes through the brain, resulting in a fractional incorporation that is inverse to circulation time (14, 68).
A model including one tissue compartment, Vb (blood volume), and K* was used to analyze the time activity curve. The model assumes that aqueous metabolites are not biologically important in the analysis. This and other assumptions underlying Equation 1 are approximately satisfied in our model. Studies at 5 min after intravenous injection of [1-14C]DHA or [1-14C]ARA in unanesthetized rats showed that ∼90% of brain radioactivity is incorporated by then in stable brain lipids, mainly phospholipids, and that the remainder is in the aqueous brain pool (23, 69). In rats and mice injected with radiolabeled DHA and ARA, after a short time, some aqueous plasma metabolites do appear and could potentially contribute to brain radioactivity. Directly injecting the aqueous pool and other studies show that its contribution to brain radioactivity is minimal (it may account for some of the brain aqueous radioactivity) (70). In our studies, ∼5% of plasma radioactivity 10 min after intravenous [1-11C]DHA injection was in the aqueous plasma pool. Furthermore, since brain radioactivity does not increase beyond 5 min after radiotracer PUFA injection in either animals or humans (36, 70), any aqueous contribution to brain radioactivity beyond this time is biologically unimportant.
The HPLC procedure that we performed on a subsample of four subjects allowed us to verify the accuracy of the rapid solid phase extraction procedure that is used to compute the [1-11C]DHA input function. Using the HPLC procedure at the 10 min time point, we found that all but ∼10% of the radioactivity in the isolated fatty acid fraction was DHA. It was not feasible to perform the HPLC procedure later in the scan because the signal from the [1-11C]DHA was too low for detection. However, the fact that most (∼60%) of the [1-11C]DHA availability occurs within the first 10 minutes limits the magnitude of any resulting error. Therefore, we consider 10–20% to be the upper limit to any overestimation of the input function (and therefore overestimation of K*).
In summary, with the help of PET and intravenously injected [1-11C]DHA, this study demonstrates that circulating unesterified DHA is taken up by the human brain at a global rate in healthy subjects of 3.8 ± 1.7 mg/day. Our PET method provides a functional image of regional incorporation of circulating unesterified DHA into brain and could be used in the future to investigate effects of age, gender, disease, genetic variation, traumatic injury, and nutritional status on brain DHA utilization. The method also could be used to examine the regional brain DHA consumption when activated by physiological or pharmacological stimulation (22), using two scans with two injections of [1-11C]DHA, as has been done with [1-11C]ARA (37).
Acknowledgments
The authors thank David Ted George and Giampiero Giovacchini for helpful discussions and Jane Bell, Margaret Der, Christopher Geyer, Debby Hill, Cheryl Jones, Monte Phillips, Merel Schollnberger, Nancy Sebring, and the staff of the National Institutes of Health PET Department for technical assistance. This study used the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health (http://biowulf.nih.gov).
Abbreviations
ARA, arachidonic acid
DHA, docosahexaenoic acid
MR, magnetic resonance
PET, positron emission tomography
PLA2, phospholipase A2
PVE, partial volume error
rCBF, regional cerebral blood flow
ROI, regions of interest
APPENDIX
Under a protocol approved by the Institutional Animal Care and Use Committee of the National Institutes of Health Clinical Center, whole-body PET scans were performed in three isofluorane-anesthetized Rhesus monkeys, weighing 9.0–10.9 kg, to estimate organ biodistribution and human radiation absorbed doses of intravenously injected [1-11C]DHA (36). Scans were performed in the two-dimensional mode using a General Electric Advance Tomograph (Waukesha, WI). A 2-h dynamic scan sequence was collected following intravenous injection of 1,480–1,887 MBq (40–51 mCi) [1-11C]DHA. ROI were drawn over organs on contiguous slices on the images summed over time.
Time activity curves (not corrected for radioactive decay) from the ROI for each organ were averaged, and the average curves extrapolated by fitting the tail to a monoexponential. The curve for each organ was integrated to infinity, and the result multiplied by organ volume and divided by injected dose to calculate the organ residence time that was then scaled to reference man. For the remainder of the body, residence time was obtained by subtracting the sum of organ residence times from the maximum residence time for a 11C-labeled tracer, 29.4 min. Radiation absorbed doses were calculated from the average residence times with MIRDOSE 3.1 (71) using tissue weighting and other relevant assumptions from the International Commission on Radiological Protection Publications 23 and 80 as well as other sources (72–74).
Radioactivity was estimated with PET in the liver, heart wall, kidney, lungs, spleen, skeletal muscle, brain, testes, and urinary bladder contents of each of the three monkeys. High activity levels were rapidly achieved in the liver without apparent washout of activity over 2 h (Appendix Fig. A1). There was no hepatobiliary excretion of tracer, and tracer elimination via the urine was modest. Appendix Table 1 shows that the highest human absorbed radiation doses (mrad/mCi) are to liver (156), heart wall (50), kidney (37), gall bladder wall (25), spleen (24), and lungs (23), with an effective dose of 17 mrem/mCi. Under Radioactive Drug Research Committee regulations (maximum dose of 5 rads to the critical organ per administration, 15 rads per 12 months), human research subjects may receive up to 1184 MBq (32 mCi) of [1-11C]DHA per study. Our study involved a maximum exposure of 4.68 rem to the liver as the critical organ.
Appendix Fig. A1.

Maximum intensity projection of transverse slices of the entire monkey body, of [1-11C]DHA images summed from 0–6 min. The viewer is looking at the front of the animal, with the animal's right to the viewer's left. There was very rapid, prominent appearance of activity in the liver. Scanning started at the beginning of tracer injection into the left femoral vein; it and the inferior vena cava are visualized. Extrapolating monkey data to humans, only ∼0.5% of the injected [1-11C]DHA goes into the brain.
Appendix Table 1.
Dosimetry tabulation for [1-11C]DHA
| Organ | Dose (rad per mCi) |
|---|---|
| Adrenals | 1.61E-02 |
| Brain | 2.87E-03 |
| Breasts | 6.22E-03 |
| Esophagus | 7.50E-03 |
| Gallbladder wall | 2.50E-02 |
| Lower large intestine | 5.27E-03 |
| Small intestine | 7.77E-03 |
| Stomach | 9.10E-03 |
| Upper large intestine | 9.29E-03 |
| Colon | 7.56E-03 |
| Heart wall | 5.04E-02 |
| Kidney | 3.69E-02 |
| Liver | 1.56E-01 |
| Lungs | 2.30E-02 |
| Muscle | 7.33E-03 |
| Ovaries | 6.03E-03 |
| Pancreas | 1.47E-02 |
| Red marrow | 7.09E-03 |
| Bone surfaces | 6.29E-03 |
| Skin | 4.56E-03 |
| Spleen | 2.37E-02 |
| Testes | 5.81E-03 |
| Thymus | 7.50E-03 |
| Thyroid | 4.74E-03 |
| Urinary bladder wall | 6.72E-03 |
| Uterus | 5.93E-03 |
| Effective dose | 1.7E-02 |
Thymus dose is used for Esophagus (as per ICRP 80); colon dose = 0.57 ULI + 0.43 LLI (as per ICRP 80).
This research was supported by the Intramural Research Programs of the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism, and National Institute on Aging.
Published, JLR Papers in Press, December 26, 2008.
References
- 1.Vaidyanathan V. V., K. V. Rao, and P. S. Sastry. 1994. Regulation of diacylglycerol kinase in rat brain membranes by docosahexaenoic acid. Neurosci. Lett. 179 171–174. [DOI] [PubMed] [Google Scholar]
- 2.Poling J. S., J. W. Karanian, N. Salem, Jr., and S. Vicini. 1995. Time- and voltage-dependent block of delayed rectifier potassium channels by docosahexaenoic acid. Mol. Pharmacol. 47 381–390. [PubMed] [Google Scholar]
- 3.Delion S., S. Chalon, D. Guilloteau, J. C. Besnard, and G. Durand. 1996. alpha-Linolenic acid dietary deficiency alters age-related changes of dopaminergic and serotoninergic neurotransmission in the rat frontal cortex. J. Neurochem. 66 1582–1591. [DOI] [PubMed] [Google Scholar]
- 4.Litman B. J., and D. C. Mitchell. 1996. A role for phospholipid polyunsaturation in modulating membrane protein function. Lipids. 31 (Suppl): S193–S197. [DOI] [PubMed] [Google Scholar]
- 5.Chalon S., S. Delion-Vancassel, C. Belzung, D. Guilloteau, A. M. Leguisquet, J. C. Besnard, and G. Durand. 1998. Dietary fish oil affects monoaminergic neurotransmission and behavior in rats. J. Nutr. 128 2512–2519. [DOI] [PubMed] [Google Scholar]
- 6.Green J. T., S. K. Orr, and R. P. Bazinet. 2008. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. J. Lipid Res. 49 939–944. [DOI] [PubMed] [Google Scholar]
- 7.Lukiw W. J., J. G. Cui, V. L. Marcheselli, M. Bodker, A. Botkjaer, K. Gotlinger, C. N. Serhan, and N. G. Bazan. 2005. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 115 2774–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Contreras M. A., R. S. Greiner, M. C. Chang, C. S. Myers, N. Salem, Jr., and S. I. Rapoport. 2000. Nutritional deprivation of alpha-linolenic acid decreases but does not abolish turnover and availability of unacylated docosahexaenoic acid and docosahexaenoyl-CoA in rat brain. J. Neurochem. 75 2392–2400. [DOI] [PubMed] [Google Scholar]
- 9.Strokin M., M. Sergeeva, and G. Reiser. 2003. Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and Ca2+. Br. J. Pharmacol. 139 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Innis S. M., and R. W. Friesen. 2008. Essential n-3 fatty acids in pregnant women and early visual acuity maturation in term infants. Am. J. Clin. Nutr. 87 548–557. [DOI] [PubMed] [Google Scholar]
- 11.Florent-Bechard S., C. Malaplate-Armand, V. Koziel, B. Kriem, J. L. Olivier, T. Pillot, and T. Oster. 2007. Towards a nutritional approach for prevention of Alzheimer's disease: biochemical and cellular aspects. J. Neurol. Sci. 262 27–36. [DOI] [PubMed] [Google Scholar]
- 12.Freeman M. P., J. R. Hibbeln, K. L. Wisner, J. M. Davis, D. Mischoulon, M. Peet, P. E. Keck, Jr., L. B. Marangell, A. J. Richardson, J. Lake, et al. 2006. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J. Clin. Psychiatry. 67 1954–1967. [DOI] [PubMed] [Google Scholar]
- 13.Murphy E. J., T. A. Rosenberger, C. B. Patrick, and S. I. Rapoport. 2000. Intravenously injected [1-14C]arachidonic acid targets phospholipids, and [1-14C]palmitic acid targets neutral lipids in hearts of awake rats. Lipids. 35 891–898. [DOI] [PubMed] [Google Scholar]
- 14.Robinson P. J., J. Noronha, J. J. DeGeorge, L. M. Freed, T. Nariai, and S. I. Rapoport. 1992. A quantitative method for measuring regional in vivo fatty-acid incorporation into and turnover within brain phospholipids: review and critical analysis. Brain Res. Brain Res. Rev. 17 187–214. [DOI] [PubMed] [Google Scholar]
- 15.Rapoport S. I., D. Purdon, H. U. Shetty, E. Grange, Q. Smith, C. Jones, and M. C. J. Chang. 1997. In vivo imaging of fatty acid incorporation into brain to examine signal transduction and neuroplasticity involving phospholipids. Ann. N. Y. Acad. Sci. 820 56–74. [DOI] [PubMed] [Google Scholar]
- 16.Chang M. C., T. Arai, L. M. Freed, S. Wakabayashi, M. A. Channing, B. B. Dunn, M. G. Der, J. M. Bell, T. Sasaki, P. Herscovitch, et al. 1997. Brain incorporation of [1-11C]-arachidonate in normocapnic and hypercapnic monkeys, measured with positron emission tomography. Brain Res. 755 74–83. [DOI] [PubMed] [Google Scholar]
- 17.Rapoport S. I., M. C. Chang, K. Connolly, D. Kessler, A. Bokde, R. E. Carson, P. Herscovitch, M. Channing, and W. C. Eckelman. 2000. In vivo imaging of phospholipase A2-mediated signaling in human brain using [11C]arachidonic acid and positron emission tomography (PET). J. Neurochem. 74 S21. [Google Scholar]
- 18.Rapoport S. I., M. C. Chang, and A. A. Spector. 2001. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J. Lipid Res. 42 678–685. [PubMed] [Google Scholar]
- 19.Wakabayashi S., L. M. Freed, J. M. Bell, and S. I. Rapoport. 1994. In vivo cerebral incorporation of radiolabeled fatty acids after acute unilateral orbital enucleation in adult hooded Long-Evans rats. J. Cereb. Blood Flow Metab. 14 312–323. [DOI] [PubMed] [Google Scholar]
- 20.Wakabayashi S., L. M. Freed, M. C. J. Chang, and S. I. Rapoport. 1995. In vivo imaging of brain incorporation of fatty acids and of 2-deoxy-D-glucose demonstrates functional and structural neuroplastic effects of chronic unilateral visual deprivation in rats. Brain Res. 679 110–122. [DOI] [PubMed] [Google Scholar]
- 21.Jones C. R., T. Arai, and S. I. Rapoport. 1997. Evidence for the involvement of docosahexaenoic acid in cholinergic stimulated signal transduction at the synapse. Neurochem. Res. 22 663–670. [DOI] [PubMed] [Google Scholar]
- 22.DeGeorge J. J., T. Nariai, S. Yamazaki, W. M. Williams, and S. I. Rapoport. 1991. Arecoline-stimulated brain incorporation of intravenously administered fatty acids in unanesthetized rats. J. Neurochem. 56 352–355. [DOI] [PubMed] [Google Scholar]
- 23.Nariai T., J. J. DeGeorge, N. H. Greig, S. Genka, S. I. Rapoport, and A. D. Purdon. 1994. Differences in rates of incorporation of intravenously injected radiolabeled fatty acids into phospholipids of intracerebrally implanted tumor and brain in awake rats. Clin. Exp. Metastasis. 12 213–225. [DOI] [PubMed] [Google Scholar]
- 24.Jones C. R., T. Arai, J. M. Bell, and S. I. Rapoport. 1996. Preferential in vivo incorporation of [3H]arachidonic acid from blood into rat brain synaptosomal fractions before and after cholinergic stimulation. J. Neurochem. 67 822–829. [DOI] [PubMed] [Google Scholar]
- 25.Grange E., O. Rabin, J. Bell, and M. C. Chang. 1998. Manoalide, a phospholipase A2 inhibitor, inhibits arachidonate incorporation and turnover in brain phospholipids of the awake rat. Neurochem. Res. 23 1251–1257. [DOI] [PubMed] [Google Scholar]
- 26.Chen C. T., D. W. Ma, J. H. Kim, H. T. Mount, and R. P. Bazinet. 2008. The low density lipoprotein receptor is not necessary for maintaining mouse brain polyunsaturated fatty acid concentrations. J. Lipid Res. 49 147–152. [DOI] [PubMed] [Google Scholar]
- 27.Purdon D., T. Arai, and S. I. Rapoport. 1997. No evidence for direct incorporation of esterified palmitic acid from plasma into brain lipids of awake adult rat. J. Lipid Res. 38 526–530. [PubMed] [Google Scholar]
- 28.DeMar J. C., Jr., H. J. Lee, K. Ma, L. Chang, J. M. Bell, S. I. Rapoport, and R. P. Bazinet. 2006. Brain elongation of linoleic acid is a negligible source of the arachidonate in brain phospholipids of adult rats. Biochim. Biophys. Acta. 1761 1050–1059. [DOI] [PubMed] [Google Scholar]
- 29.Demar J. C., Jr., K. Ma, L. Chang, J. M. Bell, and S. I. Rapoport. 2005. alpha-Linolenic acid does not contribute appreciably to docosahexaenoic acid within brain phospholipids of adult rats fed a diet enriched in docosahexaenoic acid. J. Neurochem. 94 1063–1076. [DOI] [PubMed] [Google Scholar]
- 30.Lefkowitz W., S. Y. Lim, Y. Lin, and N. Salem, Jr. 2005. Where does the developing brain obtain its docosahexaenoic acid? Relative contributions of dietary alpha-linolenic acid, docosahexaenoic acid, and body stores in the developing rat. Pediatr. Res. 57 157–165. [DOI] [PubMed] [Google Scholar]
- 31.Menard C. R., K. J. Goodman, T. N. Corso, J. T. Brenna, and S. C. Cunnane. 1998. Recycling of carbon into lipids synthesized de novo is a quantitatively important pathway of alpha-[U-13C]linolenate utilization in the developing rat brain. J. Neurochem. 71 2151–2158. [DOI] [PubMed] [Google Scholar]
- 32.DeMar J. C., Jr., K. Ma, J. M. Bell, and S. I. Rapoport. 2004. Half-lives of docosahexaenoic acid in rat brain phospholipids are prolonged by 15 weeks of nutritional deprivation of n-3 polyunsaturated fatty acids. J. Neurochem. 91 1125–1137. [DOI] [PubMed] [Google Scholar]
- 33.Channing M. A., L. Freed, S. Wakabayashi, R. Carson, N. Simpson, B. B. Dunn, and S. I. Rapoport. 1992. [1-11C]Labeled polyhomoallyilic fatty acids: phospholipid metabolic tracers for the brain. Am. J. Nucl. Med. 13 1093. [Google Scholar]
- 34.Channing M. A., and N. Simpson. 1993. Radiosynthesis of 1-[11C]polyhomoallylic fatty acids. J. Labeled Compounds Radiopharmacol. 33 541–546. [Google Scholar]
- 35.Giovacchini G., A. Lerner, M. T. Toczek, C. Fraser, K. Ma, J. C. DeMar, P. Herscovitch, W. C. Eckelman, S. I. Rapoport, and R. E. Carson. 2004. Brain incorporation of 11C-arachidonic acid, blood volume, and blood flow in healthy aging: a study with partial-volume correction. J. Nucl. Med. 45 1471–1479. [PubMed] [Google Scholar]
- 36.Giovacchini G., M. C. Chang, M. A. Channing, M. Toczek, A. Mason, A. L. Bokde, C. Connolly, B. K. Vuong, Y. Ma, M. G. Der, et al. 2002. Brain incorporation of [11C]arachidonic acid in young healthy humans measured with positron emission tomography. J. Cereb. Blood Flow Metab. 22 1453–1462. [DOI] [PubMed] [Google Scholar]
- 37.Esposito G., G. Giovacchini, M. Der, J. S. Liow, A. K. Bhattacharjee, K. Ma, P. Herscovitch, M. Channing, W. C. Eckelman, M. Hallett, et al. 2007. Imaging signal transduction via arachidonic acid in the human brain during visual stimulation, by means of positron emission tomography. Neuroimage. 34 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herscovitch P., J. Markham, and M. E. Raichle. 1983. Brain blood flow measured with intravenous H2(15)O. I. Theory and error analysis. J. Nucl. Med. 24 782–789. [PubMed] [Google Scholar]
- 39.Bligh E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37 911–917. [DOI] [PubMed] [Google Scholar]
- 40.Agren J. J., A. Julkunen, and I. Penttila. 1992. Rapid separation of serum lipids for fatty acid analysis by a single aminopropyl column. J. Lipid Res. 33 1871–1876. [PubMed] [Google Scholar]
- 41.Aveldano M. I., M. VanRollins, and L. A. Horrocks. 1983. Separation and quantitation of free fatty acids and fatty acid methyl esters by reverse phase high pressure liquid chromatography. J. Lipid Res. 24 83–93. [PubMed] [Google Scholar]
- 42.Folch J., M. Lees, and G. H. Sloane Stanley. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226 497–509. [PubMed] [Google Scholar]
- 43.Skipski V. P., J. J. Good, M. Barclay, and R. B. Reggio. 1968. Quantitative analysis of simple lipid classes by thin-layer chromatography. Biochim. Biophys. Acta. 152 10–19. [DOI] [PubMed] [Google Scholar]
- 44.Makrides M., M. A. Neumann, R. W. Byard, K. Simmer, and R. A. Gibson. 1994. Fatty acid composition of brain, retina, and erythrocytes in breast- and formula-fed infants. Am. J. Clin. Nutr. 60 189–194. [DOI] [PubMed] [Google Scholar]
- 45.Jenkinson M., and S. Smith. 2001. A global optimisation method for robust affine registration of brain images. Med. Image Anal. 5 143–156. [DOI] [PubMed] [Google Scholar]
- 46.Smith S. M. 2002. Fast robust automated brain extraction. Hum. Brain Mapp. 17 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pham D. L., and J. L. Prince. 1999. Adaptive fuzzy segmentation of magnetic resonance images. IEEE Trans. Med. Imaging. 18 737–752. [DOI] [PubMed] [Google Scholar]
- 48.Kargas G., T. Rudy, T. Spennetta, K. Takayama, N. Querishi, and E. Shrago. 1990. Separation and quantitation of long-chain free fatty acids in human serum by high-performance liquid chromatography. J. Chromatogr. 526 331–340. [DOI] [PubMed] [Google Scholar]
- 49.Martinez M. 1992. Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders. Brain Res. 583 171–182. [DOI] [PubMed] [Google Scholar]
- 50.Demar J. C., Jr., K. Ma, J. M. Bell, M. Igarashi, D. Greenstein, and S. I. Rapoport. 2006. One generation of n-3 polyunsaturated fatty acid deprivation increases depression and aggression test scores in rats. J. Lipid Res. 47 172–180. [DOI] [PubMed] [Google Scholar]
- 51.Maes M., and R. S. Smith. 1998. Fatty acids, cytokines, and major depression. Biol. Psychiatry. 43 313–314. [DOI] [PubMed] [Google Scholar]
- 52.Beltz B. S., M. F. Tlusty, J. L. Benton, and D. C. Sandeman. 2007. Omega-3 fatty acids upregulate adult neurogenesis. Neurosci. Lett. 415 154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ge S., K. A. Sailor, G. L. Ming, and H. Song. 2008. Synaptic integration and plasticity of new neurons in the adult hippocampus. J. Physiol. 586 3759–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banasr M., and R. S. Duman. 2007. Regulation of neurogenesis and gliogenesis by stress and antidepressant treatment. CNS Neurol. Disord. Drug Targets. 6 311–320. [DOI] [PubMed] [Google Scholar]
- 55.Salem, N. J. 1989. Omega-3 fatty acids: molecular and biochemical aspects. In New Protective Roles of Selected Nutrients in Human Nutrition. G. Spiller and J. Scala, editors. Alan R. Liss, New York. 109–228.
- 56.Salem N., Jr., P. Serpentino, J. S. Puskin, and L. G. Abood. 1980. Preparation and spectroscopic characterization of molecular species of brain phosphatidylserines. Chem. Phys. Lipids. 27 289–304. [DOI] [PubMed] [Google Scholar]
- 57.Diau G. Y., A. T. Hsieh, E. A. Sarkadi-Nagy, V. Wijendran, P. W. Nathanielsz, and J. T. Brenna. 2005. The influence of long chain polyunsaturate supplementation on docosahexaenoic acid and arachidonic acid in baboon neonate central nervous system. BMC Med. 3 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ibanez V., P. Pietrini, G. E. Alexander, P. Millet, A. L. W. Bokde, D. Teichberg, M. B. Schapiro, B. Horwitz, and S. I. Rapoport. 2001. Different patterns of age-related metabolic brain changes during healthy aging and AD, using atrophy correction. Neurology. 56 (Suppl.): A373. [Google Scholar]
- 59.Ibanez V., P. Pietrini, M. L. Furey, G. E. Alexander, P. Millet, A. L. Bokde, D. Teichberg, M. B. Schapiro, B. Horwitz, and S. I. Rapoport. 2004. Resting state brain glucose metabolism is not reduced in normotensive healthy men during aging, after correction for brain atrophy. Brain Res. Bull. 63 147–154. [DOI] [PubMed] [Google Scholar]
- 60.Ito H., I. Yokoyama, H. Iida, T. Kinoshita, J. Hatazawa, E. Shimosegawa, T. Okudera, and I. Kanno. 2000. Regional differences in cerebral vascular response to PaCO2 changes in humans measured by positron emission tomography. J. Cereb. Blood Flow Metab. 20 1264–1270. [DOI] [PubMed] [Google Scholar]
- 61.Serhan C. N. 2005. Novel eicosanoid and docosanoid mediators: resolvins, docosatrienes, and neuroprotectins. Curr. Opin. Clin. Nutr. Metab. Care. 8 115–121. [DOI] [PubMed] [Google Scholar]
- 62.Bazan N. G. 2006. The onset of brain injury and neurodegeneration triggers the synthesis of docosanoid neuroprotective signaling. Cell. Mol. Neurobiol. 26 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salem N., Jr., B. Litman, H. Y. Kim, and K. Gawrisch. 2001. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 36 945–959. [DOI] [PubMed] [Google Scholar]
- 64.Reivich M. 1974. Blood flow metabolism couple in brain. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 53 125–140. [PubMed] [Google Scholar]
- 65.Purdon A. D., and S. I. Rapoport. 1998. Energy requirements for two aspects of phospholipid metabolism in mammalian brain. Biochem. J. 335 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sokoloff L. 1999. Energetics of functional activation in neural tissues. Neurochem. Res. 24 321–329. [DOI] [PubMed] [Google Scholar]
- 67.Esposito G., G. Giovacchini, J. S. Liow, A. K. Bhattacharjee, D. Greenstein, M. Schapiro, M. Hallett, P. Herscovitch, W. C. Eckelman, R. E. Carson, et al. 2008. Imaging neuroinflammation in Alzheimer's disease with radiolabeled arachidonic acid and PET. J. Nucl. Med. 49 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson P. J., and S. I. Rapoport. 1986. Kinetics of protein binding determine rates of uptake of drugs by brain. Am. J. Physiol. 251 R1212–R1220. [DOI] [PubMed] [Google Scholar]
- 69.Washizaki K., Q. R. Smith, S. I. Rapoport, and A. D. Purdon. 1994. Brain arachidonic acid incorporation and precursor pool specific activity during intravenous infusion of unesterified [3H]arachidonate in the anesthetized rat. J. Neurochem. 63 727–736. [DOI] [PubMed] [Google Scholar]
- 70.DeGeorge J. J., J. G. Noronha, J. M. Bell, P. Robinson, and S. I. Rapoport. 1989. Intravenous injection of [1-14C]arachidonate to examine regional brain lipid metabolism in unanesthetized rats. J. Neurosci. Res. 24 413–423. [DOI] [PubMed] [Google Scholar]
- 71.Stabin M. G. 1996. MIRDOSE: personal computer software for internal dose assessment in nuclear medicine. J. Nucl. Med. 37 538–546. [PubMed] [Google Scholar]
- 72.ICRP. 1975. Report of Task Group on Reference Man, No. 23. Pergamon, Oxford, UK.
- 73.Kirschner A. S., R. D. Ice, and W. H. Beirwaltes. 1975. Radiation dosimetry of 131I–19-iodocholesterol: the pitfalls of using tissue concentration data (Authors' reply). J. Nucl. Med. 16 248–249. [Google Scholar]
- 74.Roedler, H. D. 1980. Accuracy of internal dose calculations with specific consideration of radiopharmaceutical kinetics. In Third International Radiopharmaceutical Symposium. E. E. Watson, A. T. Schlafke-Stelson, J. L. Coffey, and R. J. Cloutier, editors. FDA, Oak Ridge.